

Summary
In CBA/J mice, susceptibility to Mycobacterium tuberculosis (M.tb) is associated with low interferon-gamma (IFN-γ) responses to antigens (Antigen 85 (Ag85) and Early Secreted Antigenic Target-6 (ESAT-6)) that have been defined as immunodominant. Here, we asked whether the failure of CBA/J mice to recognize Ag85 is a consequence of M.tb infection or whether CBA/J mice have a general defect in generating specific T cell responses to this protein antigen. We compared CBA/J mice during primary M.tb infection, Ag85 vaccination followed by M.tb challenge, or M.tb memory immune mice for their capacity to generate Ag85-specific IFN-γ responses and to control M.tb infection. CBA/J mice did not respond efficiently to Ag85 in the context of natural infection or re-infection. In contrast, CBA/J mice could generate Ag85-specific IFN-γ responses and protective immunity when this antigen was delivered as a soluble protein. Our data indicate that although M.tb infection of CBA/J mice does not drive an Ag85 response, they can fully and protectively respond to Ag85 if it is delivered as a vaccine. The data from this experimental model suggest that the Ag85-containing vaccines in clinical trials should protect M.tb susceptible humans.
Keywords: Tuberculosis, Antigen 85, vaccination, CBA/J, murine
1. Introduction
Up to one-third of the world’s population may be infected with the bacterium Mycobacterium tuberculosis (M.tb), of which a small fraction develop pulmonary tuberculosis disease (TB) [1]. The underlying mechanisms of susceptibility to M.tb and TB disease progression in most adult human patients are poorly understood [2]. It is possible that subtle differences in the responses to immunodominant antigens may predispose the host to M.tb susceptibility and TB disease progression. Here, we focus on immune responses to Antigen 85 (Ag85) using a mouse model of M.tb aerosol infection.
Ag85 is detectable in TB patients [3] and is an immunodominant antigen recognized by TB patients [3, 4]. Contacts of TB patients also respond variably to Ag85 [5] but the cause(s) or significance of the variation are unknown. There is evidence that TB patients [4] do not respond as strongly to Ag85 as do individuals who control M.tb infection. Together, these data show variability in human immune responses to Ag85 and associate weak Ag85 immune responses with M.tb susceptibility and TB disease. It is not known whether poor Ag85-specific IFN-γ responses cause TB disease in humans, or whether poor Ag85-specific responses are a marker of increased risk for TB disease. Studies in mice suggest the latter [6, 7].
Responses to Ag85 are important to understand because it is a vaccine component in human clinical trials [8]. The efficacy results from human Ag85-containing vaccine clinical trials are not available, and whether M.tb susceptible humans respond efficiently to Ag85 vaccination and M.tb challenge is unknown. We address these questions experimentally to determine whether the context of Ag85 exposure (natural infection/re-infection versus soluble protein) affects Ag85-specific IFN-γ responses and protective immunity.
We use a low-dose aerosol infection of M.tb susceptible CBA/J mice [9, 10] to model M.tb susceptible humans. We have shown that CBA/J mice have low IFN-γ responses to other M.tb antigens as compared to C57BL/6 mice [11] and to Ag85 during early M.tb infection [7]. Additional M.tb susceptible C3H substrains ([6], our unpublished observations) and DBA/2 mice (our unpublished observations) also respond poorly to Ag85 as determined by IFN-γ production during primary M.tb infection. Here, we extend our previous findings to show that CBA/J mice have low Ag85-specific IFN-γ responses throughout early and chronic M.tb infection and to M.tb re-infection. Given the very low IFN-γ responses to Ag85 during infection, we questioned whether CBA/J mice were capable of recognizing and responding to Ag85. Delivery of Ag85 as a soluble antigen (vaccine plus adjuvant format) successfully generated Ag85-specific IFN-γ responses and protected CBA/J mice against M.tb to the same degree as C57BL/6 mice. These results indicate that although CBA/J mice can respond to purified Ag85 protein, they do not respond well to Ag85 in its native form in vivo.
Overall, our results suggest that CBA/J mice may model M.tb susceptible humans who do not respond strongly to Ag85 [4]. A low Ag85-specific response during natural M.tb infection, however, does not correspond to a failure to respond to Ag85 vaccination during challenge infection. Therefore, we predict that the Ag85-containing vaccines in clinical trials will afford some protection for M.tb susceptible humans.
2. Results
Ag85-specific IFN-γ production from lung cells and blood of M.tb-infected mice
To determine whether CBA/J mice generated Ag85-specific IFN-γ responses during primary M.tb infection, we cultured lung and blood cells with M.tb Ag85, concanavalin-A (Con-A) or ovalbumin (not shown), and compared the responses to C57BL/6 mice, which are resistant to M.tb [9, 10] and respond vigorously to Ag85 [6, 7]. Similar to M.tb Culture Filtrate Protein (CFP) and ESAT-6 [11], lung cells from CBA/J mice produced significantly less Ag85-specific IFN-γ than did lung cells from C57BL/6 mice (Figure 1A). In both mouse strains, peak Ag85-specific IFN-γ production occurred at day 21 of M.tb infection (Figure 1A), which corresponds to Ag85 gene expression [12], peak Ag85-specific T cell numbers [12] and stabilization of M.tb growth [11, 12]. CBA/J mice harbor elevated M.tb burdens [11] and their lungs contain more Ag85 protein (Supplemental Figure 1) than C57BL/6 mice. It seems unlikely, therefore, that the low Ag85-specific IFN-γ responses in CBA/J mice result from limited antigen availability due to inadequate amounts of Ag85 in vivo. PBMCs from CBA/J mice also produced less Ag85-specific IFN-γ than did C57BL/6 PBMCs, but there was no peak of Ag85-specific IFN-γ from CBA/J PBMCs (Figure 1B). Our positive control, the potent T-cell activator Con-A, showed that cells from CBA/J mice were not inherently defective in IFN-γ production (Figure 1C and Figure 1D).
Figure 1. Antigen 85-specific IFN-γ responses in mice.
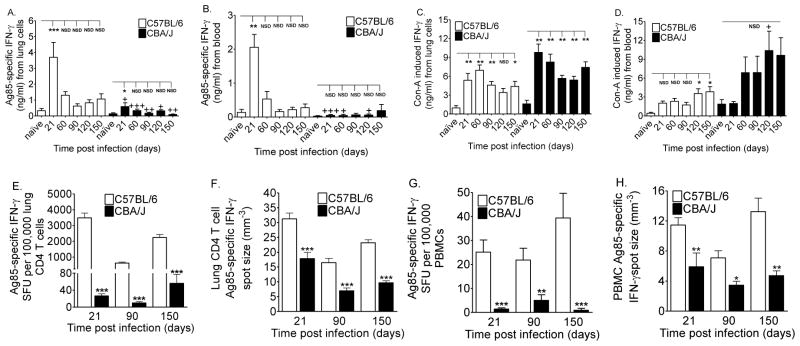
C57BL/6 and CBA/J mice were infected with 50–100 CFU of M.tb. 2 × 105 lung cells (A,C) and 1:10 diluted whole blood (B,D) were cultured with Ag85 (A, B) or concanavalin-A (C, D) for 72 hours. IFN-γ in the supernatants was quantified by ELISA. Serial dilutions of purified (93.7 ± 1.7%) lung CD4 T cells (E, F) and PBMCs (G, H) were cultured with Ag85-pulsed bone marrow-derived APCs for 36 hours on anti-IFN-γ coated ELISPOT plates to quantify the frequency (E, G) of responding cells and the amount (F, H) of IFN-γ produced by each responding cell. Results (A, B, C, D) are the average ± SEM of 4 independent experiments. Each experiment had 4 to 5 mice per strain per time point, total n = 16 to 20 mice per group. Data were analyzed by mouse strain with comparison to non-infected controls by one-way ANOVA with Dunnett’s posttest, *p<0.05; **p<0.01, ***p<0.001, NSD = no significant difference. Pair wise comparisons between C57BL/6 and CBA/J mice at each time point were analyzed by Student’s t-tests, +p<0.05, ++p<0.01, +++p<0.001. Results (E, F, G, H) are the average ± SEM of 3 (E, G) or 2 (F, H) independent experiments. Each experiment had 5–6 mice per strain per time point, total n = 15 to 18 mice per group (A, C) or total n = 10 to 12 mice per group (B, D). Data were analyzed by one-way ANOVA with Tukey’s posttest, *p<0.05; **p<0.01, ***p<0.001.
Overall, CBA/J mice had poor Ag85-specific IFN-γ responses throughout early and chronic asymptomatic primary M.tb infection. The same is true for other M.tb antigens due to low frequencies of responding T cells [11]. We therefore investigated the numbers and potency of Ag85-specific IFN-γ producing cells from CBA/J mice.
Numbers and potency of Ag85-specific IFN-γ producing cells from M.tb-infected mice
ELISPOTs were used to determine whether low Ag85-specific IFN-γ production by CBA/J mice reflected a reduced frequency of responding cells or reduced secretion by individual cells. The lungs and blood of CBA/J mice contained significantly lower frequencies (Figures 1E and 1G) and absolute numbers (data not shown) of Ag85-specific IFN-γ secreting cells than did M.tb resistant C57BL/6 mice. The individual Ag85-specific IFN-γ secreting cells from CBA/J mice also produced significantly less than the cells from C57BL/6 mice (Figure 1F and 1H). Together, these results identified two reasons for poor Ag85-specific IFN-γ responsiveness in CBA/J mice: a 50–100 fold reduction in the frequency of cells and a 2-fold reduction in Ag85-specific IFN-γ output at the single-cell level. The mechanisms underlying poor Ag85-specific IFN-γ responses in CBA/J mice are unknown, but likely involve H-2k haplotype-dependent and -independent effects [7].
We next asked whether CBA/J mice could respond to Ag85 by delivering exogenous Ag85 as a pre-exposure vaccine and whether CBA/J mice could generate Ag85-specific IFN-γ memory recall responses as a result of prior exposure to M.tb.
Exogenous delivery of Ag85 induced IFN-γ responses in CBA/J mice
We delivered Ag85 as a soluble protein with an adjuvant (Adj) to determine whether Ag85 could be recognized by M.tb challenged CBA/J mice in vivo. We hypothesized that the poor capacity of CBA/J mice to respond to Ag85 during primary infection would impair their ability to respond to Ag85 as a soluble protein when challenged with M.tb.
CBA/J mice were vaccinated with PBS, Adj, Ag85 plus Adj, or M. bovis BCG and challenged with M.tb by aerosol at least 10 weeks after the first injection. Mice were euthanized 28 days later. Contrary to our expectations, but similar to M.tb resistant mice [13, 14], vaccination increased Ag85-specific IFN-γ responses in M.tb challenged CBA/J mice. In the lungs, the frequency of Ag85-specific IFN-γ secreting cells in Ag85 vaccinated CBA/J mice was significantly higher than all other groups (Figure 2A). Ag85 vaccination did not affect the frequency of circulating Ag85-specific IFN-γ producing cells (Figure 2C), nor did Ag85 vaccination affect IFN-γ output by individual lung or blood cells (Figures 2B and 2D). M. bovis BCG vaccination of CBA/J mice increased IFN-γ output by individual Ag85-specific IFN-γ producing cells (Figure 2B).
Figure 2. Ag85 responses from vaccinated CBA/J mice.

Mice were injected subcutaneously with PBS, adjuvant (Adj), Ag85 plus Adj, or M. bovis BCG. At least 10 weeks after the first injection, mice were challenged with 50–100 CFU of M.tb, and euthanized 28 days later. Serial dilutions of lung cells (A, B) and PBMCs (C, D) were cultured with Ag85, or ovalbumin (not shown) or anti-CD8/anti-CD28 (not shown) for 36 hours on anti-IFN-γ coated ELISPOT plates to quantify the frequency of responding cells and the amount of IFN-γ produced by each responding cell. Results are the average ± SEM of 3 independent experiments. Each experiment had 5–6 mice per strain per time point; total n = 15 to 18 mice per group. Data were analyzed by one-way ANOVA with Tukey’s posttest, *p<0.05, ***p<0.001, NSD = no significant difference.
These results show that CBA/J mice can successfully prime Ag85-specific cells when Ag85 is administered exogenously. Together with the previous results, this suggests that CBA/J mice can process and present Ag85 protein, but they do not recognize Ag85 efficiently in the context of an in vivo M.tb infection. Interestingly, M. bovis BCG increased the potency of responding Ag85-specific IFN-γ cells, suggesting that CBA/J mice recognize and respond to Ag85 from M. bovis BCG better than Ag85 from M.tb.
Ag85 vaccination protected CBA/J mice against pulmonary M.tb challenge
To determine whether exogenous Ag85 plus Adj provided protection, CBA/J mice were vaccinated, challenged, and M.tb burden quantified in the lungs and mediastinal (lung-draining) lymph nodes 28 days later. Ag85 plus Adj vaccination significantly reduced M.tb lung burden as compared to the controls (PBS and Adj) (Figure 3A), but protection was not as good as M. bovis BCG vaccination (Figure 3C). In the mediastinal lymph nodes, Ag85 plus Adj vaccination had little effect on M.tb burden, while M. bovis BCG vaccination significantly reduced M.tb load (Figures 3B, 3D).
Figure 3. M.tb burden in vaccinated CBA/J mice.

Mice were injected subcutaneously with PBS, adjuvant (Adj), Ag85 plus Adj, or M. bovis BCG. At least 10 weeks after the first injection, mice were challenged with 50–100 CFU of M.tb Erdman by aerosol, and euthanized 28 days later. Lungs (A) and mediastinal lymph nodes (B) were homogenized, diluted, and plated onto supplemented 7H11 agar plates. M.tb CFU were counted after 21 days at 37°C. Protection in the lungs (C) and mediastinal lymph nodes (D) was calculated by subtracting the Log10 CFU from vaccinated groups from the average Log10 CFU of the PBS group. Results are the average ± SEM of 2 independent experiments. Each experiment had 5 mice per group, total n = 10 mice per group. Data were analyzed by one-way ANOVA with Tukey’s posttest, *p<0.05, **p<0.01, ***p<0.001.
These results show that CBA/J mice can successfully develop lung-specific protection against M.tb due to exogenous Ag85 plus Adj, possibly mediated by increased numbers of Ag85-specific IFN-γ producing cells. M. bovis BCG vaccination also protected CBA/J mice, as has been shown in other M.tb susceptible mice [15].
Immune cells and granulomas in the lungs of vaccinated CBA/J mice
Vaccination reduces the size of M.tb granulomas [16], but few Ag85-containing vaccine and challenge studies in mice have quantified immune cell subpopulations or granulomas. This was an important question to address because in addition to reducing M.tb burden an optimal vaccine should minimize non-specific inflammation.
We enumerated and phenotyped the lung immune cells in vaccinated and challenged CBA/J mice. The flow cytometry gating strategy is shown in Supplemental Figure 2. Adj and Ag85 plus Adj groups had fewer total cells (Figure 4A) and fewer immune cells (Figures 4B–4H) than did PBS and M. bovis BCG groups. Vaccination with M. bovis BCG resulted in similar cellularity and T cell activation profiles as did PBS (Figures 4A–4G). MHCII expression was enhanced on CD11c+ APCs in mice that received BCG (Figure 4H).
Figure 4. Lung cells in vaccinated CBA/J mice.

Mice were injected subcutaneously with PBS, adjuvant (Adj), Ag85 plus Adj, or M. bovis BCG. At least 10 weeks after the first injection, mice were challenged with 50–100 CFU of M.tb, and euthanized 28 days later. Lung cells were fixed and labeled with anti-CD3, anti-CD4, anti-CD8, anti-CD44, anti-CD11c, and anti-MHCII. The total lung cells (A), absolute numbers of T cells (B), CD4 T cells (C), CD8 T cells (E), CD44hi (activated) T cells (D, F), CD11c+ cells (G) and mean fluorescent intensity of MHCII expression (H) were determined. Results are the average ± SEM of 2 independent experiments. Each experiment had 5 mice per group, total n = 10 mice per group. Data were analyzed by one-way ANOVA with Tukey’s posttest, *p<0.05, **p<0.01, ***p<0.001.
We next evaluated the lungs of vaccinated and challenged CBA/J mice to determine whether the differences in immune cell numbers were associated with differences in the spatial distribution of cells to granulomas or to lymphocytic cuffs. This was important to address because distribution of cells could impact pulmonary function or control of M.tb. Immune cells were distributed differently in the lungs of each group (Figures 5A–5D) despite similarities in absolute numbers (Figure 4). The two control groups, PBS and Adj, were similar in granuloma size and number but showed moderate differences in lymphocytic cuffs, suggesting that the Adj affects lymphocyte trafficking to or within the lungs. Comparison of Adj to Ag85 plus Adj showed that Ag85 vaccination increased the size and number of granulomas and increased the number of lymphocytic cuffs, although none of the differences were statistically significant. M. bovis BCG vaccinated mice formed numerous, small granulomas. Representative lung granulomas and lymphocytic cuffs are shown (Figures 5E–H).
Figure 5. Immune cell aggregates in the lungs of vaccinated CBA/J mice.

Mice were injected subcutaneously with PBS, adjuvant (Adj), Ag85 plus Adj, or M. bovis BCG. At least 10 weeks after the first injection, mice were challenged with 50–100 CFU of M.tb, and euthanized 28 days later. Lung lobes from individual mice were formalin-fixed, paraffin-embedded, sectioned, and stained with hematoxylin and eosin. Slides were digitally scanned. Granuloma size (A) and number (B), and lymphocytic cuff size (C) and number (D) were quantified. Examples of representative granulomas (outlined in green) and lymphocytic cuffs (outlined in yellow) are shown (E-H). Results are the average and SEM from 4 slides from 1 experiment with 5 mice per group, total n = 5 mice per group. Scale bar = 100.5 μm. One-way ANOVA with Tukey’s post-test identified significant differences, *p<0.05, ***p<0.001.
In all groups at high magnification (40X), granulomas were composed of macrophages, lymphocytes, and neutrophils (not shown); and there were small foci of necrotic/apoptotic cells (not shown). As expected, intracytoplasmic acid-fast M.tb bacilli were present within macrophages in granulomas (not shown).
Overall, these results show that lung immune cells were distributed differently during primary and vaccine-challenge M.tb infections. There were also differences between Ag85 plus Adj and M. bovis BCG groups, showing that the type of vaccination affected the spatial organization of immune cells. Delivery of Ag85 as a soluble protein increased three out of four parameters measured by morphometry: granuloma size, lymphocytic cuff size, and number of lymphocytic cuffs. These data suggest that the total cellularity should have been increased in Ag85 plus Adj groups. However, by flow cytometry, the cell numbers were reduced (Figure 4). The cause of this discordance is not known. One possibility is that the large granulomas and/or lymphocytic cuffs in Ag85 plus Adj vaccinated mice contained cells which were lost during processing due to increased fragility or adherence to plastic.
M.tb memory responses to Ag85 and protection in CBA/J and C57BL/6 mice
Our previous results show that CBA/J mice responded poorly to Ag85 during primary M.tb infection, but were fully capable of responding protectively with IFN-γ production when Ag85 was delivered exogenously. We next asked whether Ag85-specific responses generated during primary M.tb infection of CBA/J mice (Figure 1) were enhanced in a re-infection model. C57BL/6 mice were included in these experiments as controls for robust Ag85-specific IFN-γ responses (Figure 1 and [7]). Mice were left untreated (primary infection), vaccinated with Ag85 plus Adj, or exposed to M.tb by aerosol (memory) as shown in the timeline (Figure 6E). After 30 days of M.tb infection in the memory mice all groups were treated with RIF and INH which reduced M.tb burdens in memory mice to undetectable levels (Figure 6A). Upon completion of the drug treatment, all groups were exposed to M.tb by aerosol and euthanized 28 days later. Ag85-specific IFN-γ responses and protection in the lungs were determined.
Figure 6. Ag85 vaccination and protective responses in C57BL/6 and CBA/J mice.

Lungs and spleens of mice treated with RIF and INH were homogenized and serial dilutions plated onto supplemented 7H11 agar plates to confirm bacterial clearance (A). Mice were treated according to the experimental timeline; “x” marks the injections for Ag85 plus Adj vaccinated mice (E). The Ag85-specific IFN-γ responses in primary infected, Ag85 plus Adj vaccinated, and memory immune mice were compared 28 days after M.tb infection (B). Lungs from identically treated groups of mice were homogenized, diluted, and plated onto supplemented 7H11 agar plates. M.tb CFU were counted after 21 days at 37°C (C). The calculated protection in the lungs is shown (D). Results are the average ± SEM of 1 experiment with 5 mice per group analyzed by one-way ANOVA with Tukey’s posttest, *p<0.05, **p<0.01, ***p<0.001.
Compared to primary infection, Ag85 plus Adj significantly increased Ag85-specific IFN-γ production from lung cells of both C57BL/6 and CBA/J mice (Figure 6B). Lung cell cultures from primary or Ag85-vaccinated CBA/J mice produced less IFN-γ than did cultures from identically treated C57BL/6 mice (Figure 6B). We anticipated that M.tb memory-immune mice would have enhanced Ag85-specific IFN-γ responses due to their prior exposure to M.tb. However, very little Ag85-specific IFN-γ was produced in memory mice of both mouse strains, possibly due to accelerated kinetics of memory antigen-specific IFN-γ responses. Alternatively, the low number of Ag85-specific cells generated from the primary infection may not expand or function protectively during recall responses.
To determine whether Ag85 vaccination and M.tb memory-immunity afforded protection, lungs were harvested from separate groups of identically treated CBA/J and C57BL/6 mice, and the M.tb burdens were calculated at day 28 of infection. In both mouse strains, Ag85 vaccination significantly decreased the M.tb burden as compared to unvaccinated mice (Figure 6C), and afforded about 1 Log10 of protection (Figure 6D). M.tb memory-induced responses were superior in reducing the M.tb load (Figure 6C), affording about 1.5 Log10 of protection in both mouse strains (Figure 6D).
Together, these results associate increasing Ag85-specific IFN-γ responses with control of M.tb growth in Ag85 plus Adj vaccinated CBA/J and C57BL/6 mice. Importantly, protection was equivalent in both mouse strains. The results from these experiments therefore demonstrate that a relative increase, rather than the absolute levels of Ag85-specific IFN-γ responses, can enhance M.tb control.
3. Discussion
M.tb Ag85 includes three related proteins (A, B, C) that share structure and function [17]. Ag85 components are immunogenic in humans [18] and in M.tb resistant mice [9, 10] including C57BL/6 ([14, 19–23]), C57BL/10 [14], C57BL/6 x BALB/c F1 hybrids [13, 24], and BALB/c mice [19–21, 25, 26]. Ag85 has been intensely investigated in animal models as a vaccine against M.tb in many formulations, including peptides or proteins [25], fusion proteins [13, 23, 24, 27], recombinant M. bovis BCG [28], recombinant vaccinia virus [29], antigen-pulsed cell-based vaccines [26, 30], and DNA vaccines [14, 20, 31, 32]. Ag85 is also a component of the vaccines in human clinical trials against M.tb [8, 29]. Efficacy results are not yet available, and it is not known whether these Ag85-containing vaccines will protect humans. It is promising that in many [13, 20, 23–26, 32] but not all [30, 31, 33] experimental animal models, Ag85 vaccination enhances IFN-γ responses and protects against M.tb.
We are interested in understanding antigen-specific responses of M.tb susceptible CBA/J mice [10] to model M.tb susceptible immunocompetent humans. M.tb susceptible C3H mice also have low responses to Ag85 during early primary M.tb infection [6, 7] due to haplotype-dependent T cell responses [6]. Little is known, however, about the ability of any M.tb susceptible strains to respond Ag85 throughout chronic asymptomatic M.tb infection or in the context of M.tb re-infection or vaccination followed by M.tb challenge. Here, we showed that CBA/J mice had low IFN-γ responses to Ag85 throughout chronic primary infection (Figure 1), yet this did not prevent CBA/J mice from recognizing and responding to Ag85 when it was delivered exogenously. In fact, delivery of Ag85 in an adjuvanted, pre-exposure vaccine reduced the M.tb burden by 2–10 fold in CBA/J lungs (Figure 3). Similar to other mouse strains [14, 25], Ag85-induced protection in CBA/J mice was associated with more Ag85-specific IFN-γ producing cells in the lungs (Figure 2 and not shown). As in other mouse strains [23, 24], M. bovis BCG vaccination of CBA/J mice provided superior protection, perhaps due to antigenic persistence or diversity. Thus, despite relatively low responses to Ag85 during primary infection or re-infection, M.tb susceptible CBA/J mice were fully capable of generating Ag85-specific protective immunity when the antigen was delivered in an appropriate context, i.e. as an exogenous soluble protein. Additional data from our laboratory also show that the contradictory responses of CBA/J mice to Ag85 in vivo during M.tb infection (low) versus responses induced by exogenous exposure and challenge (high and protective) are not limited to Ag85. Indeed, when CBA/J mice were exposed to ESAT-6 plus Adj and challenged with M.tb, they developed strong ESAT-6-specific IFN-γ responses and protective immunity in the lungs that was equivalent to protection in C57BL/6 mice (Supplemental Figure 3).
Our results demonstrate that low Ag85-specific IFN-γ responses from CBA/J mice resulted from two mechanisms: a low frequency of Ag85-specific IFN-γ producing cells, and low Ag85-specific IFN-γ output by individual responding cells. CBA/J mice do not have general deficiencies in IFN-γ production (Figure 1). The relative importance of frequency versus output by individual Ag85-responsive cells has not been determined, but we speculate that the frequency of responding cells would have a larger effect on M.tb control. Additional studies are needed to determine why CBA/J mice have such low IFN-γ responses to Ag85 in the context of M.tb infection. The mechanisms are likely complex, involving anti-inflammatory cytokines [34], presentation by the H-2k haplotype [6, 7] or diminished T cell co-stimulation in vivo, as IFN-γ output by single cells is robust when triggered with anti-CD3/anti-CD28 [11]. Specific studies are needed to identify the Ag85 peptides that fit H-2k homo- and heter-dimeric MHC molecules, how those peptides influence the generation and function of Ag85-specific T cells, and whether the Ag85 peptides present during M.tb infection (primary or re-infection) are different than the Ag85 peptides generated by Ag85 vaccination.
Ag85-containing vaccines are currently in human clinical trials [8, 29]. Our experimental results in CBA/J mice may predict that Ag85-containing vaccines will protect M.tb susceptible humans. Important limitations to be addressed in future mouse experiments include determining pre-exposure responses to exogenous Ag85, and long-term experiments to assess whether Ag85 vaccination improves TB disease outcome in CBA/J mice.
Overall, our results demonstrate four main findings. First, CBA/J mice did not respond efficiently to Ag85 in the context of M.tb primary infection or re-infection, but could produce abundant IFN-γ in response to positive controls. This suggests that CBA/J mice may recognize uncharacterized M.tb antigens, or that immunogenic epitopes of Ag85 are not available during in vivo infection. Further studies may identify other immunodominant M.tb antigens in CBA/J mice and possibly develop those antigens as vaccine candidates for M.tb susceptible humans. Second, CBA/J mice could generate robust protective immune responses to exogenous Ag85. This indicates that vaccination can be successful despite inefficient Ag85-specific IFN-γ responses during natural (primary or re-infection) M.tb infection. Third, in our memory model, M.tb-induced protection was not associated with enhanced Ag85 responses in either M.tb-resistant mice (C57BL/6) or M.tb susceptible mice (CBA/J). This surprising finding may be consistent with memory immune responses that precede M.tb control. Alternatively, Ag85 exposure during natural M.tb infection may not generate robust long-term protective Ag85-specific memory immune cells. Finally, Ag85 vaccination induced immune cell infiltrates that were larger than cell infiltrates in M. bovis BCG vaccinated CBA/J mice. Whether this has a negative impact on pulmonary function or disease progression is unknown and should be addressed in future studies.
4. Materials and methods
Mice
Specific pathogen free 8-week-old female C57BL/6 and CBA/J mice (Charles River Laboratories, Wilmington, MA) were maintained in ventilated cages within Biosafety Level-3 facilities and provided with sterile food and water ad libitum. All protocols were approved by The Ohio State University’s Institutional Laboratory Animal Care and Use Committee.
M.tb stocks
M.tb Erdman (ATCC #35801) and M. bovis BCG (ATCC #35734) were obtained from American Type Culture Collection (Manassas, VA.). Stocks were grown in Proskauer-Beck liquid medium containing 0.05% Tween 80 to mid-log phase and frozen in 1ml aliquots at −80°C. One aliquot was used for each experimental M.tb infection.
M.tb infection
Mice received 50–100 CFU of M.tb Erdman using an Inhalation Exposure System (Glas-col, Terre Haute, IN). M.tb dose and lung burden were determined as previously desribed [11]. Mice in long-term experiments (Figure 1) were monitored, euthanized, and excluded from experiments if TB-associated morbidity developed [11].
Vaccination
Vaccination protocols were modified from Deitrich et al [13] and Andersen [35]. 250 μg/ml of synthetic monophosphoryl lipid A (MPL) (Avanti Polar Lipids, Inc., Alabaster, AL) in 0.2% triethylamine in PBS was heated at 70°C and then sonicated for 30 seconds. 2.5 mg/ml of dimethyldioctadecyl ammonium bromide (DDA) (Sigma-Aldrich, Co., St. Louis, MO) in PBS was heated at 80°C for 10 minutes and then cooled. M.tb antigens at 100 μg/ml (Ag85) or equivalent volumes of PBS were added to the cooled DDA solution. Finally, equivalent volumes of the MPL solution and DDA solutions (with or without antigen) were mixed. Vaccines were agitated immediately prior to injection. Ag85 vaccinates mice were injected subcutaneously interscapularly 3 times at 2-week intervals with 200 μl (containing 25 μg MPL, 250 μg DDA, and 10 μg of antigen). Control groups received 200 μl of PBS or 200 μl of adjuvant (MPL + DDA). In some experiments, mice received one injection of 2.5 × 105 M. bovis BCG bacilli in 200 μl of PBS. Mice were rested and challenged with M.tb Erdman by aerosol at least 10 weeks after the first injection.
Antimycobacterial treatment
Mice received 250 mg/L of isoniazid (isonicotinic acid hydrazine; INH (Sigma)) and 100 mg/L of rifampin (rifampicin; RIF (Sigma)) in the drinking water for 12 weeks. The protocol was modified [36–39] and based on consumption of 6 ml of water per mouse per day [40]. The targeted doses were 25 – 50 mg/kg/day for INH and 10 – 20 mg/kg/day for RIF, which are below the carcinogenic [41] or toxic [42] levels. INH was dissolved in a small volume of water at 50 mg/ml. RIF was dissolved in a minimal volume of DMSO (Sigma) at 100 mg/ml. MQ water was added to achieve the final concentrations and the pH was adjusted to neutral.
Lung cell isolation
Cells were isolated from the lungs [43]. Briefly, lungs were perfused with heparinized PBS, diced into 1 mm2 pieces, digested with collagenase/DNase, and single cell suspensions created by pressing the pieces through cell strainers. Red blood cells were lysed. The leukocytes were counted and re-suspended in media, or fixed with 0.1 % sodium azide.
Blood collection
Heparinized blood was collected and diluted in media as previously described [11].
Bone marrow derived APCs
Bone marrow cells from the tibiae and femora of age and sex matched non-infected C57BL/6 and CBA/J mice were differentiated into APCs [11] using GM-CSF containing media derived from GM-EL4 cells [44]. The only modification was that 5 μg/ml of M.tb Ag85 (Colorado State University’s NIH NIAID Contract No. HHSN266200400091C) was added to the cultures.
Lung and blood antigen-specific responses by ELISA
100 μl of lung cells containing 2 × 105 cells or 100 μl of diluted blood from each individual mouse were cultured in duplicate or triplicate as previously described [11]. The only modification was M.tb Ag85 (5 μg/ml) was added to the cultures.
Lung and PBMC antigen-specific responses by ELISPOT
Purified CD4 T cells from the lungs of mice or PBMCs were cultured with antigen-pulsed bone marrow-derived APCs as previously described [11]. The only modification was that APCs were pulsed with 5 μg/ml of Ag85.
Flow cytometry
Conjugated and unconjugated antibodies and isotype controls were purchased from BD Biosciences, including Fc Block™ (clone 2.462), PerCP-Cy5.5 anti-CD3ε (145-2C11), APC-Cy7 anti-CD4 (GK1.5), PE-Cy7 anti-CD8 (53-6.7), APC anti-TCR-β chain (H57597), PerCP anti-CD8 (53-6.7), FITC anti-CD44 (IM7), FITC rat IgG2bκ, FITC anti-I-A/I-E (2G9), and FITC rat IgG2aκ. The staining protocol and results analyses were performed as previously described [11].
Histology and histomorphometry
5 μm sections from formalin-fixed, paraffin-embedded sections were stained with hematoxylin and eosin or for acid-fast bacilli. Blind evaluation was performed by a board-certified veterinary pathologist (PS). Histomorphometry was performed using Aperio ImageScope v8.2.5 by a second board-certified veterinary anatomic pathologist (GB) [45].
Statistics
Statistical analyses were performed using Prism 4 software (GraphPad Software, San Diego, CA). Multigroup comparisons were analyzed with one-way ANOVA using Dunnett’s post-test or Tukey’s posttest. Pairwise comparisons used the Student’s t-test. Statistical significance was defined as *p<0.05, **p<0.01, ***p<0.001.
Supplementary Material
BD Falcon high-binding 96-well ELISA plates were coated with 100 μl of mouse IgG3 monoclonal anti-Ag85 CS.90 diluted 1:80 in PBS overnight at 4°C. Wells were blocked with 1% BSA in PBS for 2 hours at room temperature, and then washed 6 times with PBS-tween. 100 μl of serially diluted, purified Ag85 in 1% BSA with a top concentration of 2.5 μg/ml; 100 μl lung homogenates from day 21 M.tb-infected C57BL/6 and CBA/J mice; 100 μl of diluent control (1% BSA) or 100 μl of lung homogenates from non-infected control mice were added to each well in duplicates and incubated for 2 hours at room temperature followed by overnight at 4°C. Plates were washed 6 times in PBS-tween and then soaked in PBS-tween for 3 hours at room temperature. 100 μl of rabbit polyclonal anti-Ag85, diluted 1:2000 in 1% BSA, were added to each well, and incubated for 2 hours at room temperature followed by overnight at 4°C. Plates were then washed 6 times with PBS-tween and then soaked in PBS-tween for 2 hours at room temperature. 100 μl of polyclonal goat anti-rabbit HRP-conjugated detection antibody diluted in 1% BSA were added to each well and incubated for 2 hours at room temperature followed by 6 washes with PBS-tween. Samples were then developed with 100 μl of TMB substrate per well for 20 minutes, stopped with 100 μl of 0.18M H2SO4 per well, and optical density immediately read at 450nm with 570nm wavelength correction. Data shown are from 1 representative experiment of 2 experiments, each with 2 M.tb-infected mice per strain (A) and the standard curve for purified Ag85 (B).
CBA/J mice were injected with PBS, Adj, Ag85 plus Adj, or M. bovis BCG, rested for at least 10 weeks after the first injection, and infected with 50–100 CFU of M.tb Erdman by aerosol exposure. After 28 days, mice were euthanized and single cell suspensions from the lungs were fixed, blocked, and labeled with fluorescent antibodies. Samples from individual mice were read on an LSRII and analyzed using FACSDiva software. First, viable cells identified by excluding lysed red blood cell membranes and autofluorescent cellular debris. Within the viable cells, CD4 T cells and CD8 T cells were then identified by CD3, CD4, and CD8 expression. Next, within the viable cells alveolar macrophages (AMMO) (CD11chiCD11bmid), dendritic cells (DC) (CD11chiCD11bhi), monocytes and small macrophages (monos/macs) (CD11cmid-loCD11bmid), and neutrophils (neutrophils) (CD11clo-negCD11bhi) were discriminated based on CD11c and CD11b expression as described in [46]. The MFI of MHCII-expressing innate immune cells was also quantified using an isotype control to set the gates.
CBA/J mice were injected subcutaneously with PBS, adjuvant (Adj), ESAT-6 plus Adj, or M. bovis BCG. At least 10 weeks after the first injection, mice were challenged with 50–100 CFU of M.tb Erdman by aerosol, and euthanized 28 days later. Serial dilutions of lung cells (A) and PBMCs (B) were cultured with ESAT-6, or ovalbumin (not shown), or anti-CD8/anti-CD28 (not shown) for 36 hours. The frequency of ESAT-6-specific IFN-γ producing cells was calculated per 100,000 cells by subtracting the number of SFU in the negative control wells (ovalbumin). C57BL/6 and CBA/J mice were assigned to groups and treated as in the timeline in Figure 6, except that ESAT-6 plus Adj was injected. After 28 days of primary or challenge M.tb infection, lungs were homogenized, diluted, and plated onto supplemented 7H11 agar plates. M.tb CFU were counted after 21 days at 37°C (C). The calculated protection in the lungs is shown (D). Results (A, B, C, D) are the average ± SEM of 1 experiment with 5 mice per group analyzed by one-way ANOVA with Tukey’s posttest, *p<0.05, **p<0.01, ***p<0.001.
Acknowledgments
Support was provided by NIH R01 (AI064522; JT), NIH T32 (RR07073; GB) and NIH K08 (AI071111; GB). M.tb Ag85 and anti-Ag85 antibodies were provided by Colorado State University’s TBVTRM Contract HH5N266200400091c NIH N01AI40091. Histology services were provided by The Ohio State University’s Department of Veterinary Biosciences.
Abbreviations
- Adj
Adjuvant
- Ag85
Antigen 85
- CFP
Culture Filtrate Protein
- Con-A
Concanavalin-A
- ESAT-6
Early Secreted Antigenic Target-6
- M.tb
Mycobacterium tuberculosis
- rm
recombinant murine
- SFU
Spot Forming Unit
Footnotes
Conflict of interest
The authors have no conflicts of interest.
References
- 1.World Health Organisation. Fact File: 10 Facts about tuberculosis. 2009 http://www.who.int/features/factfiles/tb_facts/en/index2.html.
- 2.Hunter R. Pathology of post primary tuberculosis of the lung: An illustrated critical review. Tuberculosis. 2011;91:497–509. doi: 10.1016/j.tube.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shende N, Gutpa S, Upadhye V, Kumar S, Harinth B. Isolation and analysis of circulating tuberculous antigens in Mycobacterium tuberculosis. Indian J Tuberc. 2007;54:125–129. [PubMed] [Google Scholar]
- 4.Huygen K, Van Vooren J, Turneer M, Bosmans R, Dierckx P, De Bruyn J. Specific lymphoproliferation, gamma interferon production, and serum immunoglobulin G directed against a purified 32 kDa mycobacterial protein antigen (P32) in patients with active tuberculosis. Scand J Immunol. 1988;27:187–194. doi: 10.1111/j.1365-3083.1988.tb02338.x. [DOI] [PubMed] [Google Scholar]
- 5.Demissie A, Ravn P, Olobo J, Doherty TM, Eguale T, Geletu M, Hailu W, Andersen P, Britton S. T-Cell Recognition of Mycobacterium tuberculosis Culture Filtrate Fractions in Tuberculosis Patients and Their Household Contacts. Infect Immun. 1999;67:5967–5971. doi: 10.1128/iai.67.11.5967-5971.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamath AB, Alt JM, Debbabi H, Taylor C, Behar SM. The major histocompatibility haplotype affects T-cell recognition of mycobacterial antigens but not resistance to Mycobacterium tuberculosis in C3H mice. Infect Immun. 2004;72:6790–6798. doi: 10.1128/IAI.72.12.6790-6798.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beamer G, Cyktor J, Carruthers B, Turner J. H-2 alleles contribute to Antigen 85-specific interferon-gamma responses during Mycobacterium tuberculosis infection. Cell Immunol. 2011;271:53–61. doi: 10.1016/j.cellimm.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whelan KT, Pathan AA, Sander CR, Fletcher HA, Poulton I, Alder NC, Hill AVS, McShane H. Safety and Immunogenicity of Boosting BCG Vaccinated Subjects with BCG: Comparison with Boosting with a New TB Vaccine, MVA85A. PLoS One. 2009;4:e5934. doi: 10.1371/journal.pone.0005934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medina E, North RJ. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunol. 1998;93:270–274. doi: 10.1046/j.1365-2567.1998.00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner J, Gonzalez-Juarrero M, Saunders BM, Brooks JV, Marietta P, Ellis DL, Frank AA, Orme IM. Immunological basis for reactivation of tuberculosis in mice. Infect Immun. 2001;69:3264–3270. doi: 10.1128/IAI.69.5.3264-3270.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beamer GL, Flaherty DK, Vesosky B, Turner J. Peripheral blood interferon-{gamma} release assays predict lung responses and Mycobacterium tuberculosis disease outcome in mice. Clin Vacc Immunol. 2008;15:474–483. doi: 10.1128/CVI.00408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogerson BJ, Jung YJ, LaCourse R, Ryan L, Enright N, North RJ. Expression levels of Mycobacterium tuberculosis antigen-encoding genes versus production levels of antigen-specific T cells during stationary level lung infection in mice. Immunology. 2006;118:195–201. doi: 10.1111/j.1365-2567.2006.02355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietrich J, Aagaard C, Leah R, Olsen AW, Stryhn A, Doherty TM, Andersen P. Exchanging ESAT6 with TB10 in an Ag85B Fusion Molecule-Based Tuberculosis Subunit Vaccine: Efficient Protection and ESAT6-Based Sensitive Monitoring of Vaccine Efficacy. J Immunol. 2005;174:6332–6339. doi: 10.4049/jimmunol.174.10.6332. [DOI] [PubMed] [Google Scholar]
- 14.Romano M, Roupie V, Wang X, Denis M, Jurion F, Adnet PY, Laali R, Huygen K. Immunogenicity and protective efficacy of tuberculosis DNA vaccines combining mycolyl-transferase Ag85A and phosphate transport receptor Pst-3. Immunology. 2006;118:321–332. doi: 10.1111/j.1365-2567.2006.02373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medina E, North R. Genetically susceptible mice remain proportionally more susceptible to tuberculosis after vaccination. Immunology. 1999;96:16–21. doi: 10.1046/j.1365-2567.1999.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dannenberg AM., Jr Perspectives on Clinical Preclinical Testing of New Tuberculosis Vaccines. Clin Microbiol Rev. 2010;23:781–794. doi: 10.1128/CMR.00005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiker HG, Harboe M. The antigen 85 complex: a major secretion product of Mycobacterium tuberculosis. Microbiol Mol Biol Rev. 1992;56:648–661. doi: 10.1128/mr.56.4.648-661.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Dissel JT, Arend SM, Prins C, Bang P, Tingskov PN, Lingnau K, Nouta J, Klein MR, Rosenkrands I, Ottenhoff THM, Kromann I, Doherty TM, Andersen P. Ag85B-ESAT-6 adjuvanted with IC31® promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naïve human volunteers. Vaccine. 2010;28:3571–3581. doi: 10.1016/j.vaccine.2010.02.094. [DOI] [PubMed] [Google Scholar]
- 19.D’Souza S, Rosseels V, Romano M, Tanghe A, Denis O, Jurion F, Castiglione N, Vanonckelen A, Palfliet K, Huygen K. Mapping of Murine Th1 Helper T-Cell Epitopes of Mycolyl Transferases Ag85A, Ag85B, and Ag85C from Mycobacterium tuberculosis. Infect Immun. 2003;71:483–493. doi: 10.1128/IAI.71.1.483-493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lozes E, Huygen K, Content J, Denis O, Montgomery DL, Yawman AM, Vandenbussche P, Van Vooren JP, Drowart A, Ulmer JB, Liu MA. Immunogenicity and efficacy of a tuberculosis DNA vaccine encoding the components of the secreted antigen 85 complex. Vaccine. 1997;15:830–833. doi: 10.1016/s0264-410x(96)00274-5. [DOI] [PubMed] [Google Scholar]
- 21.Ulmer JB, Liu MA, Montgomery DL, Yawman AM, Randall Deck R, DeWitt CM, Content J, Huygen K. Expression and immunogenicity of Mycobacterium tuberculosis antigen 85 by DNA vaccination. Vaccine. 1997;15:792–794. doi: 10.1016/s0264-410x(96)00255-1. [DOI] [PubMed] [Google Scholar]
- 22.Duffy D, Dawoodji A, Agger E, Andersen P, Westermann J, Bell E. Immunological memory transferred with CD4 T cells specific for tuberculosis Ag85B-TB10. 4: persisting antigen enhances protection. PLoS One. 2009;4:8272. doi: 10.1371/journal.pone.0008272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennekov T, Dietrich J, Rosenkrands I, Stryhn A, Doherty TâM, Andersen P. Alteration of epitope recognition pattern in Ag85B and ESAT-6 has a profound influence on vaccine-induced protection against Mycobacterium tuberculosis. Eur J Immunol. 2006;36:3346–3355. doi: 10.1002/eji.200636128. [DOI] [PubMed] [Google Scholar]
- 24.Dietrich J, Andersen C, Rappuoli R, Doherty TM, Jensen CG, Andersen P. Mucosal Administration of Ag85B-ESAT-6 Protects against Infection with Mycobacterium tuberculosis and Boosts Prior Bacillus Calmette-Guerin Immunity. J Immunol. 2006;177:6353–6360. doi: 10.4049/jimmunol.177.9.6353. [DOI] [PubMed] [Google Scholar]
- 25.Giri PK, Verma I, Khuller GK. Enhanced immunoprotective potential of Mycobacterium tuberculosis Ag85 complex protein based vaccine against airway Mycobacterium tuberculosis challenge following intranasal administration. FEMS Immunol Med Microbiol. 2006;47:233–241. doi: 10.1111/j.1574-695X.2006.00087.x. [DOI] [PubMed] [Google Scholar]
- 26.Malowany J, McCormick S, Santosuosso M, Xizhong Z, Aoki N, Ngai P, Wang J, Leitch J, Bramson J, Wan Y, Xing Z. Development of cell-based tuberculosis vaccines: genetically modified dendritic cell vaccine is a much more potent activator of CD4 and CD8 T cells than peptide- or protein-loaded counterparts. Mol Ther. 2006;13:766–774. doi: 10.1016/j.ymthe.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Langermans JAM, Doherty TM, Vervenne RAW, Laan Tvd, Lyashchenko K, Greenwald R, Agger EM, Aagaard C, Weiler H, Soolingen Dv, Dalemans W, Thomas AW, Andersen P. Protection of macaques against Mycobacterium tuberculosis infection by a subunit vaccine based on a fusion protein of antigen 85B and ESAT-6. Vaccine. 2005;23:2740–2750. doi: 10.1016/j.vaccine.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 28.Horwitz M, Harth G, Dillon B, Maslesa-Galic S. Recombinant bacillus Calmette-Guerin (BCG) vaccines expressing the Mycobacterium tuberculosis 30-kDa major secretory product induce greater protective immunity in a highly susceptible animal model. Proc Nat Acad Sci USA. 2000;97:13853–13858. doi: 10.1073/pnas.250480397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McShane H, Pathan AA, Sander CR, Goonetilleke NP, Fletcher HA, Hill AVS. Boosting BCG with MVA85A: the first candidate subunit vaccine for tuberculosis in clinical trials. Tuberculosis. 2005;85:47–52. doi: 10.1016/j.tube.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 30.González-Juarrero M, Turner J, Basaraba RJ, Belisle JT, Orme IM. Florid pulmonary inflammatory responses in mice vaccinated with Antigen-85 pulsed dendritic cells and challenged by aerosol with Mycobacterium tuberculosis. Cell Imunol. 2002;220:13–19. doi: 10.1016/s0008-8749(03)00010-8. [DOI] [PubMed] [Google Scholar]
- 31.Taylor J, Turner O, Basaraba R, Belisle J, Huygen K, Orme I. Pulmonary necrosis resulting from DNA vaccination against tuberculosis. Infect Immun. 2003;71:2192–2198. doi: 10.1128/IAI.71.4.2192-2198.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Souza S, Denis O, Scorza T, Nzabintwali F, Verschueren H, Huygen K. CD4+ T cells contain Mycobacterium tuberculosis infection in the absence of CD8+ T cells in mice vaccinated with DNA encoding Ag85A. Eur J Immunol. 2000;30:2455–2499. doi: 10.1002/1521-4141(200009)30:9<2455::AID-IMMU2455>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Turner J, Rhoades E, Keen M, Belisle J, Frank A, Orme I. Effective pre-exposure tuberculosis vaccines fail to protect when they are given in an immunotherapeutic mode. Infect Immun. 2000;68:1706–1709. doi: 10.1128/iai.68.3.1706-1709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beamer GL, Flaherty DK, Assogba BD, Stromberg P, Gonzalez-Juarrero M, de Waal Malefyt R, Vesosky B, Turner J. Interleukin-10 Promotes Mycobacterium tuberculosis Disease Progression in CBA/J Mice. J Immunol. 2008;181:5545–5550. doi: 10.4049/jimmunol.181.8.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersen P. Effective vaccination of mice against Mycobacterium tuberculosis infection with a soluble mixture of secreted mycobacterial proteins. Infect Immun. 1994;62:2536–2544. doi: 10.1128/iai.62.6.2536-2544.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hubbard RD, Flory CM, Collins FM. Memory T cell-mediated resistance to Mycobacterium tuberculosis infection in innately susceptible and resistant mice. Infect Immun. 1991;59:2012–2016. doi: 10.1128/iai.59.6.2012-2016.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buccheri S, Reljic R, Caccamo N, Meraviglia S, Ivanyi J, Salerno A, Dieli F. Prevention of the post-chemotherapy relapse of tuberculous infection by combined immunotherapy. Tuberculosis. 2009;89:91–94. doi: 10.1016/j.tube.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Cynamon M, Sklaney MR, Shoen C. Gatifloxacin in combination with rifampicin in a murine tuberculosis model. J Antimicrob Chemother. 2007;60:429–432. doi: 10.1093/jac/dkm200. [DOI] [PubMed] [Google Scholar]
- 39.Cardona PJ, Amat I, Gordillo S, Arcos V, Guirado E, Díaz J, Vilaplana C, Tapia G, Ausina V. Immunotherapy with fragmented Mycobacterium tuberculosis cells increases the effectiveness of chemotherapy against a chronical infection in a murine model of tuberculosis. Vaccine. 2005;23:1393–1398. doi: 10.1016/j.vaccine.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Bachmanov AA, Reed DR, Beauchamp GK, Tordoff MG. Food Intake, Water Intake, and Drinking Spout Side Preference of 28 Mouse Strains Behavior Genetics. Springer; Netherlands: 2002. pp. 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biancifiori C, Severi L. The relation of isoniazid (INH) and allied compounds to carcinogenesis in some species of small laboratory animals: a review. Br J Cancer. 1966;20:528–538. doi: 10.1038/bjc.1966.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grosset J, Leventis S. Adverse effects of rifampin. Rev Infect Dis. 1983;(Suppl 3):S440–S450. doi: 10.1093/clinids/5.supplement_3.s440. [DOI] [PubMed] [Google Scholar]
- 43.Vesosky B, Flaherty DK, Rottinghaus EK, Beamer GL, Turner J. Age dependent increase in early resistance of mice to Mycobacterium tuberculosis is associated with an increase in CD8 T cells that are capable of antigen independent IFN-gamma production. Expt Gerontol. 2006;41:1185–1194. doi: 10.1016/j.exger.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Hurwitz AA, Yu TFY, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Nat Acad Sci USA. 1998;95:10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vesosky B, Rottinghaus EK, Stromberg P, Turner J, Beamer G. CCL5 participates in early protection against Mycobacterium tuberculosis. J Leuk Biol. 2010;87:1153–1165. doi: 10.1189/jlb.1109742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. J Immunol. 2003;171:3128–3135. doi: 10.4049/jimmunol.171.6.3128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BD Falcon high-binding 96-well ELISA plates were coated with 100 μl of mouse IgG3 monoclonal anti-Ag85 CS.90 diluted 1:80 in PBS overnight at 4°C. Wells were blocked with 1% BSA in PBS for 2 hours at room temperature, and then washed 6 times with PBS-tween. 100 μl of serially diluted, purified Ag85 in 1% BSA with a top concentration of 2.5 μg/ml; 100 μl lung homogenates from day 21 M.tb-infected C57BL/6 and CBA/J mice; 100 μl of diluent control (1% BSA) or 100 μl of lung homogenates from non-infected control mice were added to each well in duplicates and incubated for 2 hours at room temperature followed by overnight at 4°C. Plates were washed 6 times in PBS-tween and then soaked in PBS-tween for 3 hours at room temperature. 100 μl of rabbit polyclonal anti-Ag85, diluted 1:2000 in 1% BSA, were added to each well, and incubated for 2 hours at room temperature followed by overnight at 4°C. Plates were then washed 6 times with PBS-tween and then soaked in PBS-tween for 2 hours at room temperature. 100 μl of polyclonal goat anti-rabbit HRP-conjugated detection antibody diluted in 1% BSA were added to each well and incubated for 2 hours at room temperature followed by 6 washes with PBS-tween. Samples were then developed with 100 μl of TMB substrate per well for 20 minutes, stopped with 100 μl of 0.18M H2SO4 per well, and optical density immediately read at 450nm with 570nm wavelength correction. Data shown are from 1 representative experiment of 2 experiments, each with 2 M.tb-infected mice per strain (A) and the standard curve for purified Ag85 (B).
CBA/J mice were injected with PBS, Adj, Ag85 plus Adj, or M. bovis BCG, rested for at least 10 weeks after the first injection, and infected with 50–100 CFU of M.tb Erdman by aerosol exposure. After 28 days, mice were euthanized and single cell suspensions from the lungs were fixed, blocked, and labeled with fluorescent antibodies. Samples from individual mice were read on an LSRII and analyzed using FACSDiva software. First, viable cells identified by excluding lysed red blood cell membranes and autofluorescent cellular debris. Within the viable cells, CD4 T cells and CD8 T cells were then identified by CD3, CD4, and CD8 expression. Next, within the viable cells alveolar macrophages (AMMO) (CD11chiCD11bmid), dendritic cells (DC) (CD11chiCD11bhi), monocytes and small macrophages (monos/macs) (CD11cmid-loCD11bmid), and neutrophils (neutrophils) (CD11clo-negCD11bhi) were discriminated based on CD11c and CD11b expression as described in [46]. The MFI of MHCII-expressing innate immune cells was also quantified using an isotype control to set the gates.
CBA/J mice were injected subcutaneously with PBS, adjuvant (Adj), ESAT-6 plus Adj, or M. bovis BCG. At least 10 weeks after the first injection, mice were challenged with 50–100 CFU of M.tb Erdman by aerosol, and euthanized 28 days later. Serial dilutions of lung cells (A) and PBMCs (B) were cultured with ESAT-6, or ovalbumin (not shown), or anti-CD8/anti-CD28 (not shown) for 36 hours. The frequency of ESAT-6-specific IFN-γ producing cells was calculated per 100,000 cells by subtracting the number of SFU in the negative control wells (ovalbumin). C57BL/6 and CBA/J mice were assigned to groups and treated as in the timeline in Figure 6, except that ESAT-6 plus Adj was injected. After 28 days of primary or challenge M.tb infection, lungs were homogenized, diluted, and plated onto supplemented 7H11 agar plates. M.tb CFU were counted after 21 days at 37°C (C). The calculated protection in the lungs is shown (D). Results (A, B, C, D) are the average ± SEM of 1 experiment with 5 mice per group analyzed by one-way ANOVA with Tukey’s posttest, *p<0.05, **p<0.01, ***p<0.001.