

Abstract
Among the four major building blocks of life, glycans play essential roles in numerous physiological and pathological processes. Due to their non-templated biosynthesis, advances towards elucidating the molecular details of glycan functions are relatively slow compared with the pace of protein and nucleic acid research. Over the past 30 years, chemical tools have emerged as powerful allies to genetics and molecular biology in the study of glycans in their native environment. This tutorial review will provide an overview of the recent technological developments in the field, as well as the progress in the application of these techniques to probe glycans in cells and organisms.
Introduction
Over the past thirty years, revolutionary advances in biosciences have produced a variety of tools for the visualization and manipulation of nucleic acids and proteins in living systems. For example, RNA and DNA can be tracked in vivo through hybridization with a complementary sequence functionalized with probes for fluorogenic or colorimetric detection. Likewise, proteins can be studied in vivo by fusion to fluorescent proteins to visualize them or photoactivatable domains to control their functions.1 The invention of these technologies has been enabled through the use of sophisticated biochemical and genetic manipulations. However, glycans, until recently, remained an elusive target for modern imaging techniques. Unlike proteins and nucleic acids that are genetically encoded, glycosylation is a posttranslational modification in which glycans are assembled in a stepwise fashion. Thus, techniques developed to label nucleic acids and proteins are not directly transferable to label glycans.
Glycans mediate diverse biological processes in a cellular context by interacting with glycan-binding proteins called lectins.2 Glycan-lectin interactions regulate many aspects of cellular functions ranging from signal transduction to cell adherence and cell-cell communications. Accordingly, aberrant glycan expression and glycan-lectin interactions are a hallmark of malignancies including cancer and autoimmune diseases. For these reasons, to determine the molecular function and regulation of glycans, tools for perturbing and detecting these biomolecules in their native environments are required.
The classical methods for probing glycans in cells and organisms involve the use of lectins and antibodies. Many lectins are commercially available as fluorescently labeled conjugates that recognize diverse glycan structures from monosaccharides, the simplest building blocks of glycans, to polysaccharides, oligomers built from monosaccharides. However, most lectins originate from plants and are toxic to the cells they are probing. Binding to their target glycans with only a millimolar affinity, the specificity of lectins is usually low. Similarly, antibody-based detection methods have been developed to target specific glycan structures such as Lewis X, Sialyl Lewis X and heparan among many others. These antibodies are tissue impermeant, and as a result, their applications in vivo are limited. Recently, Bülow and coworkers showed that a GFP-tagged single chain variable fragment antibody, when expressed in Caenorhabditis elegans, allows direct visualization of specific heparan sulfate patterns.3 However, the endogenous glycan functions may be blocked by transgenically introduced antibodies, urging further validation of the generality of this approach in living organisms. Glycans, when released from protein or lipid anchors, can be analyzed using nuclear magnetic resonance (NMR), mass spectrometry (MS) and high performance liquid chromatography (HPLC). However, these techniques can only be used in vitro and are not applicable in cells and living organisms. In this review, we will provide a brief overview of the recent technological developments that have enabled the chemical probing of glycans in living systems, including cultured bacteria, yeast, mammalian cells, C. elegans, zebrafish and mice.
Glycan remodeling via metabolic oligosaccharide engineering
First developed in the early 80’s, metabolic oligosaccharide engineering (MOE) is a method to perform structure-activity-relationship (SAR) studies to explore glycan functions in living systems. This method involves chemically synthesizing a monosaccharide analog, supplying cells with the unnatural monosaccharide, and following up with the cellular outcome. As an early example, N-acetyl mannosamine (ManNAc) analogs were synthesized with elongated or branched hydrocarbons introduced to replace the N-acetyl side chain of the natural metabolic precursor of sialic acid. When fed to cells, the unnatural substrate is metabolized and incorporated into cell surface sialylated glycoconjugates by exploiting the substrate promiscuity of CMP sialic acid synthetase and sialyltransferases, key enzymes in the sialic acid biosynthetic pathway.4
In this early work, ManNAc analogs were administrated to cells or animals directly in the unprotected form. The hydrophilic nature of these unnatural substrates prevents their free diffusion into the cytoplasm compartment. In order to achieve productive labeling, millimolar concentration was required. In the late 90’s, it was discovered that peracetylation of monosaccharides could enhance their cellular uptake.5 Once inside the cell the acetyl groups are rapidly hydrolyzed by nonspecific esterases, liberating the monosaccharides to metabolic processing. These studies laid the groundwork for the future development of MOE as a general approach to investigate glycan functions in living systems.
The year 1997 witnessed a breakthrough in the field of MOE when Carolyn Bertozzi demonstrated that a chemically reactive group, the ketone, could be incorporated into the N-acyl side chain of ManNAc to allow a covalent ligation reaction on the surface of live cells.6 The covalent reaction enabled the introduction of a biophysical probe to detect the tagged sialic acids (Scheme 1A). This pioneering work ushered in a period of active research aimed at discovering highly specific reactions as fast chemical ligation tools in complex cellular environments. Termed bioorthogonal chemistry, reactions that fall into this category proceed rapidly at physiological conditions without interference with the native biological machineries. By using a functional group that is not found in the biological system, specificity toward the tagged targets (e.g. glycans) is ensured among a myriad of biomacromolecules and metabolites presented in cells. These functional groups include aldehydes, ketones, thiols, azides and alkynes, each of which can be specifically targeted using a bioorthogonal reaction.
Scheme 1.

(A) Labeling cell-surface sialylated and fucosylated glycans using metabolic oligosaccharide engineering and bioorthogonal chemistry. Positions in sialic acid and fucose that are amenable for chemical tagging are highlighted in red. (B) condensation of ketones with aminooxy and hydrazide compounds. (C) CuAAC. (D) Copper-free click chemistry. (E) The Staudinger ligation. (F) Thiol-maleimide coupling reaction.
The coupling reaction between a carbonyl compound (i.e. aldehyde and ketone) and an N-nucleophile (i.e. hydrazide or aminooxy) is a classical example of bioconjugate chemistry (Scheme 1B). The reaction products (i.e. hydrazone or oxime) are relatively stable compared to condensation products of the carbonyl with other nucleophiles commonly found in biological systems, such as primary amines. While ketones are not present on the cell surface, metabolites inside the cell, such as pyruvate, can react with nucleophilic probes, limiting the use of this reaction in the cytoplasm.
Considered as “the cream of the crop” of click chemistry,8 azides and alkynes are not present within the cell or on the cell surface, making them ideal tags to be installed synthetically into monosaccharide building blocks without significantly disrupting their natural functions. Indeed, the Huisgen 1,3-dipolar cycloaddition reaction between azides and alkynes represents prototypic bioorthogonal click chemistry7 that has been used for rapid detection of glycans in living organisms.
To react with azides at room temperature, terminal alkynes require activation by a Cu(I) catalyst.8, 9 This reaction, discovered independently by Sharpless and coworkers and Meldal and coworkers, is termed copper-catalyzed azide-alkyne cycloaddition (CuAAC) (Scheme 1C). In 2004, Fokin and coworkers developed polytriazoles as the first generation ligands to accelerate this transformation in aqueous solutions (Figure 1A).10 The toxicity associated with Cu(I) initially prevented the application of this reaction in vivo;7 nevertheless, recent progress in ligand design has resulted in highly active catalysts that enable robust glycan ligation in living systems, including cultured bacteria, mammalian cells, and zebrafish, within a time-scale of minutes at non-toxic concentrations (Figure 1B).11–13 The development of non-toxic Cu(I) catalysts opens the possibility of applying CuAAC to track glycans in higher order mammals such as mice.
Figure 1.

The first (A) and the second generation (B) tris(triazolylmethyl)amine-based ligands that accelerate CuAAC.
Alkynes can also be activated by raising their ground-state energies using ring strain, a variant of azide-alkyne cycloadditions commonly known as copper-free click chemistry (Scheme 1D). Cyclooctynes and seven-membered thiacycloalkynes are strained alkynes that react rapidly with azides in the absence of Cu(I). The first generation strain-release-promoted cycloaddition proceed with modest reaction rates in the order of k = 10−3 M−1s−1. Further development led to gem-difluoro,14 biaryl fused cyclooctynes15 and biarylazacyclooctynones16 that exhibit considerately enhanced reactivity (rate constants ~0.1 – 1.6 M−1s−1) (Figure 2). While these rates are sufficiently fast for most applications, they are still significantly slower than the CuAAC reaction.
Figure 2.
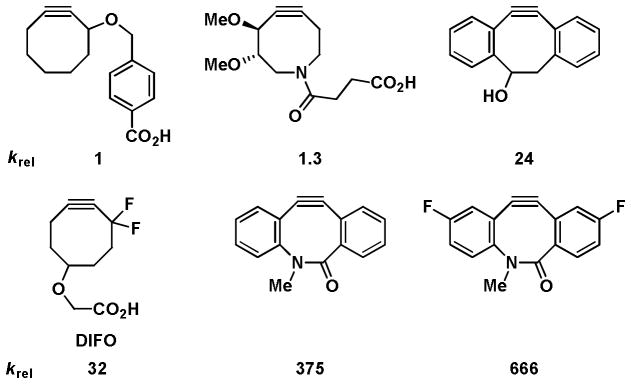
Relative reactivity of cyclooctyne and biarylazacyclooctynone probes.
The Staudinger ligation is another reaction involving the azide that utilizes a phosphine reagent carrying an electrophilic trap (Scheme 1E). With second-order rate constant in the range of 10−3 M−1 s−1 for alkyl azides, the Staudinger ligation proceeds much more slowly in aqueous media compared to the reaction of azides with cyclooctynes. In addition, phosphines are prone to air oxidation, which often necessitates the use of high micromolar (>250 μM) concentration of these reagents and hours of reaction time. Surprisingly, this reaction shows higher efficiency than the copper-free click chemistry in mice, presumably due to the superior bioavailability of the phosphine probes and nonspecific reactivity of cyclooctyne reagents towards thiol-containing biomolecules.
In the oxidative environment of the cell surface, cross-coupling reactions between thiols and electrophiles such as maleimides to generate thioethers have been considered as ‘bioorthogonal’ (Scheme 1F). Yarema and coworkers showed that thiol-containing sialic acids on the cell surface could be detected using a maleimide-functionalized biotin.17 This selectivity is achieved by the positioning of the thiol label in the outer periphery of the cell surface where the cysteine residues in membrane proteins are either oxidized to disulfides, bound to metal ions or buried under the glycocalyx.
Besides unnatural monosaccharide building blocks harboring chemical handles for bioorthogonal ligation, MOE also allows incorporation of monosaccharides containing photo-crosslinkers into cellular glycans, a strategy that has been successfully used to capture protein binding partners. Among the photo-crosslinkers available, the most popular ones that have been applied to glycan labeling are diazirine and aryl azide.
It took over 20 years before MOE was applied to label glycans other than sialic acid. By realizing that all mucin-type O-linked glycans share the core structure of N-acetylgalactosamine α linked to Thr/Ser, the Bertozzi lab introduced the peracetylated N-azidoacetylgalactosamine (Ac4GalNAz), an azido analog of GalNAc, to label mucins found on the cell surface (Scheme 2).18 In the cytoplasm, Ac4GalNAz is deacetylated and activated into UDP-GalNAz, which serves as the donor substrate for polypeptide N-acetylgalactosaminyltransferases (ppGalNAcTs), a group of enzymes that initiate the O-glycosylation of peptide backbones to produce mucins. Recently, however, it was discovered that a large portion of UDP-GalNAz is actually converted into UDP-GlcNAz by an enzyme called UDP-Glc/GlcNAc C4-epimerase and transferred to Thr or Ser residues of cytosolic and nuclear proteins by another enzyme called O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) (Scheme 2). By exploiting this pathway, Ac4GalNAz has also been used as a substrate to label O-GlcNAcylated proteins metabolically.19
Scheme 2.
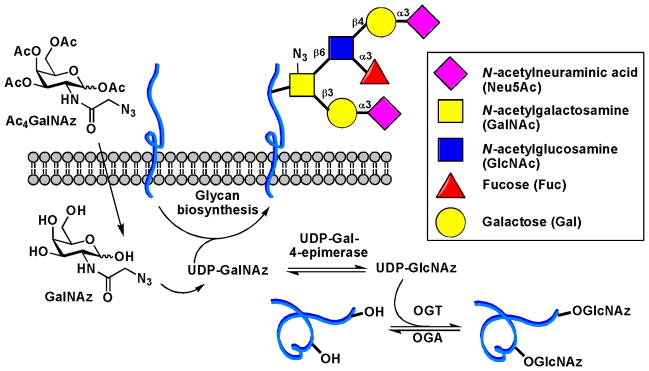
Metabolic labeling of mucin-type O-glycans and O-GlcNAcylated proteins
Within a year of each other, Wong and Bertozzi discovered independently that fucose analogs bearing an alkyne or azide tag at the C-5 position could serve as metabolic precursors to label fucosylated glycans in cultured mammalian cells by exploiting the substrate promiscuity of the GDP-Fucose salvage pathway.20, 21 Vertebrates obtain the majority of their universal fucosyl donor, GDP-Fucose, from the de novo biosynthetic pathway that converts GDP-mannose into GDP-Fucose; therefore, the labeling efficiency through the salvage pathway is low (Scheme 3). To increase the incorporation efficiency of the unnatural fucose into fucosylated glycans and extend this method in vivo, our lab11 and the Bertozzi lab22 developed a strategy to bypass the salvage pathway and use the alkyne-bearing GDP-fucose analog directly as the metabolic substrate to label fucosylated glycans in zebrafish embryos. Using the enzymatic approach developed in our lab,23 GDP-fucose analogs designed for “click” ligation were synthesized in vitro and microinjected into zebrafish embryos at one-cell stage, allowing detection by CuAAC and copper-free click chemistry at various developmental stages starting from 2.5 hours post-fertilization (hpf). With fucose being the most recent example, only four of the nine common mammalian monosaccharides have been probed using MOE thus far.
Scheme 3.

GDP-Fucose de novo biosynthetic pathway and salvage pathway in vertebrates.
So far we have discussed how to enhance cellular uptake of metabolic precursors and choose a bioorthogonal tag for your glycan of interest. The site on a monosaccharide building block to which reactive tags are introduced is another critical factor that glycan engineers must take into consideration. As exemplified by ManNAc, the committed metabolic precursor of sialic acid, only the acyl side chain and the C4-OH in this monosaccharide can be chemically modified, permitting productive incorporation into sialylated glycans. Other positions in ManNAc either participate in phosphorylation (C6-OH) or aldol condensation with pyruvate (C1), catalyzed by glycan processing enzymes, or are involved in the formation of the six-membered hemiacetal (C3-OH and C5-OH, in ManNAc and sialic acid, respectively) (Figure 3). When administered to cells, ManNAc analogs modified at the acetyl side chain are metabolized to sialic acid and incorporated in both N- and O-linked glycans, as well as in glycolipids. Interestingly, when the C4-OH of ManNAc is substituted by the azide, only the O-linked glycans are modified. Sialic acid analogs, when protected as esters, can serve the metabolic substrates directly. Reactive groups have been successfully introduced into the N-acyl and the C9 positions of these substrates (Scheme 1).
Figure 3.
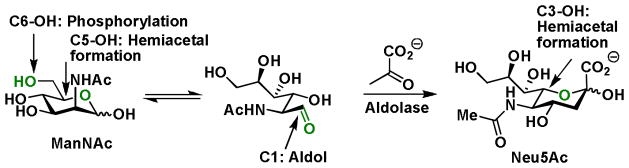
Functional groups in ManNAc participating in enzymatic transformations and hemiacetal formation.
Chemoenzymatic methods for labeling glycans
The elongation of glycan chains naturally occurs through an ordered, enzymatic addition of activated monosaccharides, i.e. nucleotide sugars, a process taking place in endoplasmic reticulum and Golgi. Inspired by nature, the Hsieh-Wilson laboratory developed an alternative method to metabolic labeling for the detection of cytosolic and nuclear proteins modified by O-GlcNAc. This method mimics the natural process of enzymatic addition and utilizes an engineered 1,4-galactosyltransferase to transfer a ketone-tagged UDP-N-acetylgalactosamine analog to the GlcNAc residues in cell lysates.24 When the ketone is replaced with an azide, this method can be extended to image O-GlcNAcylated proteins in fixed cells;25 the azide alleviates the problem of nonspecific ketone-aminooxy condensation in the cytoplasm.
In mammalian organisms, GlcNAc is the only glycan found on cytosolic and nuclear proteins. Other glycans, after being processed in the secretory pathway, are transferred to the plasma membrane or secreted as glycoproteins or glycolipids. These glycans are built from monosaccharides in various linkages. It is these specific linkages that confer glycans unique properties that enable specific molecular recognition. Until recently, methods only existed for labeling glycans that contain a particular monosaccharide motif amenable to modification by the metabolic strategy. Due to the diversity in their structures, higher-order glycans (i.e. di-, tri- and polysaccharides) of specific composition cannot be uniquely labeled by hijacking their biosynthetic pathways with unnatural monosaccharide building blocks. In 2009, our laboratory described a chemoenzymatic approach to label N-acetyllactosamine (LacNAc, Galβ1,4GlcNAc), an ubiquitous disaccharide glycan found in many cell surface glycoproteins.26 In this method, a chemically modified GDP-fucose analog containing an azide- or a terminal alkyne is transferred specifically to the LacNAc disaccharide using an α1,3 fucosyltransferase, the enzyme involved in the generation of the Lewis X trisaccharide. The labeled LacNAc can then be detected using CuAAC or copper-free click chemistry (Scheme 4). We showed that the modified fucose could be used to analyze the changes of LacNAc during lymphocyte activation processes and image LacNAc in live zebrafish embryos. Following this initial work, the Hsieh-Wilson group showed that a combination of BgtA, bacterial homolog of the human blood group A antigen glycosyltransferase, and UDP-GalNAz allows the labeling of cell-surface Fuc α(1–2)Gal, a disaccharide epitope involved in many physiological and pathological processes (Scheme 4).27 To our knowledge, the works published by our laboratory and Hsieh-Wilson represent the only chemical methods for probing polysaccharides in living systems.
Scheme 4.

Chemoenzymatic labeling of cell-surface polysaccharides.
Direct chemical remodeling of glycans on the cell surface
Glycans containing vicinal diols (e.g. sialic acid) can be oxidized efficiently by periodate to generate aldehydes directly on the cell surface, allowing ligation with hydrazide and aminooxy nucleophiles. Traditionally an acidic pH of 5.5 is required to achieve productive reactivity.28 This condition is toxic to most cells, severely limiting the use of this technique to label glycans in living systems. Recently, Dawson discovered that aniline catalyzes the formation of oxime in near quantitative yield.29 Further improvement of this protocol enabled Paulson and coworkers to label sialylated glycoproteins on the surface of live cell with aminooxy probes upon periodate treatment under neutral conditions.30
Imaging glycans in living organisms
The debut of glycan labeling methods based on bioorthogonal chemistry paved the way for imaging glycans in living organisms. With the success of pilot studies in cultured mammalian cells, Bertozzi and coworkers demonstrated mucin-type O-linked glycans in zebrafish embryos can be tagged with the azide by metabolic treatment with Ac4GalNAz.31 Covalent labeling with DIFO-conjugated fluorescent probes via the copper-free click chemistry enabled the labeled glycans be visualized using fluorescence microscopy in subcellular resolution. The jaw region, pectoral fins, and olfactory organs were identified as the hotspots of O-glycan biosynthesis at 60 hpf. This study represents the first example of visualizing dynamic glycosylation in a living organism. Following this pioneering work, analogous approaches have been developed to follow the biosynthesis of sialylated and fucosylated glycans in developing zebrafish (Figure 4A),31,11, 22 and the trafficking of mucin O-glycans in C. elegans (Figure 4B).32
Figure 4. Imaging of glycans in living organisms.
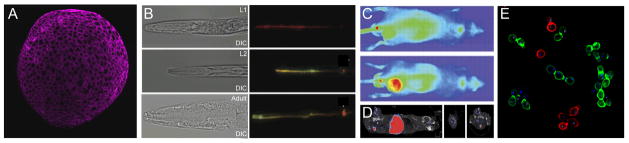
(A) A fluorescent image of Alexa Fluor 647-labeled fucosides in a zebrafish embryo (10 hpf). (B) Fluorescent images of old (green) and newly synthesized O-glycans (red) in C. elegans at different developmental stages (right panels), corresponding differential interference contrast (DIC) images (left panels) (adapted from reference 32). (C) Far-red fluorescence imaging of sialylated glycans in a mouse tumor model. Upper image shows signals from a vehicle control, lower image shows NA649 signal from a metabolically labeled mouse co-localizing with flanked tumor. (D) SPECT and X-ray CT fusion images of the same tumor model as in (C) labeled with DOTA-111In-conjugated neutravidin (adapted from reference 33). (E) Fluorescence images of S. cerevisiae with cell surface N-linked glycans labeled with Alexa Fluor 488 (green) or 647 (red) dye (adapted from reference 34).
Zebrafish and C. elegans are ideal models for fluorescence imaging because of their optical clarity. To image glycans in non-transparent organisms, far-red fluorescence imaging and single-photon-emission computed tomography (SPECT) have been applied. As demonstrated by Brindle and coworkers, sialylated glycans in tumor-implanted nude mice can be labeled by Ac4ManNAz introduced through intraperitoneal injection33. The Staudinger ligation was then employed to ligate a biotinylated phosphine probe, allowing detection of the labeled sialic acids by Neutravidin-Dylight-649 (NA649; Figure 4C) or DOTA-111In-conjugated neutravidin (DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid). Positive signal produced by the azido-dependent labeling was primarily located in tumors (Figure 4D). The upregulation of sialylated glycans is strongly correlated with malignancy for many tumors; therefore, this technique has the potential to be translated into a clinical setting to monitor the progression of cancer.
The glycans produced by Saccharomyces cerevisiae are relatively simple to engineer as compared to vertebrate organisms due to the well-established genetic approaches to screen for glycosylation mutants in this species. Breidenback et al. genetically modified the yeast to disrupt the de novo synthesis of UDP-GlcNAc, introduce a GlcNAc salvage pathway, and force the cells to scavenge the exogenously delivered GlcNAc analogs to survive. As verified by enzymatic digestion and MS analysis, azide or alkyne-bearing GlcNAc was incorporated into the core structure of N-linked glycoproteins, allowing labeling via CuAAC and copper-free click chemistry and subsequent fluorescence visualization (Figure 4E).34 By incorporating a stoichiometrically defined mixture of GlcNAc isotopologs into the same position, this method also allows the identification of 133 N-glycosites in 58 yeast proteins, nearly doubling the number of experimentally observed N-glycosites in the yeast proteome.35
To date, much of the research in this field has been centered on glycans in eukaryotic organisms, in part because prokaryotes were historically believed to be non-glycosylated beyond their cell walls. The discovery of protein glycosylation machineries in bacteria has inspired the development of tools to detect glycoconjugates of bacterial origin in their native environment.36 The metabolic labeling approach coupled with the recent advances in chemoenzymatic preparations of unnatural glycan precursors has allowed Davis and coworkers to develop a brilliant method to detect Mycobacterium tuberculosis in infected macrophages.37 This method employs a fluorescein-tagged trehalose analog as a metabolic substrate to label live M. tuberculosis (Scheme 5). Trehalose is a disaccharide found on the outer membrane of the cell envelope of the bacterium in free form or as esters of mycolic acids. The team showed that by treating infected macrophage with the unnatural trehalose, the fluorescently labeled disaccharide entered the host cell and was selectively integrated into the membrane of M. tuberculosis. This method may be transformed in a new diagnostic assay to detect and track tuberculosis through the body.
Scheme 5.
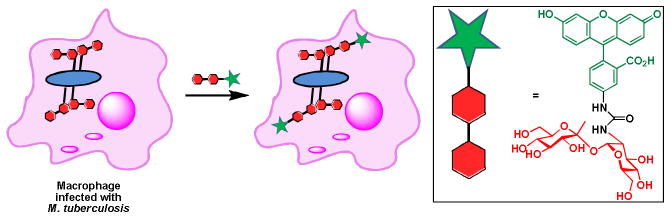
Detection of M. tuberculosis in infected macrophages.
Identification of glycoproteins via bioorthogonal tagging
Protein glycosylation is the most complex and prevalent form of posttranslational modification. Bioinformatic analyses of eukaryotic genomes have suggested that over 50% of predicted proteins are likely to be glycosylated.38 These glycoproteins are involved in a variety of physiological processes, including angiogenesis, fertilization and neuronal development. Changes in cell surface glycosylation mark the onset of cancer and inflammation. Glycoproteins are also found in certain prokaryotes,36 including Campylobacter jejuni, a pathogenic bacterium that causes inflammation of the gastrointestinal tract, and human gut symbionts Bacteroides.39 Defects in protein glycosylation not only abrogate the competitiveness of Bacteroides in colonizing mammalian intestine, it also hinders the in vitro growth of the bacteria.40 Despite their broad biological significance, the vast majority of glycoproteins in both eukaryotic and prokaryotic systems have not been characterized at the molecular level, and the functions of their glycans remain largely unknown.
Traditional methods to enrich glycoproteins for MS identification rely on lectin affinity chromatography, a method that is often complicated by nonspecific binding to irrelevant glycans and low affinity toward the desired glycan epitope.41 The chemical reporter strategy based on metabolic or chemoenzymatic labeling provides a more specific and reliable alternative to capture glycoproteins for the downstream MS analysis. By tagging the glycan of interest with a bioorthogonal functional group as a reactive handle, a biotin probe can be introduced in the subsequent step using the aforementioned ligation reactions, permitting enrichment of the glycoprotein targets using immobilized streptavidin. As demonstrated by Wong and coworkers, sialylated glycoproteins in mammalian cells labeled with peracetylated N-(4-pentynoyl) mannosamine (Ac4ManNAl) can be efficiently enriched using this method.42 Following on-bead trypsin digestion and PNGase-F treatment, N-linked glycoproteins that are sialylated were selectively released and identified by liquid chromatography coupled to tandem mass spectrometry (LC-MS2). By exploiting the fucose transporter found on the bacterial membrane, our group developed a method that allows an alkyne bearing fucose analog to be metabolically incorporated into Bacteroidales species for glycoproteomic analysis.43
Similarly, Hsieh-Wilson and coworkers used the chemoenzymatic approach developed in their laboratory to probe O-GlcNAcylated proteins in crude cell lysates.44 They discovered that phosphofructokinase 1(PFK1), a major regulatory enzyme of glycolysis, is modified by O-GlcNAc under conditions of rapid tumor growth. Hyper O-GlcNAcylation of PFK1 was shown to decrease its enzymatic activity and reshuffle metabolic flux to produce metabolites essential for DNA biosynthesis and antioxidants to quench reactive oxygen species, thus supporting tumor growth. These results implicate the blocking of O-GlcNAcylation on PFK1 as a novel cancer therapeutic.
Recently, the Hsieh-Wilson group extended the application of this chemoenzymatic strategy to quantify the stoichiometry of O-GlcNAc modification of cyclic AMP-response element binding proteins (CREB), a transcription factor involved in the formation of long-term memories.45 O-GlcNAcylated proteins in cell lysates were labeled with 2-keto galactose analog and reacted with a 2kD aminooxy-functionalized PEG tag to shift the molecular weight of the labeled species. Immunoblot analysis confirmed that a large fraction of CREB (44–48%) was monoglycosylated in cultured cortical neurons under basal conditions. Ser40 was found to be the major site of glycosylation, and this modification repressed basal transcription level of the CREB-mediated genes. Mutation of Ser40 to Ala in CREB enhanced both axonal and dendritic growth and long-term memory consolidation. Importantly, CREB requires phosphorylation at Ser133 for its activity. Neuronal stimuli enhanced O-GlcNAcylation of the Ser133-phosphorylated CREB subpopulation. As the result, CREB-induced gene expression was attenuated.
As discussed previously, glycans mediate biological recognition events by their dynamic interactions with lectins. For example, NeuAca2-6Gal epitope interacts with receptor CD22 on the same cell (in cis) and opposing cells (in trans) to regulate B-cell receptor signaling. To identify glycoproteins containing NeuAca2-6Gal that interact with CD22, the Paulson group exploited the strategy of MOE to incorporate an aryl-azide moiety to the C-9 position of cell-surface sialic acid (Scheme 6A).46 Upon the UV radiation, glycans were crosslinked to their receptors in situ. Using this method, they discovered that CD22 itself serves as the cis-ligand and forms homomultimetric complexes. By photo crosslinking cell-surface NeuAca2-6Gal that contains the aryl-azide with a CD22-Fc chimera, B-cell receptor IgM along with a group of 27 glycoproteins was found to be a major in situ trans-ligand of CD22. As showed by immunofluoresent staining, IgM is selectively redistributed to the site of cell contact upon binding to CD22 of the opposing cells. Recently, this photocrosslinking strategy also found applications beyond probing sialic acid-binding proteins. For example, Kohler and coworkers installed diazirin into O-GlcNAc as the photoactivable crosslinker to capture proteins that bind to O-GlcNAcylated proteins (Scheme 6C)47. Using this method, they observed that O-GlcNAcylated neucleoporins containing phenylalanine-glycine repeats were associated with nucleus transporter factor, suggesting that O-GlcNAc residues are involved in essential recognition events in nuclear transport.
Scheme 6.
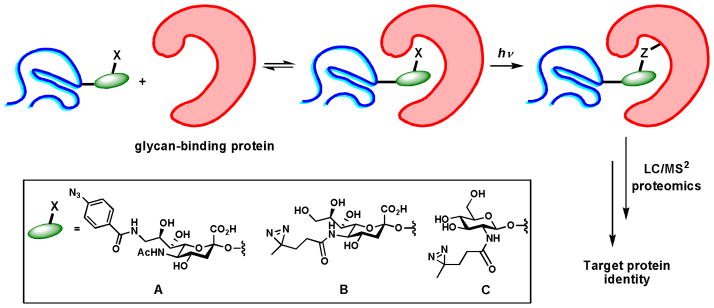
Identification of glycan-binding protein partners via photo crosslinking. (A, B) Unnatural sialic acid analogs used to capture sialic acid binding proteins. (C) The unnatural GlcNAc analog used to capture proteins that bind to O-GlcNAc-modified proteins.
Chemically engineered glycans for therapeutic applications
The chemically modified unnatural glycans have primarily been used for imaging and proteomics studies. However, there is sufficient evidence to suggest that these analogs may also be employed as therapeutics.
Several studies have shown that subtle structural changes in glycans could have profound impact on their interactions with cell-surface receptors. For example, early work done by the Pawlita group showed that modifications of the N-acyl group could drastically enhance or abolish host susceptibility to sialic acid-dependent viral infections.48 They treated mammalian cells with ManNAc analogs including N-propanoyl mannosamine (ManNProp), N-butanoyl mannosamine (ManNBut) and N-pentanoyl mannosamine (ManNPent), and confirmed that about 50% of cell surface sialic acids were replaced by the corresponding analogs with elongated N-acyl side chains. The presence of the modified sialic acids efficiently blocked the infection with B-lymphotropic papovavirus. Interestingly, cells treated with ManNProp and ManNBut showed enhanced infection by human polyomavirus whereas the viral infection was abolished in cells treated with ManNPent.
The C9 position of sialic acid is another hotspot frequently sought for potential therapeutic applications. For example, studies have shown that acetylation of the C9 hydroxyl group reduces the interaction of sialic acid with factor H,49 a regulator of complement activation, and siglec-9, a sialic acid binding lectin.50 Given the importance of both interactions in immunosignaling, the C9 modified sialic acid may find use in immunotherapy. Recently Paulson and coworkers discovered that by incorporating bulky substituents into the same position of sialic acid, high affinity ligands of CD22, a member of the siglecs family of sialic acid binding lectins, were generated.51 The restricted expression of CD22 on B cells inspired the team to evaluate the feasibility of using these glycan ligands as targeting epitopes for selective drug delivery to treat B lymphoma.52 They formulated liposomes functionalized with a high affinity glycan ligand of CD22 and found that these nanoparticles efficiently delivered encapsulated doxobubicin to tumors in a systemic Daudi B lymphoma model in NOD/SCID (non-obese diabetic/severe combined immunodeficient) mice (Scheme 7). Mice treated with the glycan-functionalized liposomes showed significantly extended mean time of survival compared to their counterparts treated with the unfunctionalized liposomes.
Scheme 7.
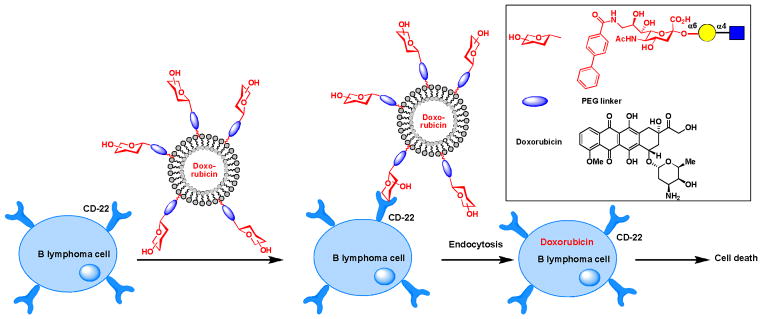
Using glycans as targeting epitopes to deliver drugs selectively to B lymphoma cells in mice models.
Glycans make up a majority of the extracellular matrix on stem cells and play important roles in maintaining the stem cell niche. As early as 1998, Schmidt et al. showed that stem cell development could be manipulated by metabolically introduced unnatural glycans.53 When they fed ManNProp to various progenitor cells, it was discovered that this unnatural sugar stimulates the proliferation of astrocytes and microglia in vitro. In a recent example, Yarema and coworkers showed that by introducing a thiol residue to sialic acids, human embryoid body-derived stem cells were differentiated into cells morphologically similar to neurons.17 These studies suggest that MOE may have therapeutic application in tissue engineering.
Historically, upregulation of sialic acid-containing glycans is strongly correlated with the transformed phenotype of many cancers.54 Moreover, a strong correlation between the level of cell-surface sialic acids and metastatic potential has been observed in several different tumor types.55 These observations prompted glycobiologists to explore the possibility of engineering non-human, sialic acid-like epitopes on the surface of tumor cells that can be targeted by the immune system as a means to eradicate cancer.56 Recently, the metabolic approach has been exploited to treat a mouse model of myopathy, a muscular disease resulted from sialic acid deficiency, in which peraceylated ManNAc was administered to the animals systemically.57 However, for cancer treatment, targeted delivery of metabolic precursors specifically to cancer tissues is desired.
Two popular approaches have been developed to achieve cancer specific targeting: caged substrates that are liberated by enzymes produced by the cancer target and substrates conjugated to targeting elements that bind to cancer-specific surface receptors. In 2010, the Bertozzi lab exploited the first approach to achieve targeting specificity of metabolically introduced sialic acid in cultured cells.58 A caged ManNAz was synthesized that was attached to a peptide substrate for prostate-specific antigen protease specifically expressed on the surface of prostate tumors (Scheme 8A). When the caged substrate came into contact with the protease, the peptide was removed, setting free ManNAz to enter the metabolic cycle. Earlier this year, the Chen lab used the second strategy to realize the same objective.59 They encapsulated azido-modified sialic acid in liposomes functionalized with folic acid to target folate receptors upregulated in tumor cells (Scheme 8B). Targeted delivery of the azido sialic acid to cancer cells expressing folate receptors was achieved with high specificity as verified by fluorescence microscopy following the CuAAC-based labeling. With the success of these two in vitro studies, it is foreseeable that similar approaches may be tested in animal models for the delivery of sugar analogs selectively to tumors as a new metabolic therapy.
Scheme 8.
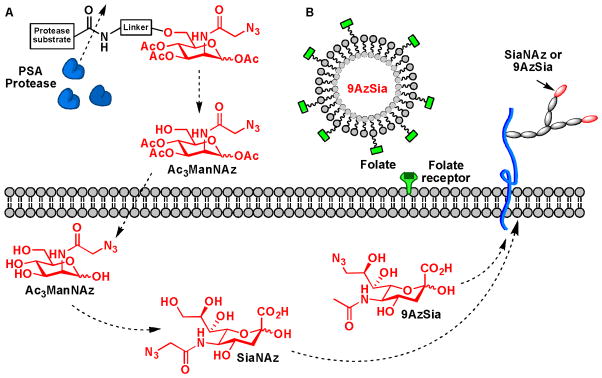
Targeted delivery of unnatural monosaccharide metabolites. (A) An uncaging strategy for targeted delivery of ManNAz to cancer cells. (B) Using folic acid as the targeting epitope.
Likewise, cell surface glycan remodeling via enzymatic approaches also holds potentials for generating therapeutic interventions. As shown by Sackstein et al., in situ fucosylation could transform the migration property of mesenchymal stem cells (MSCs) and direct their trafficking to bone.60 As a promising candidate for treating congenital and acquired bone diseases such as osteogenesis imperfect, the clinic applications of MSCs were hurdled by their poor osteotropism. Sackstein et al. discovered that sialyl Lewis X, a fucosylated glycan epitope, could be created on the cell surface of mesenchymal stem cells by fucosylation using a human α1,3 fucosyltransferase ex vivo. When introduced to the host NOD/SCID mice, the engineered MSCs infiltrated bone marrow within hours of infusion.
Future directions and concluding remarks
The relatively nascent field of chemical glycobiology has produced beneficial connections between scientists in genetics, cell biology and chemistry. Through the joint efforts of researchers from these newly connected fields, novel tools have been created to expedite the probing of glycans in cells and organisms and have provided new insight into their in vivo functions. To address untackled problems in the field, the future of glycosciences will depend heavily on the development of imaging methods for the characterization of the biosynthesis, metabolism and dynamic movement of glycans in living systems. As the repertoire of enzymes available for the chemical synthesis of complex polysaccharides expands, there will be a simultaneous need for alternative applications of these enzymes to label cell-surface polysaccharides for molecular imaging or proteomic identification. Additionally, methods to perform in vivo SAR studies beyond sialic acid will provide mechanistic insights in the design of therapeutics for the treatment of disorders caused by aberrant glycan-lectin interactions. Glycans are intrinsically involved in every major human disease; through the development of new chemical tools we are beginning to elucidate the role of glycans in the onset and progression of these pathological processes.
Acknowledgments
The author’s work that was highlighted in this review was supported by the National Institutes of Health to P.W. (GM080585 and GM093282), the Mizutani Foundation for Glycoscience, and DuPont (DuPont Young Professor Award).
Footnotes
Part of the carbohydrate chemistry themed issue.
References
- 1.Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Nat Rev Mol Cell Biol. 2005;6:885–891. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]
- 2.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attreed M, Desbois M, van Kuppevelt TH, Bulow HE. Nat Meth. 2012;9:477–479. doi: 10.1038/nmeth.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grunholz HJ, Harms E, Opetz M, Reutter W, Cerny M. Carbohydr Res. 1981;96:259–270. doi: 10.1016/s0008-6215(00)81876-5. [DOI] [PubMed] [Google Scholar]
- 5.Sarkar AK, Fritz TA, Taylor WH, Esko JD. Proc Natl Acad Sci U S A. 1995;92:3323–3327. doi: 10.1073/pnas.92.8.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahal LK, Yarema KJ, Bertozzi CR. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 7.Baskin JM, Bertozzi CR. Qsar Comb Sci. 2007;26:1211–1219. [Google Scholar]
- 8.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 9.Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 10.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 11.Soriano del Amo D, Wang W, Jiang H, Besanceney C, Yan A, Levy M, Liu Y, Marlow FL, Wu P. J Am Chem Soc. 2010;132:16893–16899. doi: 10.1021/ja106553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano Del Amo D, Wang W, Klivansky LM, Marlow FL, Liu Y, Wu P. Angew Chem Int Ed. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang W, Hong S, Tran A, Jiang H, Triano R, Liu Y, Chen X, Wu P. Chem, an Asian J. 2011;6:2796–2802. doi: 10.1002/asia.201100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ning X, Guo J, Wolfert MA, Boons GJ. Angew Chem Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J Am Chem Soc. 2012;134:9199–9208. doi: 10.1021/ja3000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sampathkumar SG, Li AV, Jones MB, Sun Z, Yarema KJ. Nat Chem Biol. 2006;2:149–152. doi: 10.1038/nchembio770. [DOI] [PubMed] [Google Scholar]
- 18.Hang HC, Yu C, Kato DL, Bertozzi CR. Proc Natl Acad Sci U S A. 2003;100:14846–14851. doi: 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyce M, Carrico IS, Ganguli AS, Yu SH, Hangauer MJ, Hubbard SC, Kohler JJ, Bertozzi CR. Proc Natl Acad Sci U S A. 2012;108:3141–3146. doi: 10.1073/pnas.1010045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawa M, Hsu TL, Itoh T, Sugiyama M, Hanson SR, Vogt PK, Wong CH. Proc Natl Acad Sci U S A. 2006;103:12371–12376. doi: 10.1073/pnas.0605418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabuka D, Hubbard SC, Laughlin ST, Argade SP, Bertozzi CR. J Am Chem Soc. 2006;128:12078–12079. doi: 10.1021/ja064619y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dehnert KW, Beahm BJ, Huynh TT, Baskin JM, Laughlin ST, Wang W, Wu P, Amacher SL, Bertozzi CR. ACS Chem Biol. 2011;6:547–552. doi: 10.1021/cb100284d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Hu T, Frantom PA, Zheng T, Gerwe B, Del Amo DS, Garret S, Seidel RD, 3rd, Wu P. Proc Natl Acad Sci U S A. 2009;106:16096–16101. doi: 10.1073/pnas.0908248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Proc Natl Acad Sci U S A. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, Hsieh-Wilson LC. J Am Chem Soc. 2008;130:11576–11577. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng T, Jiang H, Gros M, Soriano Del Amo D, Sundaram S, Lauvau G, Marlow F, Liu Y, Stanley P, Wu P. Angew Chem Int Ed. 2011;50:4113–4118. doi: 10.1002/anie.201100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaubard JL, Krishnamurthy C, Yi W, Smith DF, Hsieh-Wilson LC. J Am Chem Soc. 2012;134:4489–4492. doi: 10.1021/ja211312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gahmberg CG, Andersson LC. J Bio l Chem. 1977;252:5888–5894. [PubMed] [Google Scholar]
- 29.Dirksen A, Dirksen S, Hackeng TM, Dawson PE. J Am Chem Soc. 2006;128:15602–15603. doi: 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
- 30.Zeng Y, Ramya TN, Dirksen A, Dawson PE, Paulson JC. Nat Meth. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. Science. 2008;320:664–667. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laughlin ST, Bertozzi CR. ACS Chem Biol. 2009;4:1068–1072. doi: 10.1021/cb900254y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neves AA, Stockmann H, Harmston RR, Pryor HJ, Alam IS, Ireland-Zecchini H, Lewis DY, Lyons SK, Leeper FJ, Brindle KM. FASEB J. 2011;25:2528–2537. doi: 10.1096/fj.10-178590. [DOI] [PubMed] [Google Scholar]
- 34.Breidenbach MA, Gallagher JE, King DS, Smart BP, Wu P, Bertozzi CR. Proc Natl Acad Sci U S A. 2010;107:3988–3993. doi: 10.1073/pnas.0911247107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breidenbach MA, Palaniappan KK, Pitcher AA, Bertozzi CR. Mol Cell Proteomics. 2012;11:M111 015339. doi: 10.1074/mcp.M111.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nothaft H, Szymanski CM. Nat Rev Microbiol. 2010;8:765–778. doi: 10.1038/nrmicro2383. [DOI] [PubMed] [Google Scholar]
- 37.Backus KM, Boshoff HI, Barry CS, Boutureira O, Patel MK, D’Hooge F, Lee SS, Via LE, Tahlan K, Barry CE, 3rd, Davis BG. Nat Chem Biol. 2011;7:228–235. doi: 10.1038/nchembio.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apweiler R, Hermjakob H, Sharon N. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 39.Coyne MJ, Reinap B, Lee MM, Comstock LE. Science. 2005;307:1778–1781. doi: 10.1126/science.1106469. [DOI] [PubMed] [Google Scholar]
- 40.Comstock LE. Cell Host Microbe. 2009;5:522–526. doi: 10.1016/j.chom.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Fletcher CM, Coyne MJ, Bentley DL, Villa OF, Comstock LE. Proc Natl Acad Sci U S A. 2007;104:2413–2418. doi: 10.1073/pnas.0608797104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanson SR, Hsu TL, Weerapana E, Kishikawa K, Simon GM, Cravatt BF, Wong CH. J Am Chem Soc. 2007;129:7266–7267. doi: 10.1021/ja0724083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Besanceney-Webler C, Jiang H, Wang W, Baughn AD, Wu P. Bioorg Med Chem Lett. 2011;21:4989–4992. doi: 10.1016/j.bmcl.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, Hsieh-Wilson LC. Nat Chem Biol. 2012;8:253–261. doi: 10.1038/nchembio.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramya TN, Weerapana E, Liao L, Zeng Y, Tateno H, Yates JR, 3rd, Cravatt BF, Paulson JC. Mol Cell Proteomics. 2010;9:1339–1351. doi: 10.1074/mcp.M900461-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu SH, Boyce M, Wands AM, Bond MR, Bertozzi CR, Kohler JJ. Proc Natl Acad Sci U S A. 2012;109:4834–4839. doi: 10.1073/pnas.1114356109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keppler OT, Stehling P, Herrmann M, Kayser H, Grunow D, Reutter W, Pawlita M. J Biol Chem. 1995;270:1308–1314. doi: 10.1074/jbc.270.3.1308. [DOI] [PubMed] [Google Scholar]
- 49.Shi WX, Chammas R, Varki NM, Powell L, Varki A. J Biol Chem. 1996;271:31526–31532. doi: 10.1074/jbc.271.49.31526. [DOI] [PubMed] [Google Scholar]
- 50.Weiman S, Dahesh S, Carlin AF, Varki A, Nizet V, Lewis AL. Glycobiology. 2009;19:1204–1213. doi: 10.1093/glycob/cwp111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blixt O, Han S, Liao L, Zeng Y, Hoffmann J, Futakawa S, Paulson JC. J Am Chem Soc. 2008;130:6680–6681. doi: 10.1021/ja801052g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen WC, Sigal DS, Saven A, Paulson JC. Leuk Lymphoma. 2012;53:208–210. doi: 10.3109/10428194.2011.604755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt C, Stehling P, Schnitzer J, Reutter W, Horstkorte R. J Biol Chem. 1998;273:19146–19152. doi: 10.1074/jbc.273.30.19146. [DOI] [PubMed] [Google Scholar]
- 54.Sell S. Hum Pathol. 1990;21:1003–1019. doi: 10.1016/0046-8177(90)90250-9. [DOI] [PubMed] [Google Scholar]
- 55.Takano R, Muchmore E, Dennis JW. Glycobiology. 1994;4:665–674. doi: 10.1093/glycob/4.5.665. [DOI] [PubMed] [Google Scholar]
- 56.Guo Z, Wang Q. Curr Opin Chem Biol. 2009;13:608–617. doi: 10.1016/j.cbpa.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malicdan MC, Noguchi S, Tokutomi T, Goto Y, Nonaka I, Hayashi YK, Nishino I. J Biol Chem. 287:2689–2705. doi: 10.1074/jbc.M111.297051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang PV, Dube DH, Sletten EM, Bertozzi CR. J Am Chem Soc. 2010;132:9516–9518. doi: 10.1021/ja101080y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie R, Hong S, Feng L, Rong J, Chen X. J Am Chem Soc. 2012;134:9914–9917. doi: 10.1021/ja303853y. [DOI] [PubMed] [Google Scholar]
- 60.Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, Lin CP, Wohlgemuth R. Nat Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]