

Abstract
Internal organs such as the heart, brain, and gut develop left-right (LR) asymmetries that are critical for their normal functions1. Motile cilia are involved in establishing LR asymmetry in vertebrate embryos, including mouse, frog, and zebrafish2-6. These 'LR cilia' generate asymmetric fluid flow that is necessary to trigger a conserved asymmetric Nodal (TGF-β superfamily) signaling cascade in the left lateral plate mesoderm, which is thought to provide LR patterning information for developing organs7. Thus, to understand mechanisms underlying LR patterning, it is essential to identify genes that regulate the organization of LR ciliated cells, the motility and length of LR cilia and their ability to generate robust asymmetric flow.
In the zebrafish embryo, LR cilia are located in Kupffer's vesicle (KV)2,4,5. KV is comprised of a single layer of monociliated epithelial cells that enclose a fluid-filled lumen. Fate mapping has shown that KV is derived from a group of ~20-30 cells known as dorsal forerunner cells (DFCs) that migrate at the dorsal blastoderm margin during epiboly stages8,9. During early somite stages, DFCs cluster and differentiate into ciliated epithelial cells to form KV in the tailbud of the embryo10,11. The ability to identify and track DFCs—in combination with optical transparency and rapid development of the zebrafish embryo—make zebrafish KV an excellent model system to study LR ciliated cells.
Interestingly, progenitors of the DFC/KV cell lineage retain cytoplasmic bridges between the yolk cell up to 4 hr post-fertilization (hpf), whereas cytoplasmic bridges between the yolk cell and other embryonic cells close after 2 hpf8. Taking advantage of these cytoplasmic bridges, we developed a stage-specific injection strategy to deliver morpholino oligonucleotides (MO) exclusively to DFCs and knockdown the function of a targeted gene in these cells12. This technique creates chimeric embryos in which gene function is knocked down in the DFC/KV lineage developing in the context of a wild-type embryo. To analyze asymmetric fluid flow in KV, we inject fluorescent microbeads into the KV lumen and record bead movement using videomicroscopy2. Fluid flow is easily visualized and can be quantified by tracking bead displacement over time.
Here, using the stage-specific DFC-targeted gene knockdown technique and injection of fluorescent microbeads into KV to visualize flow, we present a protocol that provides an effective approach to characterize the role of a particular gene during KV development and function.
Keywords: Developmental Biology, Issue 73, Genetics, Cellular Biology, Neurobiology, Neuroscience, Molecular Biology, Bioengineering, Biophysics, Anatomy, Physiology, Cilia, Zebrafish, Danio rerio, Gene Knockdown Techniques, Left-right asymmetry, cilia, Kupffer's Vesicle, morpholinos, microinjection, animal model
Protocol
Overview of Stage-Specific Zebrafish Embryo Injections
Antisense morpholino oligonucleotides (MO), which bind to a targeted mRNA and disrupt protein expression from that transcript, are widely used in gene knockdown (loss-of-function) studies in zebrafish13,14. Gene Tools, LLC offers MOs that are tagged with either carboxyfluorescein (emits green fluorescence) or lissamine (emits red fluorescence) to detect MO in injected embryos using fluorescent microscopy. By injecting MO into the yolk cell at different stages of zebrafish development, it is possible to deliver the MO to specific compartments of the embryo (Figures 1A-F). MO injected into the yolk between the 1-4 cell stages (0-1 hpf) enters all embryonic cells (Figures 1A, D, D') via connections with the yolk that persist until the 32-cell stage15 to facilitate global knockdown. MO injected into the yolk during midblastula stages (2.5-3 hpf) can enter the progenitors of the DFCs (Figures 1B, E, E') likely through cytoplasmic bridges8 and knockdown gene function specifically in the DFC/KV cell lineage12, without entering most other embryonic cell lineages. As an important control to test whether gene function is required in DFC/KV or also in yolk12,16, it is also possible to restrict MO to the yolk cell by injecting between the dome-30% epiboly stages (~4.5 hpf) after all cytoplasmic bridges have closed (Figures 1C, F, F'). These injections have been used in combination to analyze gene function in DFC/KV cells17-24. To assess fluid flow in KV, fluorescent microbeads are injected into the KV lumen between the 6-10 somite stages (12-14 hpf) and then immediately imaged using videomicroscopy (Figures 1G-J). Microbeads are available that emit red or green fluorescence (or both), so it is possible to use different channels to image fluorescent microbeads and MO.
1. Stage-specific Injection of Morpholinos (MO)
Injection of MO into zebrafish embryos has been demonstrated previously25,26. Here, we briefly describe stage-specific MO injections. Following all injections, embryos are transferred to a Petri dish and incubated at 28.5 °C.
Global MO injection: Collect embryos immediately after fertilization and load them into an injection plate. Load fluorescent MO into a capillary needle and mount the needle onto a microinjector (e.g. Harvard Apparatus PLI-90 Pico-injector). Break the needle tip and adjust injection settings (pressure and/or time) to create an injection drop that has a volume of 1 nl as described25,26.Using a dissecting stereomicroscope, inject 1 nl of MO into the yolk (Figure 1A) of ≥50 embryos per experiment. Work quickly to complete injections by the 4-cell stage.
DFC-targeted MO injection: Collect embryos immediately after fertilization and incubate at 28.5 °C. When the embryos have reached the 256-cell stage (~2.5 hpf), quickly load them into an injection plate and then inject 1 nl of fluorescent MO into the yolk of ≥100 embryos between the 256-cell and 1,000-cell stages (Figure 1B).
Yolk-targeted MO injection: Collect embryos immediately after fertilization and incubate at 28.5 °C. At the dome stage (~4 hpf), load the embryos into an injection plate and then inject 1 nl of fluorescent MO in the yolk of ≥100 embryos between the dome and 30% epiboly stages (Figure 1C).
2. Selecting Injected Embryos for Analysis
- When MO injected embryos reach the 75%-epiboly stage (8 hpf), remove unfertilized and dead embryos and then screen living embryos under a fluorescent dissecting microscope for distribution of the fluorescent MO. Then allow selected embryos to develop at 28.5 °C.
- For global MO injections, select embryos that have fluorescence evenly distributed in throughout all embryonic cells (Figure 1D; Figure 2A).
- For DFC-targeted MO injections, select embryos in which fluorescent MO has diffused throughout the yolk and appears concentrated at the dorsal blastoderm margin (DFCs) (Figure 1E; Figure 2B). At this stage, it can be difficult to visualize fluorescent DFCs due to bright fluorescence in the underlying yolk. Take care to exclude embryos in which the fluorescent MO has incorporated into embryonic cells other than DFCs or remains aggregated at the injection site (Figure 2D).
- For yolk-targeted MO injections, select embryos in which MO fluorescence is evenly distributed throughout the yolk and not observed in any embryonic cells (Figure 1F;Figure 2C). Again, remove embryos in which the fluorescent MO remains aggregated at the injection site.
- Between the 2-4-somite stages (~11 hpf), screen embryos a second time under a fluorescent microscope using higher magnification. Then allow selected embryos to develop at 28.5 °C.
- For global MO injections, ensure selected embryos have MO fluorescence evenly distributed in KV and all embryonic cells (Figure 1D'; Figure 2E).
- For DFC-targeted MO injections, carefully select embryos that have MO fluorescence only in KV cells and the yolk (Figure 1E'). Transgenic embryos that express GFP in KV cells, such as Tg(Dusp6:d2EGFP)27, can aid in the identification of embryos in which MO has been successfully delivered to KV cells (Figure 2F). Discard embryos with MO fluorescence in embryonic cells other that KV cells.
- For yolk-targeted MO injections, select embryos that have MO fluorescence exclusively in the yolk (Figure 1F': Figure 2G).
3. Mounting Embryos to Analyze Fluid Flow in KV
To analyze asymmetric fluid flow in KV of global MO, DFC-targeted MO or yolk-targeted MO injected embryos, carefully dechorionate ~20 embryos between 4-6 somite stages (~11-12 hpf) with sharp forceps.
Prepare 50 ml of 1% low-melting point agarose and aliquot into 5 ml tubes to be maintained as a liquid in a dry bath at 42 °C.
Transfer one embryo to a glass depression slide (United Scientific Supplies, Inc.) and remove as much of the water as possible. Immobilize the embryo by adding enough warm 1% low-melting point agarose to just cover the embryo (too much agarose can interfere with subsequent imaging). As the agarose solidifies, use forceps to position the embryo such that KV is facing up (dorsal view) (Figure 1H). Mount 10 embryos with one embryo per slide. Work quickly to ensure all injections in the next step are completed by the 10 somite stage (14 hpf).
4. Injection of Fluorescent Microbeads into KV
Dilute Fluoresbrite Multifuorescent 0.5 micron diameter Microspheres (Polysciences, Inc.) to 1:50 in sterile water in a 1.5 ml Eppendorf tube.
Mix the 1:50 dilution of microbeads thoroughly by tapping the tube and load 3 μl into a capillary needle. Using the same microinjector used for MO injections (e.g. Harvard Apparatus PLI-90 Pico-injector), break the needle tip with forceps and adjust the injection settings to generate the smallest possible (<0.5 nl) injection drop.
Under a dissecting microscope, align the needle with the KV lumen of the first mounted embryo. Insert the needle into the lumen and inject a small volume (< 0.5 nl) of beads (Figure 1G). A small needle tip and small injection volume are critical to avoid damaging KV.
Inject all mounted embryos. Once an embryo has been injected, a drop of sterile water can be added on top of the agarose to prevent it from drying out. During the course of the injections, the beads will often obstruct the needle opening. This requires re-breaking the needle tip and adjusting the injection volume by modulating the pressure or time setting of the microinjection apparatus. Replace the needle if it becomes blunt enough to cause significant damage during injection.
Screen the injected embryos for successful delivery of beads under an upright fluorescent compound microscope (e.g. Zeiss AxioImager M1) using a 20X objective. First, determine whether KV structure is intact using brightfield illumination (Figure 1J), and then use fluorescence to observe the beads. Select embryos in which beads are present and moving inside the KV lumen. Discard embryos with KV damage or no floating beads in KV. It is easy to miss KV and deliver beads to nearby tissue or the underlying yolk. If unsuccessful, mount another 10 embryos and repeat the injection procedure.
5. Visualization and Analysis of KV Fluid Flow
For each selected embryo with beads inside KV, add a drop of sterile water to cover the agarose and then observe KV under the upright fluorescent compound microscope using a 63X water dipping objective (Figure 1I). Alternatively, a coverslip can be applied for use with non-water dipping objectives and/or inverted microscopes.
Use a high-speed camera mounted on the microscope (e.g. Zeiss AxioCamHSm) to record a 10 sec movie. Record both the beads using the fluorescent channel (approximately 70 frames/sec) and the KV lumen using a brightfield or differential interference contrast (DIC) channel.
To visualize all bead movements over time, import the movie of fluorescent beads into ImageJ software (free download at http://rsb.info.nih.gov/ij/) and create a maximum projection of all fluorescent signals in the movie. To perform the maximum projection in ImageJ, click "Image→Stacks→ Z project." In the "Z project" window, project all slices with projection type of "Max intensity". This image can be superimposed on a DIC image of the KV in which the beads were imaged (Figures 3A-B).
The movements of individual beads can be tracked using ImageJ. A plugin ("Manual Tracking") can be downloaded at http://rsbweb.nih.gov/ij/plugins/track/track.html. Open the fluorescent beads movie in ImageJ and run the Manual Tracking function. In the window of Manual Tracking, check "show parameters" and input parameters for"time intervals" and "x/y calibration". Manually select beads that remain in the focal plane of the movie for ≥50 frames and then track ≥5 beads per embryo. The path and velocity of each bead is generated by the software (Figures 3 C-D).
Representative Results
Stage-specific MO injections provide a useful approach to analyze gene function in specific compartments of the embryo. Figure 1 presents a flow chart of the injection strategies used to test gene function in DFC/KV cells and how to introduce fluorescent beads to visualize fluid flow in KV. The distribution of fluorescent MO in successful stage-specific injected embryos is shown schematically in Figures 1D-F and in live embryos in Figure 2. An unsuccessful MO injection, in which the MO remains aggregated in the yolk cell, is shown in Figure 2D.
Successfully injected embryos-selected based on the localization of fluorescent MO-can be mounted at the 4-6 somite stages for delivery of fluorescent microbeads into the KV lumen (Figures 1G-H) to analyze fluid flow. Videomicroscopy is used to record bead movements in KV (Figures 1I-J), which can be analyzed qualitatively (Figures 3A-B) or quantitatively (Figures 3C-E) using ImageJ software. In a control embryo with normal flow, beads follow counterclockwise paths that can be visualized by making a maximum projection of fluorescent bead positions over time (Figure 3A) or by tracking individual beads over time (Figure 3C). To demonstrate loss of coordinated flow, we injected embryos with MO to knockdown Rho kinase 2b (Rock2b), which we have previously shown disrupts flow18. In Rock2b MO injected embryos, beads move randomly in KV (Figures 3B, D). ImageJ software was used to calculate the velocity of individual bead tracks. The average velocity of 5 beads from control and Rock2b MO embryos is shown in Figure 3E.
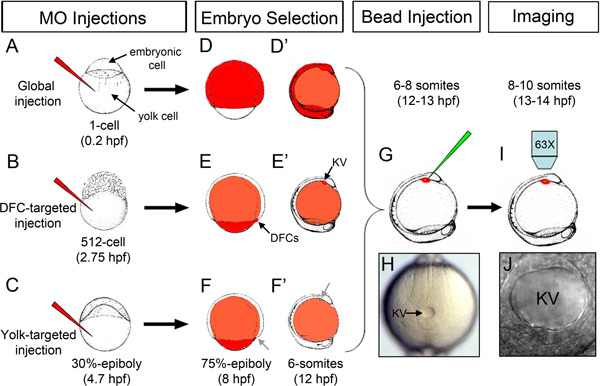 Figure 1. Overview of stage-specific MO injections and microbead injection into KV. (A) Injection of fluorescent MO (red) into the yolk at the 1-cell stage for global distribution of MO throughout the embryo. (B) DFC-targeted MO injection at the 512-cell stage to load MO into DFC/KV cells. (C) Yolk-targeted MO injection at 30%-epiboly stage to restrict MO to the yolk. (D-F') Schematic representation of the distribution of fluorescent MO (red) in successfully injected embryos at the 75% epiboly stage (D, E, F) and the 6 somite stage (D', E', F') following stage-specific injections. MO accumulates in the DFC/KV cell lineage when injected at the 512-cell stage (arrows in E, E'), but not when injected at 30% epiboly (arrows in F, F'). (G) Injection of fluorescent microbeads (green) into the KV lumen. (H) A properly mounted embryo with KV (arrow) facing up for microbead injection. (I) Imaging beads movement in KV using an upright microscope. (J) High magnification of an intact KV lumen injected with microbeads. Embryonic stages and embryo drawings are based on ref. 28.
Figure 1. Overview of stage-specific MO injections and microbead injection into KV. (A) Injection of fluorescent MO (red) into the yolk at the 1-cell stage for global distribution of MO throughout the embryo. (B) DFC-targeted MO injection at the 512-cell stage to load MO into DFC/KV cells. (C) Yolk-targeted MO injection at 30%-epiboly stage to restrict MO to the yolk. (D-F') Schematic representation of the distribution of fluorescent MO (red) in successfully injected embryos at the 75% epiboly stage (D, E, F) and the 6 somite stage (D', E', F') following stage-specific injections. MO accumulates in the DFC/KV cell lineage when injected at the 512-cell stage (arrows in E, E'), but not when injected at 30% epiboly (arrows in F, F'). (G) Injection of fluorescent microbeads (green) into the KV lumen. (H) A properly mounted embryo with KV (arrow) facing up for microbead injection. (I) Imaging beads movement in KV using an upright microscope. (J) High magnification of an intact KV lumen injected with microbeads. Embryonic stages and embryo drawings are based on ref. 28.
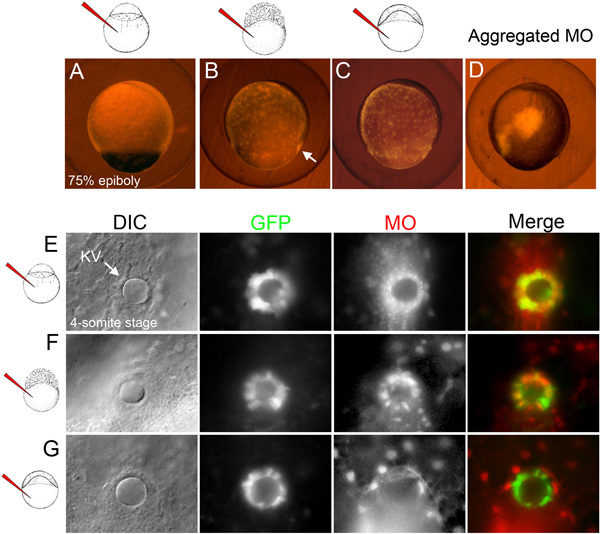 Figure 2. Selection of embryos following stage-specific MO injections. (A-C) Examples of selected embryos at the 75% epiboly stage (8 hpf) in which fluorescent MO (red) has either incorporated in all embryonic cells following global MO injection (A), diffused through the yolk and DFCs (arrow) following DFC-targeted MO injection (B) or remained in the yolk following yolk-targeted MO injection (C). (D) Example of an excluded embryo in which the fluorescent MO aggregated at the injection site. (E-G) Examples of fluorescent MO (red) distribution in selected embryos at the 4-somite stage (11.5 hpf). DIC images identify the KV lumen (arrow) in transgenic Tg(Dusp6:d2EGFP) embryos that express GFP in KV cells27. In the global MO injected embryo, MO was observed in KV and all surrounding cells (E). In the DFC-targeted MO injected embryo, MO co-localized with GFP in most KV cells and was also present in underlying yolk nuclei (F). In the yolk-targeted embryo, MO was found exclusively in yolk nuclei (G).
Figure 2. Selection of embryos following stage-specific MO injections. (A-C) Examples of selected embryos at the 75% epiboly stage (8 hpf) in which fluorescent MO (red) has either incorporated in all embryonic cells following global MO injection (A), diffused through the yolk and DFCs (arrow) following DFC-targeted MO injection (B) or remained in the yolk following yolk-targeted MO injection (C). (D) Example of an excluded embryo in which the fluorescent MO aggregated at the injection site. (E-G) Examples of fluorescent MO (red) distribution in selected embryos at the 4-somite stage (11.5 hpf). DIC images identify the KV lumen (arrow) in transgenic Tg(Dusp6:d2EGFP) embryos that express GFP in KV cells27. In the global MO injected embryo, MO was observed in KV and all surrounding cells (E). In the DFC-targeted MO injected embryo, MO co-localized with GFP in most KV cells and was also present in underlying yolk nuclei (F). In the yolk-targeted embryo, MO was found exclusively in yolk nuclei (G).
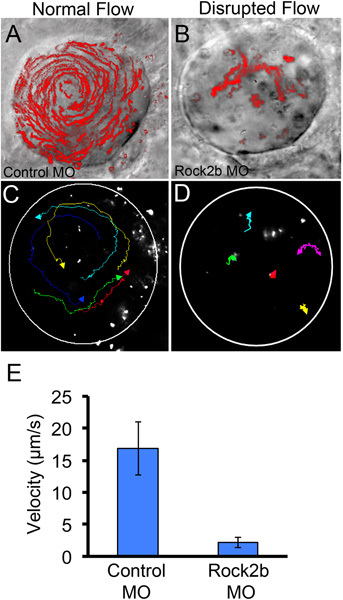 Figure 3. Qualitative and quantitative analyses of fluid flow in KV. (A-B) For qualitative analysis of flow, a maximum projection of a 10 sec movie showing movement of all fluorescent microbeads injected into KV has been superimposed on a DIC image of the KV lumen in a global control MO (A) or Rock2b MO (B) injected embryo. (C-E) To quantify flow, movement of individual microbeads (n=5) was tracked in a control embryo (C) and Rock2b MO injected embryo (D) and used to calculate an average bead velocity (E). Circles (C-D) approximate the KV lumen boundaries. Error bars (E) represent one standard deviation.
Figure 3. Qualitative and quantitative analyses of fluid flow in KV. (A-B) For qualitative analysis of flow, a maximum projection of a 10 sec movie showing movement of all fluorescent microbeads injected into KV has been superimposed on a DIC image of the KV lumen in a global control MO (A) or Rock2b MO (B) injected embryo. (C-E) To quantify flow, movement of individual microbeads (n=5) was tracked in a control embryo (C) and Rock2b MO injected embryo (D) and used to calculate an average bead velocity (E). Circles (C-D) approximate the KV lumen boundaries. Error bars (E) represent one standard deviation.
Discussion
Using stage-specific injections to target MO to the DFC/KV cell lineage is a useful approach to study cell-autonomy of gene function and avoid pleiotropic phenotypes caused by global gene knockdown. However, these injections can be technically challenging. Injection of MO between the 256-cell and 1,000-cell stages can result in three possible outcomes: 1) the MO remains aggregated at the injection site, 2) the MO diffuses throughout the yolk and enters DFC/KV cells or 3) the MO diffuses throughout the yolk and enters DFC/KV cells and other embryonic cells. Therefore, selecting embryos in which MO has moved exclusively to the DFC/KV cells is a key step in this experiment. It is important to note that MO can load into all DFC/KV cells but often enters only a subset of these cells10. The reasons underlying this mosaic targeting remain unclear, but may reflect variability in how efficiently the MO moves through the yolk and/or the timing of closure of individual bridges between the yolk and DFC progenitors. We have not found conditions that increase targeting efficiency, but rather have relied on selecting embryos in which most of the KV cells have taken up MO. The success rate for generating embryos with MO in the majority of DFC/KV cells is typically ~20-30%, but this can be quite variable from day to day. For this reason, we recommend injecting as many embryos as possible (≥100), and then carefully selecting the embryos that have the appropriate distribution of fluorescent morpholino.
Control experiments are vital for interpreting the results of DFC-targeted injections. A mismatch MO or standard negative control MO (from Gene Tools, LLC) should be used to control for non-specific effects. In addition, since DFC-targeted injections deliver MO to both the yolk and DFC/KV cell lineage (Figure 2F), it is important to determine whether phenotypes in these embryos are due to loss of gene function in DFC/KV cells or in the yolk cell. Comparing phenotypes in DFC-targeted embryos with yolk-targeted embryos (Figures 2F-G) will identify DFC/KV cell-specific affects. Of course, some genes may function in both the DFC/KV cells and the yolk16. Although we focus of MO-mediated loss-of-function analyses here, the DFC-targeted injection technique and associated control experiments can be used to assess a gain-of-function by injecting synthetic mRNA to overexpress a particular protein in the DFC/KV lineage17,22,24,29. One clever adaption of this approach used DFC-targeted mRNA injections to rescue global MO knockdown only in DFC/KV cells30. However, it should be noted that not all mRNAs injected in this manner result in expression of the encoded protein in DFCs and KV cells, and important controls should be included to assess the level of protein expression and distribution.
Successful delivery of microbeads into the KV lumen to assess asymmetric flow depends on injecting the beads at optimal stages during the rapid development of the transient KV organ. We begin mounting embryos at the 4-6 somite stages (~12 hpf) and then inject microbeads into KV up to the 10 somite stage (14 hpf). Prior to the 4 somite stage, the lumen is often too small to inject, and at later stages (>10 somites) the injection becomes difficult due to KV moving deeper into the embryo. One limitation of this technique is that requires the KV lumen to expand to a size that can be successfully injected with microbeads. In cases where MO knockdown of a particular gene severely disrupts KV organization and/or reduces KV lumen size (reviewed in ref. 31) it will be difficult, if not impossible, to assay flow using this approach.
When preparing embryos for injection of microbeads into KV, it is important to mount the embryo straight along the embryonic axis with KV facing up (Figure 1H), as embryo position can affect the angle that the needle enters KV and the angle of imaging beads in KV. It is also important to use a small-bore injection needle and small injection volume of microbeads to avoid damaging KV. We recommend practicing to develop a smooth injection stroke to insert the needle into KV, release the beads and then quickly retract the needle from the embryo. When conditions are optimal, we are able to successfully deliver microbeads to ~50% (e.g. n=5/10) of the injected embryos.
In summary, stage-specific MO injections and visualization of microbeads in KV provide an opportunity to study the function of candidate genes in LR ciliated cells.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank Fiona Foley for excellent lab support and zebrafish care. This work was supported an AHA predoctoral fellowship to G.W. (11PRE5730027) and NHLBI grants to H.J.Y. (R01HL66292) and J.D.A. (R01HL095690).
References
- Sutherland MJ, Ware SM. Disorders of left-right asymmetry: heterotaxy and situsinversus. Am. J. Med. Genet. C Semin. Med. Genet. 2009;151C(4):307–317. doi: 10.1002/ajmg.c.30228. [DOI] [PubMed] [Google Scholar]
- Essner JJ, Amack JD, Nyholm MK, Harris EB, Yost HJ. Kupffer's vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development. 2005;132(6):1247–1260. doi: 10.1242/dev.01663. [DOI] [PubMed] [Google Scholar]
- Nonaka S, et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95(6):829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Essner JJ, et al. Conserved function for embryonic nodal cilia. Nature. 2002;418(6893):37–38. doi: 10.1038/418037a. [DOI] [PubMed] [Google Scholar]
- Kramer-Zucker AG, et al. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer's vesicle is required for normal organogenesis. Development. 2005;132(8):1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- Schweickert A, et al. Cilia-driven leftward flow determines laterality in Xenopus. Curr. Biol. 2007;17(1):60–66. doi: 10.1016/j.cub.2006.10.067. [DOI] [PubMed] [Google Scholar]
- Tabin CJ. The key to left-right asymmetry. Cell. 2006;127(1):27–32. doi: 10.1016/j.cell.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Cooper MS, D'Amico LA. A cluster of noninvolutingendocytic cells at the margin of the zebrafish blastoderm marks the site of embryonic shield formation. Dev. Biol. 1996;180(1):184–198. doi: 10.1006/dbio.1996.0294. [DOI] [PubMed] [Google Scholar]
- Melby AE, Warga RM, Kimmel CB. Specification of cell fates at the dorsal margin of the zebrafish gastrula. Development. 1996;122(7):2225–2237. doi: 10.1242/dev.122.7.2225. [DOI] [PubMed] [Google Scholar]
- Amack JD, Wang X, Yost HJ. Two T-box genes play independent and cooperative roles to regulate morphogenesis of ciliated Kupffer's vesicle in zebrafish. Dev. Biol. 2007;310(2):196–210. doi: 10.1016/j.ydbio.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Oteiza P, Koppen M, Concha ML, Heisenberg CP. Origin and shaping of the laterality organ in zebrafish. Development. 2008;135(16):2807–2813. doi: 10.1242/dev.022228. [DOI] [PubMed] [Google Scholar]
- Amack JD, Yost HJ. The T box transcription factor no tail in ciliated cells controls zebrafish left-right asymmetry. Curr. Biol. 2004;14(8):685–690. doi: 10.1016/j.cub.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC. Effective targeted gene 'knockdown' in zebrafish. Nat. Genet. 2000;26(2):216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6(1):69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Law RD. Cell lineage of zebrafish blastomeres. I. Cleavage pattern and cytoplasmic bridges between cells. Dev. Biol. 1985;108(1):78–85. doi: 10.1016/0012-1606(85)90010-7. [DOI] [PubMed] [Google Scholar]
- Arrington CB, Yost HJ. Extra-embryonic syndecan 2 regulates organ primordia migration and fibrillogenesis throughout the zebrafish embryo. Development. 2009;136(18):3143–3152. doi: 10.1242/dev.031492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron A, Xu X, Lin X. Wnt/beta-catenin signaling directly regulates Foxj1 expression and ciliogenesis in zebrafish Kupffer's vesicle. Development. 2012;139(3):514–524. doi: 10.1242/dev.071746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, et al. The Rho kinase Rock2b establishes anteroposterior asymmetry of the ciliated Kupffer's vesicle in zebrafish. Development. 2011;138(1):45–54. doi: 10.1242/dev.052985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aamar E, Dawid IB. Sox17 and chordin are required for formation of Kupffer's vesicle and left-right asymmetry determination in zebrafish. Dev. Dyn. 2010;239(11):2980–2988. doi: 10.1002/dvdy.22431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer JM, Amack JD, Peterson AG, Bisgrove BW, Yost HJ. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature. 2009;458(7238):651–654. doi: 10.1038/nature07753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider I, et al. Zebrafish Nkd1 promotes Dvl degradation and is required for left-right patterning. Dev. Biol. 2010;348(1):22–33. doi: 10.1016/j.ydbio.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, et al. Canopy1, a positive feedback regulator of FGF signaling, controls progenitor cell clustering during Kupffer's vesicle organogenesis. Proc. Natl. Acad. Sci. U.S.A. 2011;108(24):9881–9886. doi: 10.1073/pnas.1017248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X, et al. Na,K-ATPase alpha2 and Ncx4a regulate zebrafish left-right patterning. Development. 2007;134(10):1921–1930. doi: 10.1242/dev.02851. [DOI] [PubMed] [Google Scholar]
- Esguerra CV. Ttrap is an essential modulator of Smad3-dependent Nodal signaling during zebrafish gastrulation and left-right axis determination. Development. 2007;134(24):4381–4393. doi: 10.1242/dev.000026. [DOI] [PubMed] [Google Scholar]
- Rosen JN, Sweeney MF, Mably JD. Microinjection of Zebrafish Embryos to Analyze Gene Function. J. Vis. Exp. 2009. p. e1115. [DOI] [PMC free article] [PubMed]
- Yuan S, Sun Z. Microinjection of mRNA and Morpholino Antisense Oligonucleotides in Zebrafish Embryos. J. Vis. Exp. 2009. p. e1113. [DOI] [PMC free article] [PubMed]
- Molina G, et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 2009;5(9):680–687. doi: 10.1038/nchembio.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203(3):253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Schneider I, Houston DW, Rebagliati MR, Slusarski DC. Calcium fluxes in dorsal forerunner cells antagonize beta-catenin and alter left-right patterning. Development. 2008;135(1):75–84. doi: 10.1242/dev.004713. [DOI] [PubMed] [Google Scholar]
- Clement A, Solnica-Krezel L, Gould KL. The Cdc14B phosphatase contributes to ciliogenesis in zebrafish. Development. 2011;138(2):291–302. doi: 10.1242/dev.055038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Bessho Y. Left-right asymmetry in zebrafish. Cell Mol. Life Sci. 2012. [DOI] [PMC free article] [PubMed]