

Abstract
While drug discovery scientists take heed of various guidelines concerning drug-like character, the influence of acid/base properties often remains under-scrutinised. Ionisation constants (pKa values) are fundamental to the variability of the biopharmaceutical characteristics of drugs and to underlying parameters such as logD and solubility. pKa values affect physicochemical properties such as aqueous solubility, which in turn influences drug formulation approaches. More importantly, absorption, distribution, metabolism, excretion and toxicity (ADMET) are profoundly affected by the charge state of compounds under varying pH conditions. Consideration of pKa values in conjunction with other molecular properties is of great significance and has the potential to be used to further improve the efficiency of drug discovery. Given the recent low annual output of new drugs from pharmaceutical companies, this review will provide a timely reminder of an important molecular property that influences clinical success.
Introduction
From an economic perspective it has become abundantly clear that pharmaceutical companies are struggling despite the increases they have made to research and development expenditure. Fortunately, extensive pre-clinical profiling has helped reduce drug failures due to poor human pharmacokinetics. More recently however, the reasons for drugs falling out of clinical development have been based on commercial decisions as well as problems with formulation and toxicity. Other factors such as more rigorous regulatory scrutiny and the paucity of simple (and druggable) targets have also contributed to the reduced productivity of the industry.1 Some commentators have even questioned the long term viability of the pharmaceutical industry.
To address the problem of high attrition rates and in keeping with good management practice, analysis of the entire drug discovery process has been essential to help companies compete in the current environment. Medicinal chemists have also been actively involved in understanding drug failures by examining and defining the physicochemical properties of compounds that predict successful outcomes.1 The aim of these analyses is to improve the quality of compounds that enter clinical trials and yet there is some debate to the extent these guidelines are being followed.2 Property-based optimisation is of extreme importance to the industry as there is clear evidence that working with large and lipophilic molecules is related to problems concerning promiscuity, metabolism, bioavailability, efflux, solubility and plasma protein binding.3-5
Of key importance to how drugs behave is their acid/base character which affects their biopharmaceutical properties and how they are formulated. In drug discovery, many physicochemical properties are frequently analysed and renewed interest has been placed on ionisation profiles.1, 4-8 In this review an outline will be presented regarding the properties associated with clinical success and their relationship to acid/base character. Specifically, we will examine how charge state affects drug-receptor interactions, pharmacokinetic parameters and the biopharmaceutical properties of drug candidates.
Drug discovery
Having a particular disease state in mind, the drug discovery process typically commences with the identification and validation of a specific macromolecular target. The development of screens allows compounds to be tested in order to identify hits and leads that meet a set of predefined criteria relating to attributes such as potency, functional activity and physicochemical properties. If oral bioavailability is required then testing at this stage can help determine the quality of these ligands. The subsequent process of developing a lead compound into a drug involves a multifactorial optimisation of potency, selectivity and biopharmaceutical properties such as absorption, distribution, metabolism, excretion and toxicity1, 4 (ADMET) (Figure 1).
Figure 1.
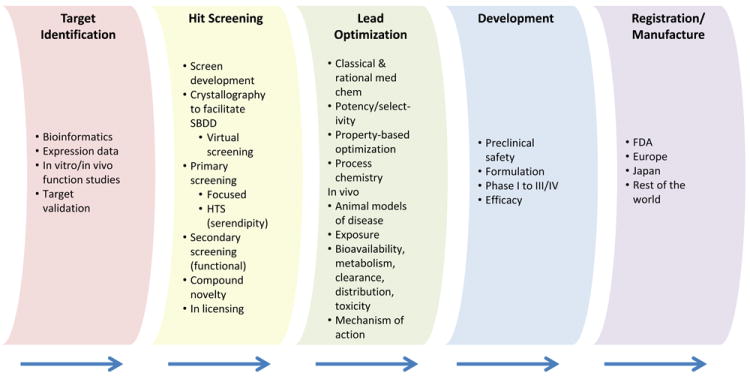
The drug discovery process.
Crucially, the acid/base profile of a compound has a direct effect on the lipophilicity of a substance as governed by the ionisation constants (pKa values) of key functional groups. As such, further studies and an awareness of acid/base profiles for research compounds, clinical candidates and drugs is absolutely required if we wish to understand and monitor lipophilicity. Figure 2 illustrates four important facets of drug development that are influenced by drug acid/base equilibria. Indeed, acid/base character affects drug potency and selectivity, and has a great impact on both pharmacokinetic and biopharmaceutical properties.
Figure 2.
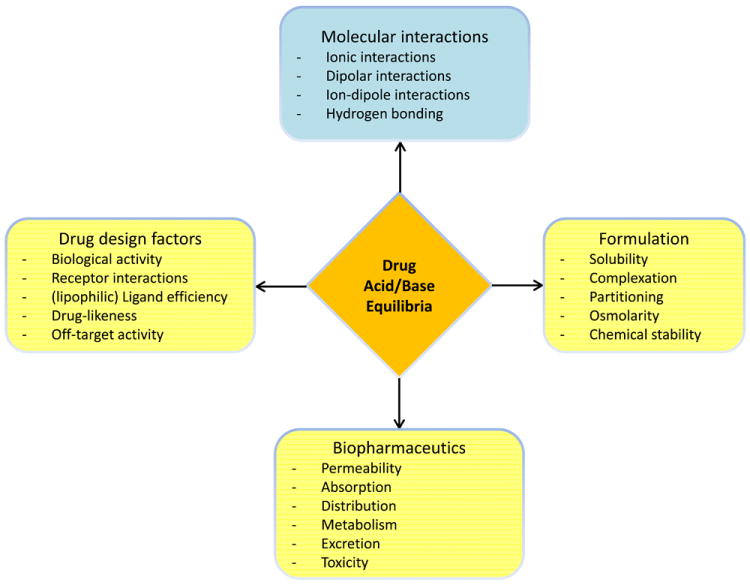
Diagram illustrating the key properties that are influenced by the acid/base character of drugs. Each node highlights an important facet of drug discovery and development.
Biopharmaceutical properties
Efforts to determine the physicochemical properties that relate to long-term compound viability have been conducted in concert with gathering biology data on attributes such as: cell toxicity, efflux liability, metabolic stability or inhibition, cell permeability, bioavailability, CNS permeability, protein binding, brain tissue binding, promiscuity, clearance and volumes of distribution. Armed with this data, relationships have been sought with a range of simple physicochemical properties. Most of this attention has been placed on easily calculated parameters such as molecular weight (MW), ClogP, polar surface area (PSA), the number of hydrogen bond donors/acceptors, aromatic character and the number of rotatable bonds.
One of the first general studies to emerge is the well-known work of Lipinski and co-workers9 who stressed that problems are likely to be encountered with oral absorption if a compound meets two or more of the following criteria: molecular weight > 500, ClogP > 5, H-bond donors > 5 and H-bond acceptors > 10. More recent work has been able to refine these guidelines and this review will only highlight a subset of this research. For a comprehensive analysis of this topic we refer the reader to the review of Meanwell.1
Simple ClogP calculations can be improved by considering charge state in order at biologically relevant pH values. This usually takes the form of an estimate of the octanol/water partition coefficient at physiological pH (7.4), known as ClogD7.4. For example, Pfizer also used a large body of in-house data on cell permeability and in vitro clearance to provide useful associations with physicochemical properties.10 This involved mapping the biology data onto a plot of logD versus MW to demonstrate that compounds having acceptable values fell into an area termed the ‘golden triangle’. Significantly, as the molecular weight increased there was a much narrower band of lipophilicity that could satisfy the attributes of acceptable permeability and clearance.
A further analysis by Wager et al. on CNS drugs resulted in a scoring method that took into account the following properties: MW, ClogP, ClogD7.4, number of hydrogen bond donors, PSA and most basic pKa.11 The Central Nervous System Multiparameter Optimisation (CNSMPO) scoring technique gave an indication of the likelihood of successfully taking a compound into the clinic. This was primarily aimed at CNS drugs but there was a general (drug-like) relationship between the score and key in vitro attributes such as metabolic stability, permeability, toxicity and efflux. In the study, specific attention was given to the basicity of the compounds and basic pKa values above 8.4 did not contribute to the overall CNSMPO score. Importantly, to determine ClogD7.4 values for each substance, estimates of pKa values were required. Basic molecules with high pKa values were shown to have two potential drawbacks as CNS drugs. Firstly, charged cationic drugs show reduced penetration through the blood-brain barrier (BBB). Furthermore, these basic compounds have a higher probability of blocking hERG channels. In this study by Wager and co-workers11, ClogD7.4 values were given an optimal range between 2.0 and 4.0 for CNS drug-likeness.
The guidelines and observations outlined above cover broad drug-like characteristics and most pharmaceutical companies apply variations of these rules to assess their compounds. This approach aims to improve the probability of success by working in low-risk areas and to flag potential problem compounds early on. The danger of providing rules is that they can be mindlessly followed without fully understanding how they should be applied or their limitations. Clearly the acid/base characteristics of compounds within drug discovery have been looked at, however ionisation constants appear to have been largely under-scrutinised. The sections below specifically examine the evidence where acid/base properties have been shown to affect particular attributes of compound behaviour.
Charge state
In order to adequately discuss charge state and pKa values there is a need to outline what is meant by acidic, basic and neutral. Most studies discussed in this review that have examined the charge state properties of drugs classify compounds as acids, bases, neutral or zwitterionic. This is usually sufficient to generalise about the behaviour of compounds in relation to their physicochemical properties.4-6, 8, 12, 13 While this previous work has used pKa values to classify each compound,4-6, 8, 12, 13 the focus was on the nature of compounds at physiological pH. With respect to the term ‘neutral’, a compound may be charge neutral at its isoelectric point, even if the molecule contains an acidic or a basic functional group. In contrast, a compound is classified here as neutral if it does not have a (physiologically) relevant ionizable group (acidic or basic). The emphasis therefore is on the presence of acidic and basic groups within a molecule and the pH under discussion. An acid has been simply classified as a species HA which at a pH above the pKa will dissociate into the anionic A- form and a proton (for a simple monoprotic case). Similarly a basic substance can be depicted as species B that will accept a proton below the pKa value to generate the cationic species.
For our own studies into charge states and pKa distributions7 we also defined various ionisation categories using pKa values. Our definitions were intended to be broader to encompass charge states within the range of pH values seen both physiologically and those encountered for drug formulation. In addition to the categories above we classified certain compounds as ‘always ionised’ such as quaternary bases and acids with pKa values below 0.0, or bases with pKa values above 12.0. Ampholytes were also classified into groups according to the number of acidic and basic groups. Simple ampholytes contain one acid and one base while complex ampholytes contain other combinations of acids and bases.
Common acidic groups include: carboxylates, phenols, sulfonamides, heterocyclic nitrogen atoms, hydroxamates and less frequently, carbon acids, phosphates, tetrazoles, thiols, alcohols, acidic amides, acidic anilines, carbamates, hydrazides, imides and sulfates. Bases on the other hand include heterocyclic nitrogen atoms, aliphatic amines, guanidines, amidines, anilines and basic amides. Further information providing a definition of pKa is given in Box 1.
Box1. Definition of pKa.
When a weak acid dissociates in solution according the scheme below (1), we can express the acid ionisation constant (Ka) using equation (2). The negative logarithmic form of the acid ionisation constant (3) is more commonly used. Rearrangement of equation (3) affords the Henderson-Hasselbalch equation (4). In a similar manner we can employ these equations for the conjugate acid forms of basic functional groups (5).
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
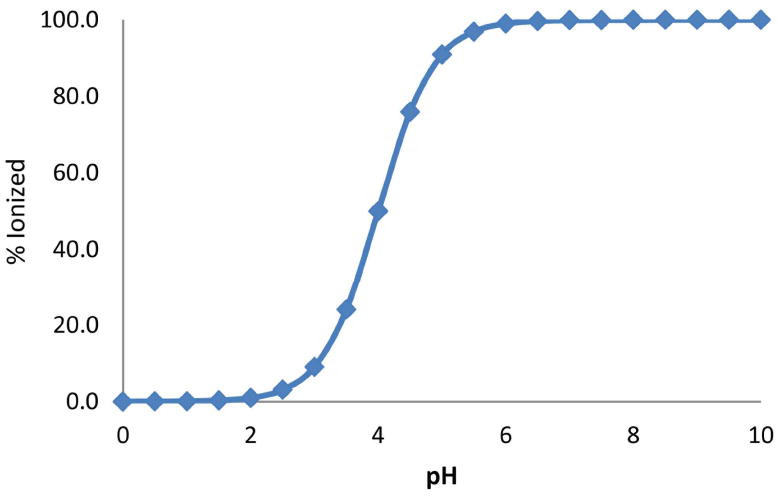
Plot showing the % protonated species against pH for a compound with a single acidic group (pKa 4.0).
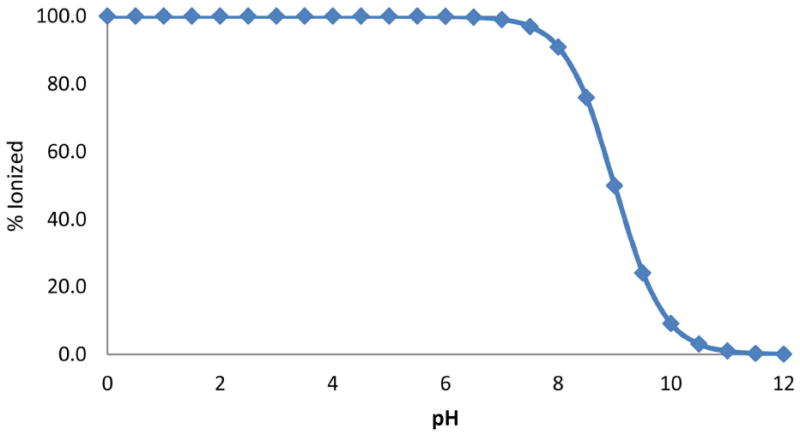
Plot showing the % protonated species against pH for a compound with a single basic group (p Ka 9.0).
Absorption, permeability and bioavailability
Considerable research has been aimed at understanding the factors affecting the absorption of drugs across intestinal membranes. Early studies by Brodie, Hogben and co-workers14 detailed their ‘pH partition hypothesis’ that clearly demonstrated the effect of ionisation state on rates of absorption of drugs from the small intestine. In this pioneering work, they showed that acids with pKa values below 3 and bases with pKa values above 8 were poorly absorbed. Since this study, other research has been undertaken where reference has been made to pKa and ADME behaviour. For example, Palm et al.15 extended the work above showing that the transport of molecules across membranes is more rapid for the uncharged species.
An example demonstrating this principle was shown with a series of three structurally related AT1 receptor antagonists possessing subtle differences in their ionisation state at pH 6.0 and 7.4.16 At a pH of 7.4 all the molecules were ionised resulting in poor permeability in model systems employing Ussing chambers, perfused jejunum loop or Caco-2 transport studies. Conversely at pH 6.0, only one molecule was largely uncharged and this compound alone was significantly absorbed. As well as highlighting the caution needed when using these model systems, this work also suggested that careful consideration should be given to the ionisation profiles of research compounds.16
In a further example, Castro and co-workers17 produced a series of compounds to address a problem of poor oral absorption in a lead compound. The lead had a good in vitro profile, however less than 5% was absorbed by the oral route. A basic amine group with a pKa value close to 9.7 was thought to be the problem. Fluorinated analogues were synthesised which lowered the pKa values of the amine to between 8.0 and 8.8. Subsequent testing showed that there was a considerable improvement in oral absorption for the fluorinated derivatives. The increase in absorption was attributed to the higher proportion of the neutral species in the gut.
Charge state was also highlighted by Martin as an important factor in predicting bioavailability in rats.12 In a larger study, Gleeson4 examined both permeability, using an artificial membrane assay (PAMPA), and bioavailability for a large number of in-house GSK compounds. The rank order of permeability was found to be: neutrals > bases > zwitterions > acids. This was explained in terms of the anionic nature of the membranes used, and was thus rationalised on electrostatic grounds. Gleeson’s study also showed that bioavailability was harder to interpret than permeability as in the former, there are both absorption and clearance components. Ionisation state only had a minor impact on bioavailability yet acids were found to be more bioavailable on average which was in contrast to acid permeability data in general. His research interestingly concurred with previous studies to show that acids had higher oral bioavailability and was likely to be the result of better solubility and lower clearance.4 Bases tend to be protonated in the gastrointestinal tract and therefore have higher polarity and reduced lipophilicity, thus limiting passive absorption across biomembranes.
Simple classification of compounds into acids and bases does not take into account the extent of ionisation which has been considered by previous workers employing pKa values.14-16 Kubinyi18 summarised this concept with the following statement,
“medicinal chemists, who did not care about the pKa values of their acids or bases, are now well aware of the risks that arise from those values being too far away from 7, the neutral pH value.”
Clearly, when we consider the nature of membranes and their lipophilic character, the generalisations above regarding non-ionised states and permeability are fully understandable. Neutral molecules are more readily able to traverse non-polar lipidic membrane environments, unlike charged compounds, where this process is energetically disfavoured.
Despite these studies and the general dogma that the neutral species is greatly favoured in passive transfer across membranes, evidence exists that shows that a proportion of charged molecules are able to be absorbed. In one study, monolayers of Caco-2 cells were used to suggest that ionic species may contribute to overall drug transport.19 While Palm et al. also showed distinct increases in permeability coefficients with increasing fraction of un-ionised (f(u)) drug, they also stressed that in cases where the f(u) was below 0.1 the contribution of the ionised form was significant.15 Quite clearly absorption involves a number of processes and complicates our ability to generalise about drug properties. Acid/base character and pKa values are important determinants for absorption and permeation, however other factors need to be taken into account such as: lipophilicity, size, metabolic lability, efflux mechanisms and hydrophilicity, which have been reviewed previously.9, 20
Volume of distribution, plasma protein binding
While a drug may be adequately absorbed, other attributes can render it poorly distributed or rapidly cleared, such that it is unable to elicit an adequate pharmacological response. The parameter, volume of distribution (Vd), is a theoretical property where large values indicate that a drug is widely distributed, while small values (e.g. 0.1 to 0.2 l/kg) suggest that the compound resides primarily in the systemic circulation. Volume of distribution is a key determinant of pharmacokinetic properties and together with clearance information, the biological half-life of a drug can be determined (half-life = 0.693 × Vd/clearance). Compounds with higher lipophilicity tend to have higher values of Vd. More importantly, binding to blood plasma proteins has a significant effect on the volume of distribution. Human plasma contains over 60 proteins, however only three of these account for the majority of drug binding: albumin (carries mostly anionic drugs, some cationic and neutral drugs), α1-acid glycoprotein (AAG) (cationic and neutral drugs) and lipoproteins (cationic and neutral drugs).21 Most computational studies that link plasma protein binding and acid/base characteristics have been limited to modelling the compounds using the following broad categories at physiological pH: acidic, basic, neutral or zwitterionic.22, 23 In general, basic compounds have large values of Vd while acidic compounds exhibit smaller values. The amount of drug exposed to the liver and the kidney thus varies considerably between acids and bases. Tissue bound compounds such as bases, tend to form interactions with the acidic head groups of (phospho)lipids whereas acids will readily bind to lysine residues in blood plasma proteins. A recent study at GlaxoSmithKline4 concurred with these established findings, demonstrating that basic compounds are more widely distributed throughout the body.23 This investigation also showed that acids had lower values of Vd than either neutral or zwitterionic compounds. Once again, other factors play a role to the extent of protein binding of drugs, particularly, lipophilicity and together with acid/base properties they greatly affect clearance and target organ exposure.
Brain tissue binding, blood-brain barrier permeability
Companies involved in new medicines research are interested in the CNS permeability of their research compounds and how well they bind to brain tissue. Companies developing drugs aimed at CNS targets are aware that the pharmacological response of a compound is directly related to the free fraction in the CNS that is available to bind to the requisite macromolecule.24 If a compound is highly protein bound, then it needs to have sufficient potency to compensate for the small fraction of freely available drug. With regard to brain tissue binding, minor differences were found between the binding of acidic, basic, neutral and zwitterionic substances, in contrast to the distinct pattern observed for plasma protein binding. Instead, brain tissue binding was found to be mostly influenced by non-specific phenomena, especially, lipophilicity.4
Another widely studied aspect of CNS drug research concerns the ability of drugs to pass through the BBB. In physiological terms, the BBB presents itself as a significant obstacle to drugs entering the CNS. Tight junctions exist between the epithelial cells of the BBB and there are significant populations of efflux pumps to counteract drug entry. Simple models that estimate the extent of penetration of drugs into the CNS are limited by data quality, and as above, most studies segregate compounds into various charge states for research purposes.25 While MW, lipophilicity and hydrogen bond donor ability are significant factors affecting BBB penetration26, ionisation state also plays a key role. Fischer and co-workers27 emphasised the lack of CNS drugs with an acidic pKa < 4 or basic groups with pKa > 10. Fan et al.25 showed that acidic drugs were the least penetrant (mean logBB value -2.0) while basic and neutral compounds showed similar mean values ca. -0.5. (note, logBB = log([brain]/[blood])). Broccatelli et al.28 also found that acidic compounds with pKa values below 5.5 were less likely to be CNS penetrant in accord with our findings.7 Their modelling work which was aimed at predicting CNS penetration, was able to deal with ionisation states using VolSurf molecular descriptors (http://www.moldiscovery.com/soft_volsurf.php). Taken together, the information above can be exploited for the design of ligands where CNS penetration is required, or for cases where CNS exclusion is needed to minimise side effects.
Efflux mechanisms
There are numerous efflux systems in place to protect the body from harmful substances. A key example is P-glycoprotein (P-gp), which is a membrane bound protein residing on the apical surface of the intestinal epithelium and astrocyte membranes of the BBB. The interest in transporters such as P-gp by the pharmaceutical industry is considerable, as this efflux pump can severely limit the oral absorption of compounds, or counteract access to the CNS. A useful predictive model for P-gp inhibitor affinity has been generated using molecular interaction fields29 and clear relationships exist between affinity for P-gp and molecular size4, 30 as well as the number of hydrogen bond donors.31 Reminiscent of many metabolism enzymes, P-gp is undiscerning regarding substrates for this protein. In the gut, both P-gp and metabolism enzymes are largely co-located performing the same function which is to avoid exposure to unwanted and potentially harmful substances.32
Ionisation states also play a role, with acids showing lower efflux ratios than neutral and basic compounds, while zwitterions possess higher efflux ratios.4 In an effort to simplify compound classification, a ‘rule of 4’ was developed to roughly describe P-gp substrate specificity.33 This rule uses the properties; MW, pKa values, and the sum of the number of nitrogen and oxygen atoms (N+O). In short, it was found that a compound with the parameters (N+O) ≥ 8, MW > 400 and acid pKa > 4, was likely to be a P-gp substrate while those with parameters (N+O) ≤ 4, MW < 400 and base pKa < 8 was less likely to be a substrate.33 These guidelines are useful and enable researchers to monitor specific physicochemical properties as well as charge states to gauge P-gp susceptibility. While these findings are important, P-gp affinity for each structural class will vary and laboratory testing is required at an early stage to establish any liabilities.
hERG binding
Of major importance to the pharmaceutical industry during drug discovery is the need to avoid compounds that block the hERG potassium channel. Serious side effects can occur from inhibiting hERG channels, such as QT prolongation, that in severe circumstances can lead to cardiac arrhythmias and death. Improved models of the hERG channel have been developed recently leading to better predictions of channel affinity.34 Of the models that have been developed to predict hERG affinity, many describe a relationship between the presence of a basic group and channel blockade.34 Indeed, the presence of a basic aliphatic nitrogen is a trigger for medicinal chemists to consider early testing for hERG affinity.35 A study of 35,200 molecules tested for hERG inhibition found that basic compounds had a higher affinity for hERG channels than zwitterionic compounds, while acidic and neutral compounds showed the weakest affinity.4
Electrophysiology studies provide a more rigorous assessment than radioligand binding and AstraZeneca tested 7,685 compounds focussing on both charge state and lipophilicity to develop guidelines for avoiding hERG inhibition.36 Molecules were classified as acidic, basic, neutral or zwitterionic based on their charge state at physiological pH while logD values were either estimated in silico or experimentally measured. In agreement with the findings at GSK4, basic compounds were found to be more likely to inhibit hERG and this inhibition was also strongly driven by lipophilicity.36 Waring and Johnstone36 defined an upper limit to lipophilicity to predict a 70% probability of producing a compound with a hERG IC50 over 10 μM (Table 1). A further 5,748 compounds were tested against this model demonstrating that the suggested logP/logD7.4 limits were able to successfully predict at the 70% level.36 Studies specifically using pKa values to assess the risk of hERG channel inhibition have yet to emerge and while no specific relationship to pKa has been shown, Wager and co-workers11 analysed in-house data to conclude that the risk of hERG channel blockade was greater when the molecule had a basic centre with a pKa above 8.4.
Table 1.
Relationship between charge state, lipophilicity and hERG inhibition
| Target upper limits of logD7.4 and ClogP to estimate >70% of compounds achieve a hERG IC50 of greater than 10 μM
| ||||
|---|---|---|---|---|
| Acids | Bases | Neutrals | Zwitterions | |
| logD7.4 | >4 | 1.4 | 3.3 | 2.3 |
| ClogP | >9 | 1.9 | 1.9 | 4.4 |
From ref 36. logD7.4 and ClogP upper limits to avoid hERG inhibition for various charge states.
Phospholipidosis
Phospholipidosis is characterised by excess phospholipids in cells giving them a foamy appearance and is known to be induced by particular compounds that are cationic amphiphiles. While this is a manageable side effect of some drugs, it is troublesome during drug development, as further studies are needed to show that the effects are reversible. Ploemen and co-workers37 developed a model to predict the potential for cationic compounds to cause phospholipidosis. This model was later updated38 to the following simple calculation focussing on both lipophilicity and the pKa of the basic group. A compound may potentially induce phospholipidosis if (ClogP)2 + (calculated basic pKa)2 ≥ 50 as long as the ClogP > 2 and pKa > 6.
Other workers have also sought simple algorithms to predict phospholipidosis. Using an in vitro model, Tomizawa et al. showed a relationship for basic compounds between ClogP, pKa and phospholipidosis.39 To extend this to other compound classes, they plotted net charge (NC) at pH 4.0 against ClogP and developed a predictive scheme for phospholipidosis (Table 2). For more advanced cases of drug development where information on the volume of distribution is available, Hanumegowda and co-workers developed a model that is superior than using pKa and ClogP values alone.40 Inducers of phospholipidosis were predicted with 82% accuracy if the most basic pKa * Vd * ClogP value was over 180. These simple models exploit pKa values to predict compound toxicity and can be easily applied in a drug discovery setting.
Table 2.
Likelihood of phospholipidosis
| ClogP | Net Charge | ||
|---|---|---|---|
| <1 | 1 | 1 < NC ≥ 2 | |
|
| |||
| < 1.61 | None | Low | High |
| 1.61 ≤ ClogP < 2.75 | None | Medium | High |
| ≥ 2.75 | None | High | High |
Based on ClogP and net charge state at pH 4.0.39
Mitochondrial dysfunction
Impairment of mitochondrial processes has recently been acknowledged as a cause of off-target drug toxicities and has resulted in the withdrawal of several compounds from the clinic. Two compounds in particular, troglitazone and cerivastatin, were delisted due to their effect on mitochondrial oxidative phosphorylation.41 The primary function of mitochondria is to produce energy for the cell in the form of adenosine triphosphate (ATP). Compounds that acutely reduce ATP production result in a range of symptoms such as lactic acidosis which presents clinically as nausea, vomiting and abdominal pain. To prevent these compounds entering animal or human testing, pre-clinical screening methods have been developed to identify molecules that interfere with mitochondrial function.42 A study of over 2,000 compounds by Naven et al. has shown that acidic drugs present the most problems.43 They also showed that there was a higher propensity for uncoupling activity with increased lipophilicity. A number of acidic functional groups were flagged as being particularly problematic such as anthranilic acids, thiazolinediones, fluoromethylsulfonanilides, salicylates and acyclindolones.
Clearance, metabolism and cytochrome P450 enzymes
A consideration of drug clearance needs to encompass both renal and hepatic pathways. There is also a need to include biliary systems and collectively these pathways influence pharmacokinetic parameters such as elimination half-life. Lipophilicity is a dominant factor in clearance as this determines the membrane permeability of a drug, particularly in renal systems. The aqueous component of blood is filtered by the kidney and reabsorption of compounds is dependent on their logD. Positive logD7.4 values predict that a compound is reabsorbed while those with values below 0.0 are more readily cleared. Ionisation state plays a role in clearance and is particularly affected by protein binding. As the anionic form of acids can be highly bound to plasma proteins they are less likely to be cleared, whereas bases tend to show higher clearance rates.4
Secondary metabolism also has to be considered as initial metabolism can often generate ionisable functional groups. For example, carboxylic acid containing compounds are liable to acyl-glucuronidation which influences their chemical reactivity and side effect profile.44 Despite this about 19% of drugs contain a carboxylate (unpublished observations of FDA approved drugs) and it remains an important functional group for medicinal chemists.
Since lipophilic compounds are reabsorbed in the kidneys, metabolism is required to render them more water soluble. Drugs and other exogenous chemicals are often metabolised by cytochrome P450 enzymes, with the majority of drugs being metabolised by the cytochrome P450 isoforms 1A2, 2C9, 2C19, 2D6 and 3A4.45 In the drug discovery field, efforts are often taken to reduce the metabolism of drugs by P450 enzymes. The reasons for this relate to reducing the production of potentially toxic metabolites and to avoid interfering with the metabolism of other drugs which can lead to drug interactions. Reducing the metabolism of a drug also allows for longer half-lives and less frequent dosing schedules. The P450 isoforms most influenced by charge state include 2C9 and 2D6, while for the remaining isoforms, ionisation state was shown to play a lesser role.4 Other factors play a role in the preference of cytochrome P450 isoforms for organic compounds and include MW and logP. Cytochrome P450 enzymes and other drug metabolising enzymes may yield biologically active metabolites, a process known as bioactivation. Fortunately, 3D protein structural information is beginning to help us appreciate substrate preferences although these enzymes are notoriously promiscuous. This is understandable remembering their broad role in metabolising both endogenous and exogenous compounds.46, 47
Acid/Base profile of oral drugs
Our analysis of the acid/base profile of small organic substances specifically examined the pKa of each functional group in order to classify each compound. The first of our studies looked at a set of published pKa values for a series of older drugs (see ref7). While this was informative, a broader set was subsequently investigated comprising compounds in current clinical use.7 This latter study provided several levels of analysis covering the proportion of charge state classes to the pKa distributions of acids and bases. Furthermore, the compounds were split into oral drugs and those that target the CNS to allow comparisons to be made between these groups. Surveys of this nature give insights into drugs in general and the observations have the potential to be applied in early stage discovery work.
Focussing on oral drugs showed that 78.6% of compounds contained an ionisable group, while 11.9% were neutral, 4.3% always ionised and the remainder (5.2%) was made up of salts, miscellaneous compounds (e.g. mixtures) and high molecular weight substances.7 Figure 3A shows that the ionisable compounds were predominantly made up of molecules with a single acidic or basic group as well as simple ampholytes containing one acid and one base. The large proportion of ionisable compounds was an interesting statistic that is a reflection of the optimisation process that provides the necessary properties suitable for binding site interactions and biopharmaceutical properties.
Figure 3.
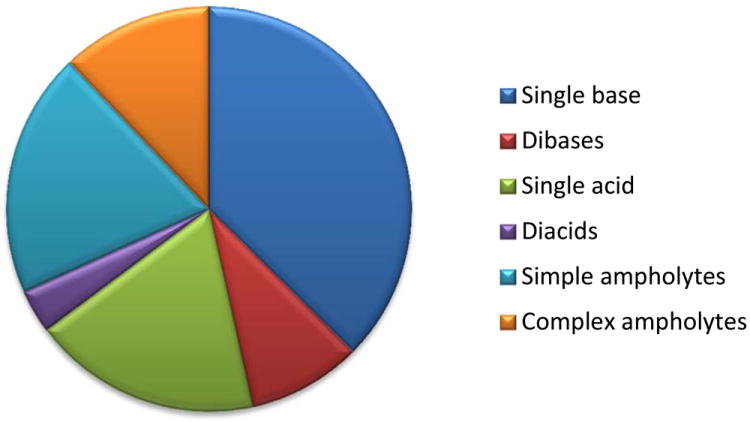
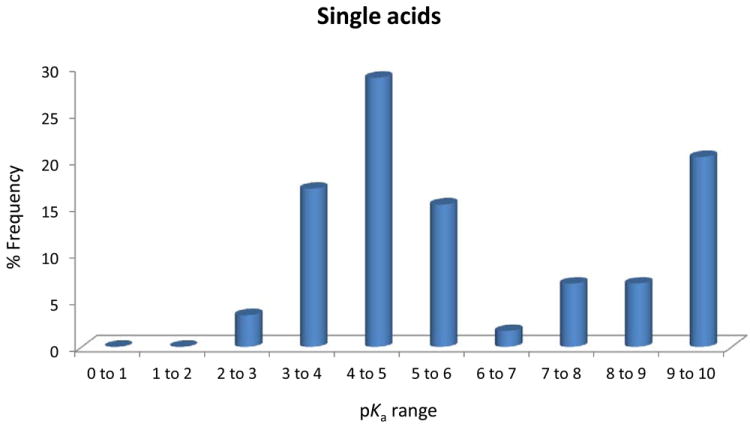
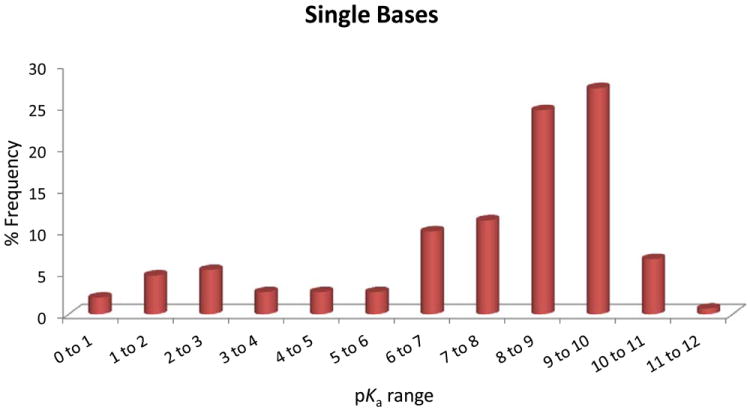
(A), Proportion of compound categories for ionisable oral drugs.7 (B), pKa distributions of oral drugs containing a single acidic functional group, or (C) single basic functional group. Compounds with the following criteria were classified as always ionised: acids with pKa values < 0 and bases with pKa values > 12, plus compounds with permanently charged groups (e.g., quaternary nitrogen atoms). Acids with pKa values above 10 or bases with pKa values below 0.0 were considered neutral. The remaining ionisable compounds (acid pKa range 0-10 and base pKa range 0-12) were divided into the following groups: single acid-containing substances, single base-containing substances, compounds with two acidic groups, compounds with two basic groups, simple ampholytes (one acidic and one basic group) and other complex combinations of acidic and basic groups (complex ampholytes). pKa values were binned into single log unit ranges (i.e. 0.0 < X ≤ 1.0, 1.0 < X ≤ 2.0, etc.). The histogram column heights are expressed as a percentage.
Plots of the pKa distributions of single acid and single base containing substances are shown in Figures 3B and 3C. Interestingly for the acids, there is a biphasic distribution showing a paucity of acids with pKa values between 6 and 7. This can be explained by the predominance of carboxylate and phenolic substances in this set and the range of pKa values usually encountered for these groups. The bases on the other hand have a distribution showing that most of these substances had pKa values above 6.0. Once again, this can be explained by the large number of aliphatic amines in this set that mostly target GPCRs. The overall makeup of the compound classes and pKa distributions is influenced by numerous factors.7 Given that these are established drugs, ADMET properties are dominant factors as well as the need to meet the necessary binding site interactions. The value of this work was to provide additional information beyond standard physicochemical properties that may influence the choice of screening collections and the diversity of ionisable functional groups within these compounds. Our analysis of screening compounds and chemogenomics datasets complements this work on drugs and a manuscript on this topic is in preparation.
Other groups have also considered the charge states of drugs which has been included in their overall analyses of physicochemical properties.6, 13 Bocker et al. specifically focussed on carboxylates but also provided a comparison of standard physicochemical properties splitting the drugs into acids, bases, neutrals and zwitterions.13 Minor differences were observed between the groups, however neutral drugs were found to have fewer rotatable bonds and basic drugs had a significantly lower number of hydrogen bond acceptors. Their study culminated in a set of observations that could predict whether a carboxylic acid containing compound would show reasonable ADME properties.13 The following list provides the desired properties, and in this case no more than one cut off value should be violated:
MW < 400
# H-bond donors < 3
# H-bond acceptors ≤ 6
# rotatable bonds ≤ 9
ClogP < 3
-1.5 < ClogD7.4 < 1.5
60 < PSA < 140
pKa (COOH) > 3
A more extensive analysis of molecular properties and charge states was conducted by Leeson and co-workers6 who also examined changes to these parameters over the past few decades. Once again compounds were split into acids, bases, neutrals and zwitterions however an additional class of ‘cations’ was included for quaternary amines. A wide range of properties were compared looking for trends to determine whether particular characteristics had reached consensus over time. For example, lipophilicity was shown to be increasing for neutral and acidic oral drugs over time, while basic drugs have already reached a lipophilicity range common to the other classes. Other findings showed that6:
MW is increasing in all classes over time
# H-bond donors is stable
# H-bond acceptors is increasing with time
Aromatic atom count – sp3 atom count (Ar-sp3) is constant with time. (Ar-sp3 describes shape or aromatic/aliphatic balance)
This form of analysis is very useful to identify trends in how medicinal chemistry and drug discovery is conducted and can be applied to examine trends between individual organisations. In the case of acids and neutral drugs it was suggested that there is the potential to tolerate greater lipophilicity. It would appear that keeping tabs on lipophilicity, the number of H-bond donors and molecular shape is vital for developing orally active compounds that can survive the rigours of drug development.
Drug-receptor interactions
Drug binding sites in macromolecular targets consist of hydrophobic and hydrophilic regions, where the latter may be involved in binding to ligands through electrostatic interactions, including ionic bonding, hydrogen bonding, dipole-dipole interactions, ion-dipole interactions and cation-π interactions. Surveys of these interactions have provided further insight into the geometry and strength of these bonds to assist drug designers.48 Ionisable groups in drugs can interact with complementary groups in macromolecules to form strong interactions, however, the strength can be reduced by competing hydration and entropic phenomena. Medicinal chemists can improve the potency of a lead by influencing either enthalpic or entropic thermodynamic components. The easy route is to add lipophilicity or increase the number of groups that make interactions. This approach has been termed ‘molecular obesity’ and leads the researcher in a direction that is likely to result in future ADMET or solubility problems.3 Seeking more specific interactions, for example, through H-bonding, requires optimal placement of the functional group in the binding site while remembering that desolvation can incur an entropic penalty. In energy terms, desolvating a non-polar group requires 10-fold less energy hence medicinal chemists find themselves adding hydrophobic groups in their efforts to improve potency. Slavishly seeking potency in these situations means that the chemist is often led down the wrong path.
To avoid molecular obesity problems and to specifically monitor the quality of lead compounds, a number of parameters have been developed. For example, during lead optimisation, a measure of the potential to develop a compound is to monitor ligand efficiency (LE).49 This parameter takes into account both the potency and MW of a compound to give a binding free energy per heavy (i.e., non-hydrogen) atom. Several ligand efficiency measures have been described and these are currently in vogue within the medicinal chemistry community.1 In accord with Andrews binding energies50, ligand efficiency in very small molecular weight compounds is influenced by charged groups which can provide large increases to LE.51 Alternatively, ligand efficiency can be traded in order to incorporate a charged functional group which may be required to improve the biopharmaceutical characteristics or solubility of a ligand.51
Off-target activity
A major concern for the scientist developing a new drug is ensuring selectivity for the intended target. While there are some instances where interactions with more than one macromolecule is sought, off target activity can lead to toxicity and side effects. A number of studies have looked at this problem attempting to find relationships with simple physicochemical properties.5, 8, 11, 52, 53 Basic compounds clearly showed that they often interact with more than one target8, 53 while acids, neutrals and zwitterions showed a reduced propensity for off-target activity.8 Although some of these studies have suggested links between MW, ClogP and promiscuity8, 11, 54, some care needs to be taken with the statistics. The increased complexity of a compound with a higher ClogP and MW, suggests that there may be an increased possibility of interacting with other targets.3 Off-target activity is yet another reason to look carefully at size and lipophilicity, and when ionisable groups are involved then logD7.4 values need to be monitored.
Formulation
The pKa value(s) of a drug can critically influence aspects of its formulation, especially for drugs that must be administered in solution. In particular, pKa values have a large effect on the aqueous solubility of the drug and in general, the ionised form of a drug is considerably more water soluble. Solubility is an extremely important physicochemical parameter that is routinely investigated in the pharmaceutical industry. Polarity, through the influence it has on solute-solvent interactions, is one of the two main contributors to aqueous drug solubility. The other is the energetic nature of the solid state, in particular the crystal lattice energy, which varies from one drug structure to the next, and also between polymorphic forms of the same structure. In general, drugs need a suitable degree of lipophilicity to: (a) reach the site of action, commencing with absorption from the GI tract, and (b) interact with the appropriate receptor. Hence, aqueous solubility is often compromised to some extent by the desired lipophilicity. The acid/base character of a drug plays an important role in determining the solubility, but this is often extensively modulated by the pH of a solution formulation causing variable polarity in aqueous solution. There is a wealth of information about drug solubility and the reader is referred to two important texts.55, 56
Since injectable solutions are preferably aqueous, a key requirement for overcoming lipophilicity-related limitations of drug design is the presence of one or more ionisable groups for which polarity, based on the extent of ionisation, can be controlled by the pH of the medium, whether in a biological environment or a formulated solution. It must be ensured however, that the key role of acid/base functional groups in receptor interactions is not compromised through adjustment of acid/base behaviour for the purpose of modifying solubility, which could also be achieved with non-ionisable polar groups.
It has long been accepted that the pH of injectable solutions should not stray outside the range 4-9, principally for reasons related to pain or tissue damage on injection. Formulation of an ionisable poorly water-soluble drug may require an extreme pH value in order to get adequate solubility. Judicious choice of the ionisable group or groups (at the design stage) can greatly assist in solving this solubility problem. To be effective, the introduced group must be significantly ionised in the formulation vehicle, and ideally also under physiological conditions. While difficult to generalise, effective pKa values for solubility enhancement should be in the range 4-9 to ensure so that there is a sufficient proportion of ionised drug to achieve adequate solubility in water. Bases should have pKa values nearer the upper end of this range, while values for acids should be nearer the lower end. Of course, a relatively water soluble drug will be easier to formulate than one with marginal solubility. A further issue relevant to injectable and ophthalmic drugs is that of osmotic effect which is controlled by the drug substance pKa and the solution pH. This is of importance where the drug concentration is high, such that the drug itself is the main osmotic determinant.
Another aspect of the influence of pKa on formulation is the potential impact of ionisation state on drug stability. For example, at pH > 6, morphine degrades through two oxidation pathways, one involving the free base form of the alicyclic 3° nitrogen, and the other involving the anionic form of the 3-OH phenolic group.57 In both cases, the oxidation rate increases with deprotonation of each functional group. For both types of degradation pathway, if drug design considerations lead to oxidizable groups with pKa values near the physiologically relevant range, then oxidative degradation may be accelerated.
pKa Prediction methods
It is important to mention that the main interest in acid/base equilibria of drugs relates to aqueous biological systems. The mechanism by which acid/base behaviour impacts these systems is through the changes that occur to aqueous solution drug properties when the polarity of the molecule is altered as a result of the changed ionisation (protonation-deprotonation reactions) of one or more functional groups. Another important aspect when considering acid/base character is to reasonably predict or accurately measure pKa values which also affects the estimation of logD values. A range of software packages is available to estimate pKa values in aqueous environments and these vary in both their accuracy and the algorithm applied. Given the breadth of this topic we refer the reader to Lee and Crippen58 who discuss the various techniques and software available.
Commercial pKa prediction software packages:
ACD Labs/pKa (ACD Labs, http://www.acdlabs.com/home/)
ADME Boxes (Pharma Algorithms, http://pharma-algorithms.com/)
ADMET Predictor (Simulations Plus, http://www.simulations-plus.com/)
CSpKa (ChemSilico, http://www.chemsilico.com/)
Marvin (ChemAxon, http://www.chemaxon.com/)
MoKa (Molecular Discovery, http://www.moldiscovery.com/)
Pipeline Pilot (Accelrys, http://accelrys.com/)
pKalc (CompuDrug, http://www.compudrug.com/)
QikProp and Epik (Schrödinger, http://www.schrodinger.com/)
Quacpac (OpenEye, http://www.eyesopen.com/)
SPARC (ARChem, http://archemcalc.com/sparc/)
pKa prediction methods have also been evaluated for their accuracy.58, 59 One problem that needs to be considered for these calculations concerns choosing the appropriate tautomeric state for a molecule as this will greatly affect pKa predictions.60 Another factor to be considered is the accuracy of experimental data that is used for validating pKa prediction models. This data is often unreliable, or else the experimental method needs careful scrutiny. A more recent compilation has provided many values where reliability of the experimental technique has been assessed.61 Finally, higher throughput instrumentation has been developed in recent years for measuring pKa values and the reader is referred to the review by Comer for more information.62
Conclusions
In any industry, there is need for continual appraisal of key processes to maintain efficiency and competitiveness. Some insights can have a long lasting and substantial impact such as the observations of Lipinski and co-workers.9 Since their publication emerged, additional guidelines have arisen regarding the nature of chemical space relevant to drug discovery.1 Pharmaceutical companies have often led this research in an attempt to understand and reduce the rate of compound attrition in drug development.1, 4, 5, 8, 9, 11, 52 This review and our recent acid/base analyses7 are intended to provide a timely reminder about the importance of acid/base profiles and pKa values. Given that they influence all aspects of drug discovery it is useful to once again analyse acid/base properties to add this to our knowledge of chemical space. Considering acid/base properties therefore represents yet another opportunity to improve drug discovery processes.
Supplementary Material
Acknowledgments
We acknowledge a range of primary sources that could not be included explicitly due to space restrictions but are cited indirectly. This work was supported in part by NIH grants GM-095952 and MH-084690 (TIO).
References
- 1.Meanwell NA. Chem Res Toxicol. 2011;24:1420–1456. doi: 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- 2.Leeson PD, St-Gallay SA. Nat Rev Drug Discov. 2011;10:749–765. doi: 10.1038/nrd3552. [DOI] [PubMed] [Google Scholar]
- 3.Hann MM. Med Chem Commun. 2011;2:349–355. [Google Scholar]
- 4.Gleeson MP. J Med Chem. 2008;51:817–834. doi: 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- 5.Gleeson MP, Hersey A, Montanari D, Overington J. Nat Rev Drug Discov. 2011;10:197–208. doi: 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leeson PD, St-Gallay SA, Wenlock MC. MedChemComm. 2011;2:91–105. [Google Scholar]
- 7.Manallack DT. SAR QSAR Environ Res. 2009;20:611–655. doi: 10.1080/10629360903438313. [DOI] [PubMed] [Google Scholar]
- 8.Leeson PD, Springthorpe B. Nat Rev Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 9.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Del Rev. 1997;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 10.Johnson TW, Dress KR, Edwards M. Bioorg Med Chem Lett. 2009;19:5560–5564. doi: 10.1016/j.bmcl.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 11.Wager TT, Hou X, Verhoest PR, Villalobos A. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin YC. J Med Chem. 2005;48:3164–3170. doi: 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- 13.Bocker A, Bonneau PR, Hucke O, Jakalian A, Edwards PJ. ChemMedChem. 2010;5:2102–2113. doi: 10.1002/cmdc.201000355. [DOI] [PubMed] [Google Scholar]
- 14.Schanker LS, Tocco DJ, Brodie BB, Hogben CA. J Pharmacol Exp Ther. 1958;123:81–88. [PubMed] [Google Scholar]
- 15.Palm K, Luthman K, Ros J, Grasjo J, Artursson P. J Pharmacol Exp Ther. 1999;291:435–443. [PubMed] [Google Scholar]
- 16.Boisset M, Botham RP, Haegele KD, Lenfant B, Pachot JI. Eur J Pharm Sci. 2000;10:215–224. doi: 10.1016/s0928-0987(00)00073-7. [DOI] [PubMed] [Google Scholar]
- 17.Castro JL, Collins I, Russell MG, Watt AP, Sohal B, Rathbone D, Beer MS, Stanton JA. J Med Chem. 1998;41:2667–2670. doi: 10.1021/jm980204e. [DOI] [PubMed] [Google Scholar]
- 18.Kubinyi H. In: Computational Approaches to Structure Based Drug Design. Stroud RM, editor. Royal Society of Chemistry; London: 2007. pp. 24–45. [Google Scholar]
- 19.Pagliara A, Reist M, Geinoz S, Carrupt PA, Testa B. J Pharm Pharmacol. 1999;51:1339–1357. doi: 10.1211/0022357991777164. [DOI] [PubMed] [Google Scholar]
- 20.Vieth M, Siegel MG, Higgs RE, Watson IA, Robertson DH, Savin KA, Durst GL, Hipskind PA. J Med Chem. 2004;47:224–232. doi: 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]
- 21.Zhang F, Xue J, Shao J, Jia L. Drug Discov Today. 2012;17:475–485. doi: 10.1016/j.drudis.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Gleeson MP. J Med Chem. 2007;50:101–112. doi: 10.1021/jm060981b. [DOI] [PubMed] [Google Scholar]
- 23.Gleeson MP, Waters NJ, Paine SW, Davis AM. J Med Chem. 2006;49:1953–1963. doi: 10.1021/jm0510070. [DOI] [PubMed] [Google Scholar]
- 24.Kalvass JC, Maurer TS. Biopharm Drug Dispos. 2002;23:327–338. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 25.Fan Y, Unwalla R, Denny RA, Di L, Kerns EH, Diller DJ, Humblet C. J Chem Inf Model. 2010;50:1123–1133. doi: 10.1021/ci900384c. [DOI] [PubMed] [Google Scholar]
- 26.Clark DE. Drug Discov Today. 2003;8:927–933. doi: 10.1016/s1359-6446(03)02827-7. [DOI] [PubMed] [Google Scholar]
- 27.Fischer H, Gottschlich R, Seelig A. J Membr Biol. 1998;165:201–211. doi: 10.1007/s002329900434. [DOI] [PubMed] [Google Scholar]
- 28.Broccatelli F, Larregieu CA, Cruciani G, Oprea TI, Benet LZ. Adv Drug Deliv Rev. 2012;64:95–109. doi: 10.1016/j.addr.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broccatelli F, Carosati E, Neri A, Frosini M, Goracci L, Oprea TI, Cruciani G. J Med Chem. 2011;54:1740–1751. doi: 10.1021/jm101421d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gombar VK, Polli JW, Humphreys JE, Wring SA, Serabjit-Singh CS. J Pharm Sci. 2004;93:957–968. doi: 10.1002/jps.20035. [DOI] [PubMed] [Google Scholar]
- 31.Seelig A. Eur J Biochem. 1998;251:252–261. doi: 10.1046/j.1432-1327.1998.2510252.x. [DOI] [PubMed] [Google Scholar]
- 32.Benet LZ. Mol Pharm. 2009;6:1631–1643. doi: 10.1021/mp900253n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Didziapetris R, Japertas P, Avdeef A, Petrauskas A. J Drug Target. 2003;11:391–406. doi: 10.1080/10611860310001648248. [DOI] [PubMed] [Google Scholar]
- 34.Raschi E, Ceccarini L, De Ponti F, Recanatini M. Expert Opin Drug Metab Toxicol. 2009;5:1005–1021. doi: 10.1517/17425250903055070. [DOI] [PubMed] [Google Scholar]
- 35.Vaz RJ, Li Y, Rampe D. Prog Med Chem. 2005;43:1–18. doi: 10.1016/S0079-6468(05)43001-5. [DOI] [PubMed] [Google Scholar]
- 36.Waring MJ, Johnstone C. Bioorg Med Chem Lett. 2007;17:1759–1764. doi: 10.1016/j.bmcl.2006.12.061. [DOI] [PubMed] [Google Scholar]
- 37.Ploemen JP, Kelder J, Hafmans T, van de Sandt H, van Burgsteden JA, Saleminki PJ, van Esch E. Exp Toxicol Pathol. 2004;55:347–355. doi: 10.1078/0940-2993-00338. [DOI] [PubMed] [Google Scholar]
- 38.Pelletier DJ, Gehlhaar D, Tilloy-Ellul A, Johnson TO, Greene N. J Chem Inf Model. 2007;47:1196–1205. doi: 10.1021/ci6004542. [DOI] [PubMed] [Google Scholar]
- 39.Tomizawa K, Sugano K, Yamada H, Horii I. J Toxicol Sci. 2006;31:315–324. doi: 10.2131/jts.31.315. [DOI] [PubMed] [Google Scholar]
- 40.Hanumegowda UM, Wenke G, Regueiro-Ren A, Yordanova R, Corradi JP, Adams SP. Chem Res Toxicol. 2010;23:749–755. doi: 10.1021/tx9003825. [DOI] [PubMed] [Google Scholar]
- 41.Dykens JA, Will Y. Drug Discov Today. 2007;12:777–785. doi: 10.1016/j.drudis.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 42.Nadanaciva S, Dykens JA, Bernal A, Capaldi RA, Will Y. Toxicol Appl Pharmacol. 2007;223:277–287. doi: 10.1016/j.taap.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Naven RT, Swiss R, McLeod JK, Will Y, Greene N. Toxicol Sci. 2012 doi: 10.1093/toxsci/kfs279. [DOI] [PubMed] [Google Scholar]
- 44.Skonberg C, Olsen J, Madsen KG, Hansen SH, Grillo MP. Expert Opin Drug Metab Toxicol. 2008;4:425–438. doi: 10.1517/17425255.4.4.425. [DOI] [PubMed] [Google Scholar]
- 45.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 46.Ekroos M, Sjogren T. Proc Natl Acad Sci U S A. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams PA, Cosme J, Ward A, Angove HC, Matak Vinkovic D, Jhoti H. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 48.Bissantz C, Kuhn B, Stahl M. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hopkins AL, Groom CR, Alex A. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 50.Andrews PR, Craik DJ, Martin JL. J Med Chem. 1984;27:1648–1657. doi: 10.1021/jm00378a021. [DOI] [PubMed] [Google Scholar]
- 51.Perola E. J Med Chem. 2010;53:2986–2997. doi: 10.1021/jm100118x. [DOI] [PubMed] [Google Scholar]
- 52.Hughes JD, Blagg J, Price DA, Bailey S, Decrescenzo GA, Devraj RV, Ellsworth E, Fobian YM, Gibbs ME, Gilles RW, Greene N, Huang E, Krieger-Burke T, Loesel J, Wager T, Whiteley L, Zhang Y. Bioorg Med Chem Lett. 2008;18:4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 53.Peters JU, Schnider P, Mattei P, Kansy M. ChemMedChem. 2009;4:680–686. doi: 10.1002/cmdc.200800411. [DOI] [PubMed] [Google Scholar]
- 54.Hopkins AL, Mason JS, Overington JP. Curr Opin Struct Biol. 2006;16:127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 55.Grant DJW, Higuchi T. Solubility behavior of organic compounds. Wiley; New York: 1990. [Google Scholar]
- 56.Avdeef A, editor. Absorption and Drug Development: Solubility, Permeability, and Charge State. Wiley; Hoboken: 2003. [Google Scholar]
- 57.Connors KA, Amidon GL, Stella VJ. Chemical stability of pharmaceuticals. 2. Wiley-Interscience; New York: 1986. [Google Scholar]
- 58.Lee AC, Crippen GM. J Chem Inf Model. 2009;49:2013–2033. doi: 10.1021/ci900209w. [DOI] [PubMed] [Google Scholar]
- 59.Manchester J, Walkup G, Rivin O, You Z. J Chem Inf Model. 2010;50:565–571. doi: 10.1021/ci100019p. [DOI] [PubMed] [Google Scholar]
- 60.Martin YC. J Comput Aided Mol Des. 2009;23:693–704. doi: 10.1007/s10822-009-9303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prankerd RJ. A critical compilation of pKa values for pharmaceutical substances. Elsevier Academic Press; Amsterdam: 2007. [DOI] [PubMed] [Google Scholar]
- 62.Comer JEA. In: Comprehensive Medicinal Chemistry. Triggle DJ, editor. Vol. 5. Elsevier; 2007. pp. 357–397. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.