

Highlights
► We investigated the occurrence of BRAF, GNAQ, and GNA11 mutations in pediatric low-grade glioma. ► We found an increase in the copy number of BRAF. ► BRAF and GNAQ mutations are mutually exclusive. ► Understanding the molecular basis of low-grade glioma may increase treatment options.
Keywords: Pediatric, Low-grade glioma, BRAF mutation, Copy number variation, GNAQ/GNA11
Abstract
Fifty-two samples of pediatric low-grade glioma (48 primary, 4 recurrent) were analyzed for BRAF copy number variation (digital PCR analysis, CopyCaller) and point mutations of BRAF V600E, and exon 5 Q209 in GNAQ, and GNA11, using the MALDI-TOF mass spectrometer with validation by direct sequencing. An increased BRAF copy number was found in 18/47 primary samples tested; 15 of them (83.3%) were pilocytic astrocytomas. A BRAF mutation was found in 3/48 primary tumors, all with a normal BRAF copy number and no GNAQ mutation. One sample had a GNAQ209 mutation (Q209P626) with a normal BRAF gene; none of the tumors had a GNA11Q209 mutation. Recurrent or progressive tumors, analyzed in four patients, had the same molecular genotype as their primary. Increased BRAF copy number and activating BRAF mutations may be involved in the development of low-grade glioma via overactivation of the Ras/Raf pathway. This is the first report of a mutation in GNAQ209 in pediatric low-grade glioma. Understanding the molecular mechanisms underlying glioma initiation and growth may assist in the development of targeted therapies.
1. Introduction
Brain tumors are the most common solid tumors in children, with a prevalence of 2.4 per 100 000 to 4 per 100 000. More than 50% are low-grade (I–II) gliomas originating from glial cells, most often astrocytes [1]. The World Health Organization (WHO) distinguishes low-grade from high-grade (III–IV) gliomas by their less aggressive biological behavior and better response to treatment. Among the various types of low-grade gliomas, pilocytic astrocytomas account for 23% of all central nervous system tumors in children. The first line of treatment of low-grade glioma is surgical resection. However, tumors located in the diencephalon, brain stem, or optic pathways cannot be completely resected [2] and require chemotherapy or radiation, which may have severe side effects, such as cognitive impairment, vascular injury, and secondary malignancies. Even surgery alone can be associated with major neurological and behavioral morbidities and recurrence. The reported 10-year survival rate is more than 95% for pilocytic astrocytoma grade I [3] but much lower for grade II [4].
Relatively little is known about the molecular changes that promote the formation or growth of low-grade gliomas in children. Research in glioma biology points to a major role of the Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) pathway, which is pivotal to the transduction of mitogenic stimuli from activated growth factor receptors during regulation of cell proliferation, survival, and differentiation [5]. Serine/threonine-protein kinase B-raf, a member of the Raf family of kinases, serves as a downstream effector in the MAPK pathway. Recently, genetic studies of low-grade glioma have focused on gains in band 7q34 [6–11] which includes part of the BRAF gene locus that encodes the kinase domain [6]. The Ras/Raf pathway may also be activated by oncogenic mutations of the RAF gene itself, such as the substitution of glutamic acid for valine at amino acid 600 (V600E). However, the low rate at which such mutations have been found in pediatric low-grade gliomas (3–10%) [6–11], suggests that other mechanisms may be involved in their development.
In the RAS pathway, the guanine nucleotide-binding protein (Gq) subunit alpha, a protein encoded by the GNAQ gene, may also play a role in tumorigenesis. Genetic, biochemical, and biological analyses have shown that GNAQ behaves like a human oncogene. The reported mutations occurred exclusively in codon 209 in the Ras-like domain and led to constitutive activation [12,13]. The glutamine at codon 209 of the GNAQ gene corresponds to residue 61 of the RAS gene and is essential for guanosine triphosphate (GTP) hydrolysis [13]. In other Ras family members, mutations at this site caused loss of GTPase activity [13]. It is noteworthy that exon 5 of GNA11, another Gq family member, contains an equivalent residue to Q209 of GNAQ. BRAF and GNAQ mutations are mutually exclusive in several tumors, such as uveal melanoma [14]. The prevalence of GNAQ or GNA11 mutations in pediatric low-grade gliomas has not yet been investigated.
The aim of the present study was to determine the occurrence of BRAF, GNAQ, and GNA11 mutations in pediatric low-grade glioma, primary and recurrent, and to analyze variations in copy number of the BRAF oncogene which identify gains in band 7q34.
2. Results
2.1. Clinical data
Fifty-two samples of pediatric low-grade glioma (WHO I–II) were collected from 48 children operated in 1995–2000 (including material from second operations) at Schneider Children’s Medical Center of Israel. Thirteen patients received chemotherapy before (one patient) or after surgery (12), and three also received radiation therapy. Table 2 summarizes the relevant clinical data. Mean patient age was 8.5 years (range, 1 month to 18 years), and the female to male ratio was 1.08. The medical histories revealed that four patients had neurofibromatosis type 1 (patient nos. 5, 8, 12 and 35, Table 2). Two patients had tuberous sclerosis (nos. 47, 48), and 1 patient had Lhermitte-Duclos disease (no. 32).
Table 2.
Clinical and demographic data of children with low-grade glioma.
| Pt. no. | Age at 1st op. (yr) | M/F | Diagnosis | Tu. location | 2nd op. | MH | Chemotherapy | Radiotherapy | Death |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.9 | F | PA | OTG | n | n | y | n | n |
| 2 | 16.9 | F | PA | chia–ht | y | n | y | n | n |
| 3 | 23.4 | F | PA | OTG | n | n | y | n | n |
| 4∗ | 2 | M | PA | chia–ht | y | n | y | y | n |
| 5 | 3 | F | PA | chia–ht | y | NF-1 | y | n | y |
| 6 | 1.1 | M | PA | chia–ht | n | n | y | n | n |
| 7 | 8.4 | M | PA | chia–ht | n | n | n | n | n |
| 8 | 4.6 | F | PA | OTG | n | NF-1 | y | n | n |
| 9 | 3.8 | F | PA | chia–ht | n | n | n | n | n |
| 10 | 1.4 | F | PA | chia–ht | n | n | y | n | n |
| 11 | 2.7 | M | PA | PF | y | n | y | n | n |
| 12 | 10.11 | M | PA | Temporal | n | NF-1 | n | n | n |
| 13 | 16.6 | F | PA | PF | n | n | n | n | n |
| 14 | 16.1 | M | PA | PF | y | n | n | n | n |
| 15 | 4.1 | M | PA | PF | n | n | n | n | n |
| 16 | 12.7 | F | PA | Temporal | y | n | n | n | n |
| 17 | 14.11 | M | PA | Pineal | n | n | n | n | n |
| 18 | 5 | F | PA | PF | n | n | n | n | n |
| 19 | 8.1 | F | PA | PF | n | n | n | n | n |
| 20 | 2 | F | PA | PF | y | n | n | n | n |
| 21 | 14.1 | M | PA | PF | y | n | n | n | n |
| 22 | 6.1 | F | PA | PF | y | n | y | n | n |
| 23 | 12.4 | M | PA | PF | n | n | n | n | n |
| 24 | 1.9 | M | PA | PF | y | n | n | n | n |
| 25 | 6.8 | F | PA | Ventricle | n | n | n | n | n |
| 26 | 15 | M | PA | PF | n | n | n | n | n |
| 27 | 2.5 | M | Astroblastoma | Temporal | y | n | n | n | n |
| 28 | 14.3 | F | PA | Temporal | n | n | n | n | n |
| 29 | 22 | F | FA | Temporal | n | n | n | y | n |
| 30 | 7 | F | PA | Ventricle | y | n | n | n | n |
| 31 | 11 | F | PA | Brainstem | n | n | y | n | n |
| 32 | 2.2 | M | Gangliocytoma | PF | n | LD | n | n | n |
| 33 | 0.1 | F | FA | Temporal | n | n | n | n | n |
| 34 | 4.2 | M | PA | Thalamus | n | n | n | n | n |
| 35 | 10.1 | F | PA | PF | n | NF-1 | y | n | n |
| 36 | 3.5 | M | Pilomyxoid ast. | PF | n | n | n | n | n |
| 37 | 17 | F | Ganglioglioma | Brainstem | y | n | n | n | n |
| 38 | 3.1 | M | Ganglioglioma | Temporal | n | n | n | n | n |
| 39 | 12.8 | M | Ganglioglioma | Temporal | n | n | n | n | n |
| 40 | 14.4 | M | Ganglioglioma | Occipital | y | n | n | n | n |
| 41 | 14 | F | Ganglioglioma | Temporal | y | n | n | n | n |
| 42 | 14.2 | F | Ganglioglioma | Temporal | n | n | n | n | n |
| 43 | 9.11 | M | Ganglioglioma | Temporal | n | n | n | n | n |
| 44∗ | 12.11 | F | PXA | Temporal | y | n | y | y | y |
| 45 | 11 | F | PXA | Temporal | y | n | n | n | n |
| 46 | 14.9 | F | PXA | Temporal | y | n | n | n | n |
| 47 | 5.8 | M | SEGA | Ventricle | n | TS | n | n | n |
| 48 | 3.4 | M | SEGA | Ventricle | y | TS | n | n | n |
MH = medical history; PA = pilocytic astrocytoma; FA = fibrillary astrocytoma; PXA = pleomorphic xanthoastrocytoma; SEGA = subependymal giant cell astrocytoma; OTG = optic tract glioma; chia-ht = chiasmatic–hypothalamic; PF = posterior fossa; NF-1 = neurofibromatosis type 1; TS = tuberosclerosis; LD = Lhermitte–Duclos. ∗Patients 4 and 44 multiple surgeries due to progression (4) and recurrence (44) (indicated by asterisk).
Thirty-one tumors (64.6%) were located supratentorially and 17 infratentorially, 15 in the posterior fossa and two in the brain stem. The molecular analysis was performed on the material from the biopsy study of the primary or from a sample at recurrent surgery (n = 4, nos. 4, 16, 37, 44, Table 2). The second surgical sample was analyzed in these four patients since in two the tumors had been technically undefined at the first biopsy (nos. 16, 37), in one there was tumor progression (no. 4) and in one with true tumor recurrence (no. 44).
The breakdown by type of tumor was as follows: grade I glioma, 41 samples – pilocytic astrocytoma (n = 31), ganglioglioma (n = 7), subependymal giant cell astrocytoma (n = 2), and gangliocytoma (n = 1), grade II gliomas, total 7 samples -- fibrillary astrocytoma (n = 2), pleomorphic xanthoastrocytoma (n = 3), pilomyxoid astrocytoma (n = 1), and astroblastoma (n = 1). In one patient (no. 5, Table 2), the primary diagnosis based on histologic biopsy study at the first surgery was pilocytic astrocytoma of the optic pathway, whereas the diagnosis based on a sample from a third operation was pilomyxoid astrocytoma (grade II).
The median duration of follow-up was 6 years (range, 6 months to 22 years). Two patients died during follow-up (nos. 5, 44, Table 2). Patient 5, who had neurofibromatosis type 1, died of acute myeloid leukemia which could have been secondary to prolonged treatment with vincristine/carboplatin for her optic glioma (2 years from onset of treatment). Patient 44 died of malignant transformation of a pleomorphic xanthoastrocytoma.
2.2. Molecular analysis
The results of the molecular analysis of the primary glioma specimens (total 48) are summarized in Table 3. BRAF copy number was analyzed in 47 samples; one sample (from patient no. 32) failed technically due to a limited amount of DNA. An increased BRAF copy number was noted in 18 of the 47 samples: three copies in 14 samples, four copies in four samples. A BRAF mutation was found in 3 of the 48 samples analyzed; a GNAQ209 mutation, in 1 of the 47 samples analyzed; and a GNA11 mutation, in none of the 27 samples analyzed. All three samples with BRAF mutations had a normal copy number (2), and none of the GNAQ-positive samples harbored a BRAF mutation or abnormal number of BRAF alleles. All four recurrent tumors showed the same molecular genotype as their primary tumor (not shown in Table 3).
Table 3.
Results of molecular studies of pediatric low-grade gliomas.
| Pt. no. | BRAF copy number | BRAF mutation | GNA11 mutation | GNAQ mutation |
|---|---|---|---|---|
| 1 | 2 | Positive | NS | neg |
| 2 | 3 | neg | NS | neg |
| 3 | 2 | neg | NS | neg |
| 4 | 3 | neg | NS | neg |
| 5 | 3 | neg | NS | neg |
| 6 | 2 | neg | neg | neg |
| 7 | 2 | neg | neg | neg |
| 8 | 3 | neg | neg | neg |
| 9 | 4 | neg | neg | neg |
| 10 | 2 | neg | neg | NS |
| 11 | 2 | neg | NS | neg |
| 12 | 2 | neg | NS | neg |
| 13 | 3 | neg | NS | neg |
| 14 | 3 | neg | NS | neg |
| 15 | 2 | neg | NS | Positive |
| 16 | 2 | neg | neg | neg |
| 17 | 3 | neg | NS | neg |
| 18 | 3 | neg | NS | neg |
| 19 | 3 | neg | NS | neg |
| 20 | 2 | neg | neg | neg |
| 21 | 2 | neg | neg | neg |
| 22 | 3 | neg | neg | neg |
| 23 | 3 | neg | neg | neg |
| 24 | 2 | neg | NS | neg |
| 25 | 2 | neg | neg | neg |
| 26 | 2 | neg | neg | neg |
| 27 | 2 | neg | neg | neg |
| 28 | 2 | neg | neg | neg |
| 29 | 2 | neg | neg | neg |
| 30 | 3 | neg | NS | neg |
| 31 | 4 | neg | neg | neg |
| 32 | NS | neg | neg | neg |
| 33 | 2 | neg | neg | neg |
| 34 | 2 | neg | neg | neg |
| 35 | 4 | neg | neg | neg |
| 36 | 3 | neg | neg | neg |
| 37 | 4 | neg | NS | neg |
| 38 | 2 | neg | neg | neg |
| 39 | 2 | neg | NS | neg |
| 40 | 2 | neg | neg | neg |
| 41 | 2 | neg | NS | neg |
| 42 | 2 | Positive | NS | neg |
| 43 | 2 | neg | neg | neg |
| 44 | 2 | neg | neg | neg |
| 45 | 2 | Positive | NS | neg |
| 46 | 3 | neg | neg | neg |
| 47 | 2 | neg | neg | neg |
| 48 | 2 | neg | neg | neg |
NS = not studied; neg = negative.
2.2.1. BRAF mutation
The V600E point mutation was identified in the BRAF gene in 3 of the 48 primary samples (6.3%) (Table 3), derived from one patient each with pilocytic astrocytoma, ganglioglioma, and pleomorphic xanthoastrocytoma. Fig. 1 demonstrates the mutation in exon 15 of the BRAF gene, in which the nucleotide thymine (T) is replaced with adenine (A), leading to the translation of glutamic acid instead of valine and, thereby, to overactivation of the Ras/Raf cascade.
Fig. 1.
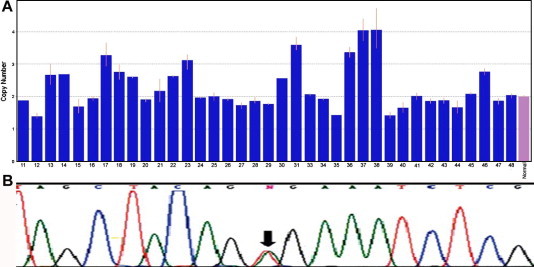
(A) Schematic illustration of the variation in copy number of the BRAF gene on digital PCR analysis (CopyCaller). The normal phenotype shows two copies. An increase in the number of copies is associated with overactivation of the Ras/Raf pathway. N = normal pediatric brain tissue. (B) Sequencing of exon 15 in the BRAF gene demonstrating the activating mutation V600E. The black arrow indicates replacement of nucleotide thymine (T) with adenine (A).
2.2.2. GNAQ209 and GNA11209 mutation
Sequencing of exon 5 of the GNAQ gene yielded one patient (no. 15, Table 3) with a mutation in codon 209, in which the nucleotide adenine (A) is replaced with cytosine (C), causing loss of GTPase activity and constitutive activation of the Ras cascade. The sample was derived from a pilocytic astrocytoma. No Q209 mutations were found in exon 5 of the GNA11 gene (Table 2).
2.2.3. BRAF copy number variation
Fig. 1 schematically summarizes the findings on quantitative real-time PCR analysis of the BRAF gene in 47 patients. The results are also shown in Table 3. More than two copies of the BRAF gene were found in 18 patients (18/47; 38.3%) and two copies, in 29 patients (61.7%). Fifteen of the 18 samples with an increased copy number (83.3%) were from pilocytic astrocytomas; the others were derived from ganglioglioma, pilomyxoid astrocytoma, and pleomorphic xanthoastrocytoma (one each).
On analysis of the clinical factors, a higher median age was noted for the patients with an increased copy number (9.5 years) than for the patients with two copies only (8.5 years), but the difference was not statistically significant. No significant differences by copy number (increased/normal) were found for sex (M/F), tumor grade (I/II), number of operations (1/more), diagnosis of neurofibromatosis-1 (yes/no), need for chemotherapy (yes/no), need for radiotherapy (yes/no), or mortality. The only statistically significant difference between the patients with and without an increased BRAF copy number was location of the glioma: 62.5% (10/16) of the infratentorial tumors had more than two copies of the gene compared to 25.8% (8/31) of the supratentorial tumors (P = 0.014).
3. Discussion
This study investigated the molecular genotype of 48 samples of primary low-grade gliomas and four samples of recurrent low-grade gliomas from children. We searched specifically for genes that are activated in the Ras/Raf (MAPK) signaling cascade (BRAF, GNAQ and GNA11). The findings showed a low rate of BRAF mutations (3/48) (one each in pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and ganglioglioma), and only one case of a GNAQ209 mutation (in a pilocytic astrocytoma). No GNA11 mutations were found. However, an increased copy number of the BRAF gene (3–4 copies) was detected in 18 patients (38.3%), compared to 29 patients (61.7%) with a normal number of copies.
These data are in line with previous studies of low-grade glioma. Pfister et al. [6] found that 7q34 duplication was the mechanism of BRAF activation in 53% of pilocytic astrocytomas, but was negative in gangliogliomas and pleomorphic xanthoastrocytomas. BRAF activation was confirmed by the study of Jones et al. [9] by overexpression of CCND1, a downstream target of the MAPK pathway, and increased phosphorylation of extracellular signal-regulated kinase (ERK) 1/2, an immediate downstream target of BRAF when phosphorylated. The tandem 2-MB duplication at the 7q34 band described in this report in 66% of pilocytic astrocytomas, led to an in-frame infusion gene incorporating the kinase domain of the BRAF oncogene. Similarly, Forshew et al. [10] reported a high frequency (64.8%) of copy number gain of about 1.9 MB at 7q34. In all the pilocytic astrocytomas located in the posterior fossa, the resulting fusion gene lacked the auto-inhibiting domain of BRAF. Both Pfister et al. [6] and Jones et al. [9] showed an association of 7q34 duplication with a supratentorial location (62% and 63% of pilocytic astrocytomas, respectively). By contrast, in another study by Bar et al. [7], of 25 sporadic pilocytic astrocytomas (20 infratentorial and five supratentorial), an increase in copy number at the 7q34 locus was noted in 17 (68%), all located infratentorially. In the present study, pilocytic astrocytoma was the pathologic diagnosis in 15 of the 18 patients (83.3%) with more than two copies of BRAF. We identified both infratentorial and supratentorial gain, but predominantly infratentorial (62.5%). Furthermore, the incidence of increased copy number in all pilocytic astrocytoma samples was 48.4%, lower than reported previously [6,7], suggesting the involvement of other mechanisms.
Mutations in the BRAF gene and changes in its copy number have also been described in lung and colon carcinoma, thyroid cancer, and melanoma [17–19]. Interestingly, administration of a drug that inhibits the mutated activated BRAF gene reportedly led to complete or partial regression of metastatic melanoma in humans [20]. However, only a small proportion of the tumors in our study and in other studies [9–11,21–24] had a BRAF600 mutation, making this drug unlikely to be effective in these cases.
This is the first study to examine the GNAQ mutation in pediatric low-grade glioma. Although very low in frequency, the GNAQ gene might play a role in the early tumorigenesis of low-grade glioma, as reported in uveal melanoma. In a large study, Lamba et al. [25] sequenced exon 5 of the GNAQ and GNA11 genes in a panel of 922 tumors, including glioblastoma, gastrointestinal stromal tumors, acute myeloid leukemia, blue nevi, melanoma, bladder, breast, colorectal, lung, ovarian, pancreas, and thyroid carcinomas. None of the tumor types showed GNAQ or GNA11 mutations except blue nevi (50%). In accordance with findings that BRAF and GNAQ mutations are mutually exclusive [14], we found only BRAF mutations in 6.3% of the samples, and the sole GNAQ mutation in a sample that had wild-type BRAF gene. All tumors positive for either BRAF or GNAQ mutations had a normal copy number of the BRAF gene. There was no correlation of a positive finding with a medical history of neurofibromatosis-1 or tuberous sclerosis.
Interestingly, the four samples taken from secondary/recurrent tumors showed identical molecular results. Given that all these tumors were low grade with no metastases, they would not be expected to undergo malignant transformation or genetic alteration.
Inhibitors of the MAPK pathway, such as AZD6244 and sorafenib, have been approved for the treatment of several types of cancer [20,26–28] and may be suitable for children with aggressive, recurrent, or unresectable tumors. This assumption is supported by the finding of Pfister and colleagues [6] that pharmacological inhibition of Ras/B-Raf/ERK signaling decreases the proliferation of low-grade glioma cells in culture. However, as the MAPK pathway plays a crucial role in tumor development, inhibiting its activity in the pediatric brain could potentially lead to a wide range of side effects. Therefore, therapies that specifically target the relevant oncogene are preferable. Better understanding of the molecular basis of low-grade glioma would increase the treatment options.
In summary, we investigated 52 pathological samples from 48 children with low-grade glioma for mutations in three genes that regulate the MAPK pathway, a crucial pathway in tumorigenesis. The main finding was an increase in the copy number of BRAF; other genetic changes causing this activation remain indeterminate. Additional pathways that are potentially involved in the formation and growth of low-grade tumorigenesis need to be explored.
4. Materials and methods
4.1. Clinical specimens
Archival samples of low-grade gliomas (WHO I–II) from patients aged less than 18 years were obtained from the Department of Pathology, Rabin Medical Center. Only samples with sufficient excess paraffin-embedded or frozen tissue for analysis were included. Tumor classification and cellularity were reviewed by a neuropathologist. Patients’ demographic and clinical data were collected from the medical files. The procurement of the samples and the study protocol were approved by the Institutional Review Board of Rabin Medical Center and the National Genetics Review Board of the Israel Ministry of Health.
4.2. DNA extraction
Forty-two formalin-fixed, paraffin-embedded tissues and 10 frozen tissues were used for DNA isolation. In brief, hematoxylin-eosin-stained 10-μm-section slides were reviewed by a pathologist. Thereafter, areas with an estimated content of more than 75% tumor cells were separated by microdissection (no. 11 surgical blade) from five consecutive 10-μm unstained paraffin-embedded sections of each block. The tissues were deparaffinized and incubated overnight in 1% sodium dodecyl sulfate (SDS) and proteinase K 0.5 mg/ml. DNA was purified by phenol-chloroform extraction and ethanol precipitation and dissolved in 50 μl of distilled water, as previously described [15,16].
4.3. Mutation analysis
Oncogenic mutations in GNAQ (GNAQQ209, exon 5), GNA11 (GNA11Q209, exon 5) and BRAF (BRAFV600E, exon 15) were analyzed with the chip-based matrix-assisted laser desorption time-of-flight (MALDI-TOF) mass spectrometer (Sequenom, San Diego, CA). Specific primers flanking the mutation sites and extension primers that bind adjacent to the mutation site were designed with assay-design software (MassARRAY; Sequenom). After amplification of the region of interest, a primer extension reaction was carried out which included sequence-specific hybridization and sequence-dependent termination, generating different products for the mutated and wild-type alleles, each with a unique mass value. The extension products were spotted onto silicon chips preloaded with proprietary matrix (SpectroChip; Sequenom) and read by the MALDI-TOF mass spectrometer.
4.4. Validation
The mutations were validated by direct sequencing of selected samples. DNA was extracted using standard SDS/proteinase K digestion followed by phenol-chloroform extraction and ethanol precipitation. Each polymerase chain reaction (PCR) amplification was performed in a 50-μl reaction volume containing 150 ng of sample DNA as a template. The reaction was performed using specific primers for BRAF, GNAQ, and GNA11 (Table 1).
Table 1.
Primers used for validation of mutations by PCR test.
| Gene | Forward | Reverse | PCR product size |
|---|---|---|---|
| BRAF V600E | GGCACATCACTGAACATAATTATC | AGCATGATATCACAAAGGTACT | 224bp |
| GNAQ209 | TTTTCCCTAAGTTTGTAAGTAGTGC | CCTCATTGTCTGACTCCACG | 222bp |
| GNA11Q209 | CGCTGTGTCCTTTCAGGATG | CCACCTCGTTGTCCGACT | 150bp |
PCR parameters were as follows: denaturation for 5 min at 95 °C; 35 cycles of 1 min at 95 °C; annealing for 1 min at 56–60 °C and for 1 min at 72 °C with Taq polymerase. The PCR product was amplified on 2% agarose gel and visualized with ethidium bromide staining. Direct sequencing of the PCR products was performed with reagents and an analyzer (Big Dye Terminator Cycle Sequencing and ABI PRISM 3700 DNA Analyzer; Applied Biosystems, Foster City, CA).
4.5. Copy number analysis
For quantitative detection of the BRAF gene, the relative dosage was determined using an ABI 7500 real-time PCR system (Applied Biosystems). The 15-l reaction mixture contained FAM-labeled 1x TaqMan® BRAF kit (ABI, Hs05004157_CN), VIC-labeled 1x TaqMan® Human RNase P detection mix (ABI, Cat. No. 431684906018), 1x TaqMan Universal PCR Master Mix (AmpliTaq Gold® DNA Polymerase, dNTPs with dUTP, Passive Reference, No AmpErase UNG®), and 5 ng genomic DNA template. RNaseP, a single-copy gene, was used as an endogenous control. Molecules of the two genes were independently amplified. FAM and VIC signals were recorded in all chambers at the end of each PCR cycle. Multiplex PCRs were undertaken using 20-μl reactions: 10 μl PCR Master Mix (TaqMan Universal; Applied Biosystems) with 1 μl uracil N-glycosylase, 1 μl RNaseP primers and probe ×20 (VIC), 4 μl BRAF primers and probe ×20 (FAM), 4 μl of the DNA sample, and 4 μl nuclease-free water. The default setting was selected for one cycle at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and annealing-extension at 60 °C for 1 min. Each sample was tested in Triplicates, with no-template controls in each assay. After the reaction was completed, digital PCR analysis software (CopyCallerTM, Applied Biosystems) was used to process the data and count the numbers of both FAM-positive chambers (BRAF, target gene) and VIC-positive chambers (RNaseP) in each panel. A total of 47 samples were analyzed, one sample (no. 32) failed technically due to limited amount of DNA.
4.6. Statistical analysis
The Statistical Package for the Social Sciences (SPSS), version 15, was used for data analysis. Means and standard deviations were calculated for all continuous variables. Chi-square test and Fisher exact test were used to determine differences in distribution across categorical variables or between two groups for continuous variables, respectively. The level of significance was set at P < 0.05 for all statistical analyses.
Acknowledgments
This study was partially supported by the Zanvyl and Isabelle Krieger Fund, Baltimore, Maryland, USA; the Eldor-Metzner Clinician Scientist Award, Chief Scientist, Israel Ministry of Health (N.G.C., Grant No. 3-3741); and the Young Investigator Award, Rabin Medical Center, Petach Tikva, Israel (Y.L).
This work was presented in part at the annual meeting of The Israel Society for Eye and Vision Research, Tzuba, Israel, March 2011; the annual meeting of the Israel Society of Neurosurgery, Eilat, Israel, March 2011 (awarded), and the annual meeting of the Israeli Society of Pediatric Hemato-oncology, Neve Ilan, Israel, March 2011.
References
- 1.Tatevossian R.G., Lawson A.R., Forshew T., Hindley G.F., Ellison D.W., Sheer D. MAPK pathway activation and the origins of pediatric low-grade astrocytomas. J. Cell. Physiol. 2010;222:509–514. doi: 10.1002/jcp.21978. [DOI] [PubMed] [Google Scholar]
- 2.Gnekow A.K., Kortmann R.D., Pietsch T., Emser A. Low grade chiasmatic-hypothalamic glioma – carboplatin and vincristin chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy. Klin. Padiatr. 2004;216:331–342. doi: 10.1055/s-2004-832355. [DOI] [PubMed] [Google Scholar]
- 3.Ohgaki H., Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 4.Fisher P.G., Tihan T., Goldthwaite P.T., Wharam M.D., Carson B.S., Weingart J.D., Repka M.X., Cohen K.J., Burger P.C. Outcome analysis of childhood low-grade astrocytomas. Pediatr. Blood Cancer. 2008;51:245–250. doi: 10.1002/pbc.21563. [DOI] [PubMed] [Google Scholar]
- 5.Lyustikman Y.M.H., Pao W., Holland E.C. Constitutive activation of Raf-1 induces glioma formation in mice. Neoplasia. 2008;10:501–510. doi: 10.1593/neo.08206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfister S., Janzarik W.G., Remke M., Ernst A., Werft W., Becker N., Toedt G., Wittmann A., Kratz C., Olbrich H., Ahmadi R., Thieme B., Joos S., Radlwimmer B., Kulozik A., Pietsch T., Herold-Mende C., Gnekow A., Reifenberger G., Korshunov A., Scheurlen W., Omran H., Lichter P. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J. Clin. Investig. 2008;118:1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bar E.E., Lin A., Tihan T., Burger P.C., Eberhart C.G. Frequent gains at chromosome 7q34 involving braf in pilocytic astrocytoma. J. Neuropathol. Exp. Neurol. 2008;67:878–887. doi: 10.1097/NEN.0b013e3181845622. 810. http://dx.doi.org/1097/NEN.1090b1013e3181845622. [DOI] [PubMed] [Google Scholar]
- 8.Deshmukh H., Yeh T.H., Yu J., Sharma M.K., Perry A., Leonard J.R., Watson M.A., Gutmann D.H., Nagarajan R. High-resolution, dual-platform aCGH analysis reveals frequent HIPK2 amplification and increased expression in pilocytic astrocytomas. Oncogene. 2008;27:4745–4751. doi: 10.1038/onc.2008.110. [DOI] [PubMed] [Google Scholar]
- 9.Jones D.T.K.S., Liu L., Pearson D.M., Bäcklund L.M., Ichimura K., Collins V.P. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forshew T., Tatevossian R.G., Lawson A.R., Ma J., Neale G., Ogunkolade B.W., Jones T.A., Aarum J., Dalton J., Bailey S., Chaplin T., Carter R.L., Gajjar A., Broniscer A., Young B.D., Ellison D.W., Sheer D. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J. Pathol. 2009;218:172–181. doi: 10.1002/path.2558. [DOI] [PubMed] [Google Scholar]
- 11.Eisenhardt, A.E., Olbrich, H., Röring, M., Janzarik, W., Anh, T.N., Cin, H., Remke, M., Witt, H., Korshunov, A., Pfister, S.M., Omran, H. and Brummer, T. (2010) Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int. J. Cancer December 28. http://dx.doi.org/10.1002/ijc.25893 [Epub ahead of print]. [DOI] [PubMed]
- 12.Hubbard K.B., Hepler J.R. Cell signalling diversity of the Gqa family of heterotrimeric G proteins. Cell. Signal. 2006;18:135–150. doi: 10.1016/j.cellsig.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Van Raamsdonk C.D., Bezrookove V., Green G., Bauer J., Gaugleret L. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onken M.D., Worley L.A., Long M.D., Duan S., Council M.L., Bowcock A.M., Harbour J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2006;49(12):5230–5234. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen Y., Goldenberg-Cohen N., Parrella P., Chowers I., Merbs S.L., Péer J., Sidransky D. Lack of BRAF mutation in primary uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003;44:2876–2878. doi: 10.1167/iovs.02-1329. [DOI] [PubMed] [Google Scholar]
- 16.Goldenberg-Cohen N., Cohen Y., Rosenbaum E., Herscovici Z., Chowers I., Weinberger D., Péer J., Sidransky D. T1799A BRAF mutations in conjunctival melanocytic lesions. Invest. Ophthalmol. Vis. Sci. 2005;46:3027–3030. doi: 10.1167/iovs.04-1449. [DOI] [PubMed] [Google Scholar]
- 17.Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M.J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B.A., Cooper C., Shipley J. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 18.Ciampi R.Z.Z., Nikiforov Y.E. BRAF copy number gains in thyroid tumors detected by fluorescence in situ hybridization. Endocr. Pathol. 2005;16:99–105. doi: 10.1385/ep:16:2:099. [DOI] [PubMed] [Google Scholar]
- 19.Stark M., Hayward N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2007;67:2632–2642. doi: 10.1158/0008-5472.CAN-06-4152. [DOI] [PubMed] [Google Scholar]
- 20.Flaherty K.T., Puzanov I., Kim K.B., Ribas A., McArthur G.A., Sosman J.A., O’Dwyer P.J., Lee R.J., Grippo J.F., Nolop K., Chapman P.B. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasheed B.K., Stenzel T.T., McLendon R.E., Parsons R., Friedman A.H., Friedman H.S., Bigner D.D., Bigner S.H. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. 1997;57:4187–4190. [PubMed] [Google Scholar]
- 22.Yu J., Zhang H., Gu J., Lin S., Li J., Lu W., Wang Y., Zhu J. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer. 2004;4:65. doi: 10.1186/1471-2407-4-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sievert A.J., Jackson E.M., Gai X., Hakonarson H., Judkins A.R., Resnick A.C., Sutton L.N., Storm P.B., Shaikh T.H., Biegel J.A. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel braf fusion gene. Brain Pathol. 2009;19:449–458. doi: 10.1111/j.1750-3639.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorente A., Mueller W., Urdangarín E., Lázcoz P., Lass U., von Deimling A., Castresana J.S. RASSF1A, BLU, NORE1A, PTEN and MGMT expression and promoter methylation in gliomas and glioma cell lines and evidence of deregulated expression of de novo DNMTs. Brain Pathol. 2009;19:279–292. doi: 10.1111/j.1750-3639.2008.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamba S.F.L., Buttitta F., Bleeker F.E., Lamba S., Felicioni L., Buttitta F., Bleeker F.E., Malatesta S., Corbo V., Scarpa A., Rodolfo M., Knowles M., Frattini M., Marchetti A., Bardelli A. Mutational profile of GNAQQ209 in human tumors. PLoS ONE. 2009;4:e6833. doi: 10.1371/journal.pone.0006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Espinosa A.V., Porchia L., Ringel M.D. Targeting BRAF in thyroid cancer. Br. J. Cancer. 2006;96:16–20. doi: 10.1038/sj.bjc.6603520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ball D.W., Jin N., Rosen D.M., Dackiw A., Sidransky D., Xing M., Nelkin B.D. Selective growth inhibition in BRAF mutant thyroid cancer by the mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244. J. Clin. Endocrinol. Metab. 2007;92:4712–4718. doi: 10.1210/jc.2007-1184. [DOI] [PubMed] [Google Scholar]
- 28.Fecher L.A., Amaravadi R.K., Flaherty K.T. The MAPK pathway in melanoma. Curr. Opin. Oncol. 2008;20:183–189. doi: 10.1097/CCO.0b013e3282f5271c. [DOI] [PubMed] [Google Scholar]