

Graphical abstract
New insights into the locking mechanism with open, closing, closed and locked structures of two isocitrate dehydrogenases.

Highlights
► Focus is on a thermophilic (CtIDH) and cold active (DpIDH) isocitrate dehydrogenase. ► Biochemical characterization and three new IDH crystal structures are presented. ► CtIDH has many ionic interactions and hydrogen bonds, which explain the Tm of 68 °C. ► Prokaryotic IDH subfamily II seems to have a unique locking mechanism. ► The large domain is locked to the small domain and NADP+ is also directed correctly.
Abbreviations: IDH, isocitrate dehydrogenase; CtIDH, Clostridium thermocellum IDH; DpIDH, Desulfotalea psychrophila IDH; EcIDH, Escherichia coli IDH; TmIDH, Thermotoga maritima; PcIDH, porcine heart mitochondrial IDH; HcIDH, human cytosolic IDH; ScIDH, Saccharomyces cerevesiae mitochondrial IDH; DhIDH, Desulfitobacterium hafniense IDH; DSC, differential scanning calorimetry; Tm, apparent melting temperature
Keywords: Temperature adaptation, Thermophilic, Psychrophilic, NADP+ selectivity, Domain movement
Abstract
Isocitrate dehydrogenase (IDH) catalyzes the oxidative NAD(P)+-dependent decarboxylation of isocitrate into α-ketoglutarate and CO2 and is present in organisms spanning the biological range of temperature. We have solved two crystal structures of the thermophilic Clostridium thermocellum IDH (CtIDH), a native open apo CtIDH to 2.35 Å and a quaternary complex of CtIDH with NADP+, isocitrate and Mg2+ to 2.5 Å. To compare to these a quaternary complex structure of the psychrophilic Desulfotalea psychrophila IDH (DpIDH) was also resolved to 1.93 Å. CtIDH and DpIDH showed similar global thermal stabilities with melting temperatures of 67.9 and 66.9 °C, respectively. CtIDH represents a typical thermophilic enzyme, with a large number of ionic interactions and hydrogen bonds per residue combined with stabilization of the N and C termini. CtIDH had a higher activity temperature optimum, and showed greater affinity for the substrates with an active site that was less thermolabile compared to DpIDH. The uncompensated negative surface charge and the enlarged methionine cluster in the hinge region both of which are important for cold activity in DpIDH, were absent in CtIDH. These structural comparisons revealed that prokaryotic IDHs in subfamily II have a unique locking mechanism involving Arg310, Asp251′ and Arg255 (CtIDH). These interactions lock the large domain to the small domain and direct NADP+ into the correct orientation, which together are important for NADP+ selectivity.
1. Introduction
Isocitrate dehydrogenase (IDH) is an enzyme in the tricarboxylic acid cycle, which catalyzes the oxidative NAD(P)+-dependent dehydrogenation and decarboxylation of isocitrate to α-ketoglutatrate and CO2. IDH has a critical metabolic function and consequently is found in organisms from all domains of life. This enzyme has been extensively studied both kinetically and structurally from psychrophilic, mesophilic and (hyper)thermophilic organisms [1–19]. With respect to cofactor specificity and oligometric states the IDH enzyme represents a diverse family which is divided into three subfamilies based on the primary sequences [15]. Subfamiliy I includes archaeal and some bacterial IDH’s, whereas subfamily II contains eukaryotic and bacterial IDH’s but is absent from the archea. The amino acid sequence identity between subfamilies I and II is low (<20%), however the three-dimensional structures have similar folds and the key catalytically residues are conserved. The active IDH enzymes are functional as homodimers in both subfamilies, each monomer consists of a small and large, clasp domains which link the two subunits together, and the active sites being situated in the clefts between a large and a small domain from opposing subunits.
The crystal structure of the group I IDH from Escherichia coli (EcIDH) was the first to be determined, subsequently many other EcIDH structures with different substrates and conformations were resolved; leading to detailed knowledge of its catalytic mechanism [2,7,20–25].
In our earlier work, we have solved the three-dimensional structure of IDH from three different hyperthermophiles; Archaeoglobus fulgidus (AfIDH) [18], Aeropyrum pernix (ApIDH) [8] and Thermotoga maritima (TmIDH) [9], searching for stabilizing interactions and explanations for the thermal stability of these enzymes. This was explained by enlarged ionic networks and an increased number of ion pairs in ApIDH and TmIDH, but AfIDH was more similar to the mesophilic EcIDH. All of the hyperthermophilic IDHs had stabilization of the N terminus; ApIDH by a disulfide bond, TmIDH by long-range electrostatic interactions and AfIDH through an aromatic cluster. These stabilizing factors are typical for hyperthermophilic proteins together with; loop shortening, stronger inter-subunit interactions, increased number of hydrogen bonds, a reduction of hydrophobic accessible surface area, increased number of ionic interactions at the surface and reduction of the repulsive charge–charge interactions [26,27,28,29]. An opposite trend is found for psycrophilic proteins, which are reported with an increase in surface charge, particularly a negative charge, an increase in hydrophobic accessible surface area, a reduced Arg/Lys ratio and higher methionine content [30,31]. However each stabilizing or destabilizing contribution is small and dependent on size, distance and position in the structure. As a result of this there is still no complete explanation of the activity–stability–flexibility relationship in cold adapted proteins.
In the search for cold-adaptive traits we performed a biochemical characterization and solved the crystal structure of IDH from the psychrophilic Desulfotalea psychrophila (DpIDH), we found an enzyme with an unusual high thermal stability (Tm = 66.9 °C) and a moderate activity at its normal working temperature [4]. Its structure revealed stabilizing features such as; a unique ionic network in the clasp region, an aromatic cluster in the small domain, and multiple N- and C-termini ionic-interactions. This is counterbalanced by elements promoting flexibility such as a methionine cluster in the dimeric interface, destabilized negatively charged amino acids close to the active site and an acidic surface.
Structural analyses of Saccharomyces cerevesiae mitochondrial IDH (ScIDH) one in an open conformation with NADP+, two in quasi-closed conformations with isocitrate or α-ketoglutarate bound, and one fully closed quaternary complex with NADP+, α-ketoglutarate and Ca2+, representing different enzymatic states, have revealed fine-tuned conformational changes to occur during the catalytic reaction, compared to the E. coli IDH (EcIDH) from subfamily I. Especially, the alpha helices α4 and α11, which are unique in the subfamily II enzymes, act as a lid covering the top of the NADP+ and isocitrate-binding sites [32].
The availability of a range of three dimensional structures provided the opportunity to compare prokaryotic and eukaryotic IDHs within subfamily II. Therefore we have determined the crystal structure of IDH’s from the thermophilic bacterium Clostridium thermocellum, the hyperthermophilic T. maritima (TmIDH) [9], the psychrophilic D. psychrophila (DpIDH) in both native and binary form with isocitrate [4], together these span a habitat temperature range from 0 to 80 °C.
We have expressed, characterized and crystallized CtIDH, and obtained an open form (CtIDH-Open) and a closed form of CtIDH. The latter is a quaternary complex with NADP+, isocitrate and a magnesium ion (CtIDH–NADP). In addition, a structure of DpIDH with NADP+, isocitrate and Mg2+ (DpIDH–NADP) was resolved and a comparative structural analysis was performed. These quaternary structures have displayed new interactions with the cofactor which lock the enzyme into a conformation ready for catalysis. Altogether, we now possess sufficient structures and in different states, open, closing, closed and locked, to suggest a new prokaryotic subfamily II IDH locking mechanism involved in substrate binding.
2. Materials and methods
2.1. Cloning, expression and purification
Putative icd genes from C. thermocellum (YP_001036717) and D. psychrophila (YP_064514), were amplified from genomic DNA by PCR using the following primer sets; 5′-CACCATGAGTAAGATAAAAATGAAAGTTCCGTT-3′, 5′-TTATGCCATACCTTCAAAAGTTTTCCTG-3′ and 5′-GGGAATTCCATATGAAGATACAAATGAAAACTCCACTCG-3′, 5′-GCGGGATCCTTACTTGTTGAGCTGTTGCAATCTTTTGCC-3′.
The NdeI and BamHI restriction sites are underlined. The cloning and purification of DpIDH were performed as described earlier [4]. For the amplification of the CtIDH gene the following protocol was performed; primer concentration at 0.5 μM; dATP, dTTP, dCTP, and dGTP at 0.2 mM; 1× Pfu polymerase buffer; 1 mM MgSO4 and 1.25 units of PfuTurbo DNA polymerase were used for the PCR and the amplifications consisted of 30 cycles (1 min at 94 °C, 1 min at 55 °C, and 1.5 min at 72 °C) and a final extension of 10 min at 72 °C. The PCR product was ligated into a pET101 vector.
E. coli strain EB106 (icd-11 dadR1 trpA62 trpE61 tna-5 lambda-) lysogenized by λDE3 [18], was used for expression. Recombinant CtIDH was produced by growing transformed cells in Luria Broth (LB) broth containing ampicillin (100 μg/ml) at 37 °C to OD600 nm = 0.7–0.8 cell density and subsequent expression (3–4 h) after induction with 1.0 mM isopropyl-β-thiogalactopyranoside (IPTG). After expression, the cells were harvested by centrifugation (5000×g, 15 min) and frozen at −20 °C until used. Cells were resupended in pre-chilled (4 °C) 20 mM sodium phosphate buffer, pH 7.0, containing 10 mM MgCl2 and disrupted using a French pressure cell at 55 megapascals followed by centrifugation (13 000×g, 30 min, 15 °C). The purification of CtIDH was performed at room temperature, and the cell extract was loaded directly onto a Red-Sepharose column equilibrated with 20 mM sodium phosphate buffer, pH 7.0 and 10 mM MgCl2. Unbound protein was removed by washing with 0.2 M sodium chloride in 20 mM sodium phosphate buffer, pH 7.0 until A280 was zero. An additional washing step was performed using 0.25 mM NADP+ in the sodium phosphate buffer and IDH was then biospecifically eluted by 10 mM dl-isocitrate and 0.25 mM NADP+ in the phosphate buffer. The enzyme was purified to homogeneity (data not shown) as judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) with Commassie blue staining, and if the protein was not purified to homogeneity this purification step was repeated. The protein concentration was measured by the method of Bradford [33], using bovine serum albumin as a standard.
2.2. Enzyme assay and determination of the catalytic parameters
IDH activity was measured photometrically (Cary 4E UV–vis spectrophotometer) by monitoring the formation of NADPH at 340 nm (ε340 = 6.22 mM−1 cm−1). The standard reaction mixture of 1 ml contained 50 mM Tricine–KOH, pH 8.0, 2 mM dl-isocitrate, 10 mM MgCl2, and 250 μM NADP+. The temperature dependence of catalytic activity and kinetic parameters were studied by assaying the initial IDH activity at different temperatures. The dpKa/dT of the 50 mM Tricine–KOH buffer was used to ensure that pH 8 was obtained across the temperature range used. For determination of Km and Vmax values, the concentration of one substrate was varied while the second was kept at saturating concentration. Km and Vmax values were determined from the direct linear plot [34].
Thermodynamic activation parameters were calculated [35], using the following equations:
| (1) |
| (2) |
| (3) |
where kB is the Boltzmann constant (1.3805 × 10−23 J K−1), h the Planck constant (6.6256 × 10−34 J s), k (s−1) the rate constant at temperature T (K), Ea the activation energy of the reaction and R (8.314 J mol−1 K−1) the gas constant.
2.3. Thermal stability measurement
Differential scanning calorimetry was carried out with a VP-DSC MicroCalorimeter (MicroCal Inc.) with cell volumes of 0.5274 ml and at a dimer concentration of 22 μM. The calorimetric scans were carried out between 20 °C and 95 °C in 50 mM potassium phosphate buffer, pH 7.5, 0.1 M NaCl, at a scan rate of 1 K/min. A constant pressure of 30 psi was applied to avoid degassing at high temperatures.
2.4. Crystallization, data collection and model building
Two crystal forms of CtIDH were obtained; one in an open confirmation (CtIDH-Open) and one quaternary complex with NADP+, isocitrate and Mg2+ (CtIDH–NADP). The two CtIDH crystals used for X-ray data collection were both obtained using the hanging drop method, with reservoir solutions of 9–11% (w/v) polyethylene glycol (PEG) PEG3350, and 0.15 M ammonium dihydrogen citrate at 37 °C. Additionally, CtIDH–NADP had 10% (v/v) propylene glycol in the reservoir solution. The drops were made by mixing 3 μl protein with 3 μl reservoir solution. The CtIDH-Open had a protein concentration of 9.5 mg/ml and no additives, whereas the CtIDH–NADP protein solution was at 5 mg/ml also containing 10 mM NADP+ and 10 mM dl-isocitrate. The final crystal sizes were about 1.0 × 0.2 × 0.1 mm3 for CtIDH-Open and 0.1 × 0.15 × 0.3 mm3 for CtIDH–NADP. The cryo conditions were 25% glycerol in addition to their respective reservoir solutions and both crystals were flash cooled in liquid nitrogen.
X-ray data were collected at European Synchrotron Radiation Facilities (ESRF), Grenoble at ID29 for the CtIDH-Open crystal, to a final resolution of 2.35 Å The crystal belongs to the space group P21 with cell parameters a = 56.15 Å, b = 107.25 Å, c = 154.59 Å and β = 93.64° and four molecules in the asymmetric unit. The dataset on the CtIDH–NADP crystal was recorded at the macromolecular crystallography beamline BL14.2 at BESSY, Berlin to a final resolution of 2.50 Å (Table 3). The crystal belongs to the space group P3221 with cell axis a = b = 129.20 Å and c = 60.59 Å. For both the CtIDH data sets the images were integrated and scaled with XDS [36] and structure factors obtained with TRUNCATE [37].
Table 3.
Statistics from the data collection and refinement statistics for the CtIDH-Open, CtIDH–NADP and DpIDH–NADP structures. The numbers in parentheses represent values in the highest of 10 resolution shells, and the resolution limits for these are indicated.
| CtIDH-Open | CtIDH–NADP | DpIDH–NADP | |
|---|---|---|---|
| PDB code | 4AOY | 4AOU | 4AOV |
| Diffraction data statistics | |||
| Beam line | ID29 | BESSY, BL14.2 | SNBL |
| Wavelength (Å) | 1.0052 | 0.9184 | 0.8536 |
| Resolution limits (Å) | 50–2.35 (2.48–2.35) | 45–2.50 (2.64–2.50) | 40.0–1.93 (2.03–1.93) |
| Space group | P21 | P3221 | C2 |
| Unit cell parameters (Å) | a = 56.2 | a = b = 129.2 | a = 61.6 |
| b = 107.3 | b = 98.9 | ||
| c = 154.6 | c = 60.6 | c = 71.9 | |
| (°) | β = 93.6 | β = 103.9 | |
| No. unique reflections | 75 792 | 20 411 | 30 844 |
| Multiplicity | 3.4 (3.3) | 5.0 (5.0) | 3.8 (3.6) |
| Completeness (%) | 99.9 (99.7) | 100.0 (100.0) | 98.1 (98.1) |
| <I>/<σ(I)> | 10.2 (2.1) | 13.1 (2.8) | 10.7 (2.7) |
| Rmerge (%) | 8.4 (59.7) | 7.1 (61.2) | 9.4 (41.4) |
| Wilson B-factor (Å2) | 46.42 | 56.17 | 17.90 |
| Refinement statistics | |||
| Resolution range (Å) | 20–2.35 | 45–2.50 | 20–1.93 |
| Total reflections | 73 524 | 19 347 | 29 252 |
| R-factor (%) | 21.13 | 18.93 | 17.49 |
| R-free (%) | 26.32 | 24.40 | 22.54 |
| No./size (%) of R-free set | 2010 (2.7) | 1 043 (5.1) | 1563 (5.1) |
| No. of protein atoms | 11 934 | 3 210 | 3 249 |
| No. of water molecules | 88 | 41 | 227 |
| No. of other molecules | – | 1 Mg2+, 1 isocitrate, 1 NADP+ | 1 Mg2+, 1 isocitrate, 1 NADP+ |
| Average B factor (Å2) | |||
| Total | 27.0 | 45.2 | 20.1 |
| Protein A/B/C/D | 27.1/26.8/26.9/27.0 | 45.2 | 19.9 |
| Water/Mg2+/iso./NADP+ | 34.7/–/–/– | 42.9/43.0/37.9/52.1 | 25.0/6.0/12.9/13.3 |
| RMSD from ideal geometry | |||
| Bonds (Å) | 0.015 | 0.017 | 0.012 |
| Angles (°) | 1.67 | 1.88 | 1.41 |
| Ramanchandran plot | |||
| Core (%) | 89.3 | 89.0 | 93.3 |
| Allowed (%) | 9.8 | 9.6 | 6.1 |
| Generously allowed (%) | 0.5 (6 residues) | 1.1 | 0.3 |
| Disallowed (%) | 0.5 (6 residues) | 0.3 (1 residue) | 0.3 (1 residue) |
CtIDH–NADP was solved by molecular replacement using 1 monomer of native DpIDH (PDB entry 2UXQ) as search model which has 63/81% sequence identity/similarity. There is one molecule in the asymmetry unit giving a water content of 62%.
It turned out to be more challenging to solve the CtIDH-Open structure in space group P21, with four monomers in the asymmetric unit giving 52% solvent. Using the program MolRep [37] and one monomer of the open T. maritima IDH (TmIDH; PDB 1ZOR) as search model (61/81% sequence identity/similarity) gave four poorly resolved rotation and translation solutions. One model was then built by overlaying each domain from DpIDH (see Fig. 3) onto the open TmIDH structure. This DpIDH-Open-model resulted in four better resolved rotation and translation peaks, where two dimers were found in the asymmetric unit.
Fig. 3.

Observed 2Fo-Fc electron density (1σ; blue) and difference Fo–Fc electron density (3.5σ; green) for the NADP molecules in the final models of CtIDH–NADP (panels a and c) and DpIDH–NADP (panels b and d). The molecules were refined with occupancy 0 for 8 of the atoms in the nicotinamide group. A similar phosphate ADP-ribose molecules was also observed for the NADP molecule bound to alcohol dehydrogenase from Saccharomyces cerevisiae[45].
The third structure presented here is the complex of DpIDH and NADP+ (DpIDH–NADP) which was obtained by co-crystallization at 8 °C using the hanging drop method and reservoir solutions with 100 mM Tris/HCl buffer at pH 7.4, 1.7–1.9 M ammonium sulfate, 2% PEG 400 and 60 mM magnesium sulfate. The protein solution contained 20 mg/ml DpIDH, 10 mM NADP+ and 10 mM dl-isocitrate, and the drops were made by mixing equal volumes of reservoir (2 μl) and protein (2 μl). The harvested DpIDH–NADP crystal was cryo protected in a solution with 25% glycerol in addition to the reservoir components, and then flash cooled in liquid nitrogen.
Data of DpIDH–NADP were collected at the Swiss-Norwegian Beamlines (SNBL), ESRF, Grenoble, France at 100 K, to 1.93 Å resolution and the complex was found to belong to the space group C2 with cell parameters a = 61.64 Å, b = 98.91 Å, c = 71.89 Å and β = 103.94°. This is a different crystal form than the native DpIDH (PDB 2UXQ) and DpIDH with only isocitrate (DpIDH-iso; PDB 2UXR) [4]. The X-ray data were integrated and scaled with XDS [36] and the structure factors were obtained using TRUNCATE [37].
The phase problem of DpIDH–NADP was solved by the molecular replacement method using the program MolRep and the native DpIDH (PDB entry 2UXQ) as search model in space group C2. There is one molecule in the asymmetric unit giving a water content of 48%, and the crystallographic twofold axis makes up the functional dimer.
All three models were refined with Refmac5 [38], solvent molecules were added as waters applying the automated ARP_WATERS routine within REFMAC5 and the models were manually inspected in O [39].
2.5. Structural analysis and electrostatic surface calculations
Prior to these calculations all double conformations were removed. Then hydrogen bonds were calculated using the program HBPLUS [40] v3.15 and the following parameters for Donor (D), Acceptor (A), Acceptor Antecedents (AA) and (calculated) Hydrogens (H) were applied: maximum distances for D–A 3.5 Å, H–A 2.5 Å; minimum angle for D–H–A, D–A–AA and H–A–AA of 90°. All ion pairs were included in the calculations. Accessible surface areas were calculated using the program AREAIMOL [37].
Ionic interactions were analyzed using the WHAT IF server (http://swift.cmbi.kun.nl/WIWWWI/), and maximum cut off distances of 4 and 6 Å were applied. Inter-subunit and inter-domain salt bridges were found from the same server with a 4 Å cut-off distance. The qualitative analysis was done in the graphical program O [39]. For each of the three structures dimer were analyzed, for CtIDH-Open from monomer A/B, whereas for CtIDH–NADP and DpIDH–NADP by applying the crystallographic symmetry. Then all hydrogen atoms were added, the electrostatic surfaces were calculated with DelPhi [41] and displayed with PyMol (http://www.pymol.org).
3. Results and discussion
3.1. Biochemical characterization
The optimal growth temperature of the organism C. thermocellum ATCC 27405 is 60 °C [42], and it is therefore defined as a thermophile. IDH from this organism (CtIDH) was cloned, the protein expressed and purified for structure determination and biochemical characterization. The thermal stability of CtIDH was determined by differential scanning calorimetry (DSC) under the conditions used previously for DpIDH and TmIDH, which have apparent melting temperatures (Tm) of 66.9 and 98.3 °C, respectively [4,9]. Notably, CtIDH had a Tm of 67.9 °C (Table 1), a temperature only marginally higher than the psychrophilic homolog. The thermophilic CtIDH is active over a broad temperature range spanning a scale from 5 to over 75 °C (Fig. 1) with an apparent optimal enzymatic activity at 70 °C. The fact that the melting temperature is lower than the optimum temperature for activity could be due to experimental differences between the DSC and the activity assay. The DSC thermograms for the thermal denaturation and the determined Tm-values are scan rate dependent (data not shown). The kcat is determined using initial-rate measurements and the enzyme is added to a pre-incubated assay solution, but in the DSC experiments the enzyme is subjected to heat over time.
Table 1.
Thermal properties and kinetic parameters (per catalytic site) for IDHs from subfamily II.
| Protein | Tgrowtha (°C) | Toptb (°C) | Tmc (°C) | Assay temp (°C) | NADP |
Isocitrate |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (μM−1 sec−1) | Km (μM) | kcat (s−1) | kcat/Km (μM−1 sec−1) | |||||
| CtIDH | 60 | 70 | 67.9 | 5 | 7.6 | 3.6 | 0.5 | 16.8 | 3.8 | 0.2 |
| 25 | 8.9 | 35.6 | 4.0 | 10 | 27.4 | 2.8 | ||||
| 45 | 11.1 | 142 | 12.8 | 20.6 | 143 | 6.9 | ||||
| DpIDH d | 10 | 35–40 | 66.9 | 5 | 3.0 | 7.8 | 2.6 | 62.5 | 4.5 | 0.07 |
| 25 | 8.1 | 35.6 | 4.4 | 131 | 31.4 | 0.24 | ||||
| 45 | 41.1 | 118.6 | 2.9 | 4230 | 115.5 | 0.03 | ||||
| DhIDH d | 37 | 45 | 58.6 | 5 | 13.1 | 11.7 | 0.9 | 23.2 | 12.2 | 0.5 |
| 25 | 29.5 | 85.8 | 2.9 | 20.9 | 82.0 | 3.9 | ||||
| 45 | 108 | 376 | 3.5 | 387 | 275.8 | 0.7 | ||||
| PcIDH e | 39 | 50 | 59 g | 25 | 5.59 | 33.3 | 5.96 | 8.37 | 33.3 | 3.98 |
| TmIDH d | 80 | 90 | 98.3 | 25 | 8.6 | 4.7 | 0.6 | 30 | 4.7 | 0.2 |
| 45 | 12.9 | 34.2 | 2.7 | 36.4 | 35.3 | 1.0 | ||||
| 70 f | 55.2 f | 252 f | 4.6 f | - | - | - | ||||
Fig. 1.

Specific enzyme activity of the psycrophilic DpIDH (●), the mesophilic DhIDH (○), the thermophilic CtIDH (▾) and hyperthermophilic TmIDH (▽) at saturated conditions.
In order to assess the effect of temperature on the catalytic parameters; enzyme activity kcat, the Km and the catalytic efficiency kcat/Km, were determined (Table 1). CtIDH has a similar activity to cold-active DpIDH at 25 °C, but a lower activity than the mesophilic DhIDH. Moreover, at 5 °C CtIDH shows only a slightly lower activity than its psychrophilic and mesophilic homologs (Table 1). Km-values for isocitrate and NADP+ for CtIDH are lower and less temperature-dependent than the psycro- and mesophilic homologues (Table 1), suggesting a high affinity for both substrate and cofactor in the 5–45 °C temperature range.
An Arrhenius plot (lnkcat as a function of 1/T) was made of the measurements of kcat at various temperatures and the calculated activation parameters are given in Table 2. The thermodynamic parameters of the experimental data such as the free energy of activation (ΔG#) for CtIDH is similar to the psychrophilic DpIDH and lower than the hyperthermophilic TmIDH but higher than the mesophilic DhIDH. This may partly explain the overall high activity observed for CtIDH (Fig. 1). The activation energy (Ea) of the reaction increases from psychrophilic to mesophilic and thermophilic and further to hyperthermophilic. This is consistent with earlier observations in other enzyme families [35,43,44]. A low activation energy reduces the thermo-dependence of the reaction rate, an aspect highly important for the psychrophilic enzymes, where it is considered an important adaptation to cold. The activation enthalpy ΔH# for CtIDH is higher than the psychrophilic and mesophilic IDHs, giving rise to a thermo-dependent enzymatic activity (kcat) for this thermophilic IDH, displayed by the absolute difference in activity within the temperature range 5–70 °C. In comparison to other IDH enzymes CtIDH has a low activation entropy (ΔS#) value, both negative and absolute. This is reflected in the low Km observed for this enzyme, indicating a high affinity for both isocitrate and NADP+. As illustrated in Fig. 1, the CtIDH is a typical thermophilic enzyme with a high activity at its natural working temperature; it is DpIDH that displays an unusual low enzyme activity even at low temperatures. In summary, the thermophilic CtIDH and psychrophilic DpIDH, despite having very different activity rates and temperature optima, nevertheless display similar thermal stability.
Table 2.
Thermodynamic activation parameters for oxidative decarboxylation of isocitrate to α-ketoglutarate and CO2 by psychrophilic DpIDH, mesophilic DhIDH, thermophilic CtIDH and hyperthermophilic TmIDH.
| Ea (kJ mol−1) | T (°C) | Kcat (s−1) | ΔG# (kJ mol−1) | ΔH# (kJ mol−1) | TΔS# (kJ mol−1) | ΔS# (kJ K−1mol−1) | |
|---|---|---|---|---|---|---|---|
| CtIDH | 65.4 | 5 | 3.8 | 66.4 | 63.1 | −3.3 | −12.0 |
| 25 | 27.4 | 66.5 | 62.9 | −3.6 | −12.9 | ||
| DpIDH a | 54.8 | 5 | 4.5 | 66.1 | 52.5 | −13.6 | −48.8 |
| 25 | 31.4 | 66.2 | 52.3 | −13.9 | −49.8 | ||
| DhIDH a | 58.8 | 5 | 12.2 | 63.8 | 56.5 | −7.2 | −26.1 |
| 25 | 82.1 | 63.8 | 56.4 | −7.4 | −26.7 | ||
| TmIDH a | 81.6 | 25 | 4.7 | 70.9 | 79.1 | 8.2 | 29.5 |
| 70 | 282.7 | 70.3 | 78.7 | 8.4 | 30.3 |
Data from Fedøy et al. [4].
3.2. The structure determination of CtIDH and DpIDH
In order to link structural differences and properties to the measured differences in biochemical properties between thermophilic CtIDH and psychrophilic DpIDH, crystal structures of thermophilic CtIDH were resolved; one apo form of the enzyme (CtIDH-Open) and one in complex with NADP+, isocitrate and a metal ion (denoted as the CtIDH–NADP). The structure of the cold active DpIDH apo enzyme and in complex with isocitrate was previously determined [4]. Here, we present a quaternary complex crystal structure with NADP+, isocitrate and a metal ion (designated as the DpIDH–NADP). Both complexes were obtained with the same crystallization conditions as the native structures, but crystallized in different space groups.
The open apo form of the structure of CtIDH (CtIDH-Open) was obtained to 2.35 Å resolution and refined to an R-factor and R-free of 21.1% and 26.3% respectively. The space group is P21 and there are four molecules in the asymmetric unit where monomers A + B and C + D form two dimers. During the refinement two groups of non-crystallographic symmetry (NCS) were applied to the molecule A and C, and B and D, respectively. In addition 16 TLS (Translation/Libration/Screw-motion) groups were used to model anisotropic displacement for the different groups. In the final CtIDH-Open structure many residues are missing including: A1, A134-A139, A305-A321 (17 residues), A402; B1, B306–B324 (19 residues), B402; C1–C2, C134–C139, C210–C214, C306–C321 (16 residues), C402; D1, D306–D324 (19 residues) and D402. In particular, it is striking that the helix α11 is missing in all four monomers, where 16–19 residues in each helix are not defined in the crystal structure (Fig. 2). There are 88 water molecules and the overall and avarage B-factors for each monomer are lower than the Wilson B-factor (Table 3).
Fig. 2.

Calculated electrostatic surface potentials (left panels) and ribbon diagrams (right panels) of CtIDH-Open (a and b) CtIDH–NADP (c and d) and DpIDH–NADP (e and f). The colors red, white and blue indicate charge potentials of −10, 0 and +10 KbT/e, respectively.
A 2.5 Å crystal structure of CtIDH with NADP+ (CtIDH–NADP), a metal interpreted as magnesium, isocitrate and the majority of the NADP+ were clearly defined in the 2Fo-Fc electron density, except for the nicotinamide moiety of NADP+ which was not defined (Fig. 3a and c). The final model refined to an R-factor of 18.9% and an R-free of 24.4%. There are 41 water molecules, some side-chains were refined with zero occupancy and the mean B-value of the protein is 45.2 Å2, which is in the same order as the Wilson B-factor (Table 3).
The final model of DpIDH–NADP was refined to 1.93 Å resolution and to an R-factor and R-free of 17.5% and 22.5%, respectively. There is one molecule in the asymmetric unit and the functional dimer is generated by the crystallographic twofold axis. The observed electron density maps clearly define one magnesium ion, one isocitrate with reasonable B-values (Table 3) and the majority of the NADP+ co-factor is defined (Fig. 3b and d). The exception is the nicotinamide group adjacent to the isocitrate for which no electron density was seen. The resulting phosphor-ADP-ribose could then be derived from an NADP+ molecule by hydrolysis of the nicotinamide group as also observed for an NADPH dependent alcohol dehydrogenase [45]. See Table 3 for further details from the three refined structures.
The sequence alignment of the IDHs displays the conservation of secondary structural elements and active site amino acid residues (Fig. 4). CtIDH-Open is more similar to TmIDH with lower RMSD score and most of the secondary structure elements in all three domains have comparable conformations. When superimposing one monomer of CtIDH-Open onto CtIDH–NADP using the CA-atoms in the small domains and the clasp domains only, the movements include most residues in the large domain, specifically residues 2–106 and 290–402 have different conformations. Ser2 in CtIDH-Open has moved 16.9 Å relative to Ser2 in the closed conformation of CtIDH–NADP (Fig. 5a and b). In one IDH monomer there is a long beta sheet with 10 β-strands for which five strands can overlap in the two CtIDH structures and then the next five β-strands have different orientations in CtIDH-Open and CtIDH–NADP as partly illustrated in Fig. 5. The CA atom of Asn38 is displaced by 13 Å in CtIDH-Open relative to CtIDH–NADP, and the large domain of CtIDH-Open is rotated about 30° compared to the closed CtIDH–NADP as measured by the angle between Asn38 (open)-Arg109-Asn38 (closed) within one monomer. In the open structures of TmIDH and human cytosolic IDH (HcIDH; PDB 1T09) the same angle was found to be ∼30° and ∼24° respectively [9,16], thus in this sense CtIDH-Open is most similar to TmIDH.
Fig. 4.

Sequence alignment of isocitrate dehydrogenase from Clostridium thermocellum (CtIDH), Thermotoga maritima (TmIDH) and Desulfotalea psychrophila (DpIDH). The secondary structure elements of CtIDH–NADP (top) and DpIDH–NADP (bottom), the isocitrate binding residues (★), the metal binding residues (▴) and NADP binding regions ( ) are indicated.
) are indicated.
Fig. 5.
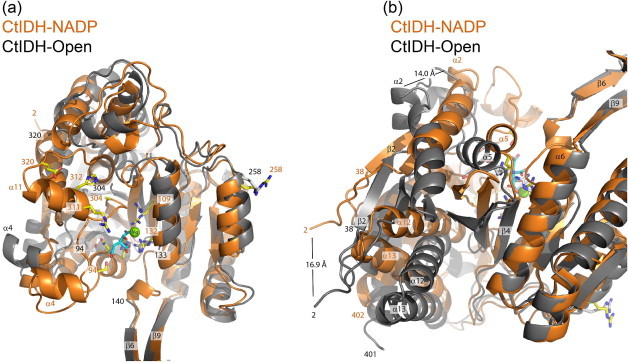
Comparison of monomer A of CtIDH–NADP (orange) and CtIDH-Open (gray) after super-positioning on the small domain (120–140, 184–283) and the clasp domain (141–183). Panels (a) and (b) have different orientations, some residues and secondary structure elements are outlined, and the distance between the CA-atoms of Ser2 and Thr52 in CtIDH–NADP and CtIDH-Open are indicated.
The new DpIDH–NADP structure presented here is most similar to DpIDH-iso (with isocitrate) for which only residues 308–318 in helix α11 are positioned closer to the small domain in the DpIDH–NADP structure. The two presented quaternary complex structures (CtIDH–NADP and DpIDH–NADP) are also similar with low RMSD, helix α11 at comparable positions and only some differences for residues at the termini of secondary structure elements.
3.3. Structural contributions to stability and flexibility
Structural analyses of hydrogen bonds, ion-pairs and inter-subunit interactions are summarized in Table 4, in order to relate structure to observed apparent melting points. Overall the number of hydrogen bonds is high at 0.995 per residue for both CtIDH–NADP and DpIDH–NADP though CtIDH has more main-chain to main-chain hydrogen bonds. There are more ion-pairs in CtIDH–NADP (28) than in DpIDH (both with NADP (25) and native (24)) and many of these are located in clusters involving three amino acids (Table 4). There are fewer inter-subunit hydrogen bonds and fewer inter-subunit ion-pairs in CtIDH–NADP than in DpIDH–NADP and DpIDH, and only two such inter-subunit ionic interactions were found in CtIDH–NADP. The different domains in CtIDH–NADP are connected with a comparable number of interactions as in DpIDH, but there are more hydrogen bonds from the clasp to the small domain in CtIDH–NADP. The dimer interface has comparable sizes in CtIDH–NADP and DpIDH–NADP (Table 4). For the CtIDH-Open structure the numbers of interactions are generally fewer, probably due to the resolution and possibly also the open conformation. By comparing CtIDH-Open to the TmIDH open structure (Tm = 98.3 °C), there are no 5-member ion-pair clusters (<4.0 Å) and a much lower number of hydrogen bonds per residue was found in CtIDH-Open (0.855) compared to TmIDH (0.980) [4]. The very high thermal stability of TmIDH is primarily explained by the abundance of large strategically placed ion pair clusters, and their absence in CtIDH probably explains its moderate Tm. What is striking is the high number of polar residues in CtIDH (30.6%) compared to DpIDH (22.9%), and many polar residues are also found in PcIDH, HcIDH and TmIDH (see Table 3 in Fedøy et. al., 2007) [4]. So totally, one CtIDH–NADP monomer is stabilized with many hydrogen bonds and more three member ion-pair networks than in DpIDH, while there are fewer inter-subunit interactions. This might account for the comparable Tm values found for CtIDH and DpIDH.
Table 4.
Structural analysis and characteristics of CtIDH-Open, CtIDH–NADP, DpIDH–NADP and DpIDH.
| CtIDH-Open | CtIDH–NADP | DpIDH–NADP | DpIDH (PDB 2UXQ)$ | |
|---|---|---|---|---|
| Apparent melting temperature (°C) | 67.9 | 66.9$ | ||
| No of amino acids in monomer A in PDB (in gene) | 378 (402) | 401 (402) | 402 (402) | 402 (402) |
| Hydrophobic residuesa (%) | 39.8 | 48.6 | ||
| Polar residuesb (%) | 30.6 | 22.9 | ||
| Charged residuesc (%) | 29.6 | 28.5 | ||
| Resolution (Å) | 2.35 | 2.50 | 1.93 | 1.75 |
| No of hydrogen bonds per residue in monomer A | 0.855 | 0.995 | 0.995 | 0.739 |
| No of SSd hydrogen bonds per residue | 0.108 | 0.150 | 0.167 | 0.070 |
| No. of SMe hydrogen bonds per residue | 0.159 | 0.137 | 0.179 | 0.107 |
| No. of MMf hydrogen bonds per residue | 0.640 | 0.708 | 0.647 | 0.562 |
| No. of ion pairs per monomer at 4/6 Å | 17/50 | 28/61 | 25/54 | 24/54 |
| No. of ion pairs per residue (4 Å) | 0.045 | 0.070 | 0.062 | 0.060 |
| No. of 2 member networks in monomer A at 4.0 Å | 12 | 14 | 15 | 18 |
| No. of 3 member networks in monomer A at 4.0 Å | 4 | 6 | 5 | 3 |
| No. of inter-subunit hydrogen bonds | 28 | 44 | 51 | 33 |
| No. of inter-subunit ion pairs at 4.0 Å | 1 | 2 | 6 | 4 |
| No. of inter-subunit 2 member network at 4.0 Å | 1 | 0 | 2 | 3 |
| No. of inter-subunit 3 member network at 4.0 Å | 0 | 2 | 4 | 0 |
| No. ion pairs < 6.0 Å (<4.0 Å)/No. hydrogen bonds between: | ||||
| Large–small domain | 3 (1)/11 | 4 (0)/12 | 5 (0)/14 | 3 (1)/11 |
| Large′–small domain | 1 (1)/11 | 4 (0) / 12 | 5 (0)/14 | 4 (1)/16 |
| Clasp–small domain | 0 (0)/14 | 0 (0)/28 | 0 (0)/22 | 0 (0)/25 |
| Accessible surface area of dimer (Å2) | 31 480 | 30 118 | 30 044 | 29 343 |
| Buried inter-subunit surface (% of dimer) | 30.2 | 30.1 | 30.1 | 30.1 |
Data from Fedøy et al. [4].
Hydrophobic residues: A,V,L,I,W,F,P,M.
Polar residues: G,S,T,Y,N,Q,C.
Charged residues: R,K,H,D,E.
SS, side-chain–side-chain hydrogen bonds.
SM, side-chain–main-chain hydrogen bonds.
MM, main-chain–main-chain hydrogen bonds.
The net charges of the CtIDH and DpIDH dimers are −12 and −24, respectively, with charged N and C termini and neutral histidines. The calculated electrostatic surface potentials of the three crystal structures are shown in Fig. 2, it is clear that both CtIDH structures are less negatively charged than DpIDH, even if the CtIDH-Open structure is slightly more negatively charged due to disordered missing residues. In particular, the NADP+ binding cleft in CtIDH is more neutral than the negatively charged one found in DpIDH (Fig. 2).
In DpIDH an enlarged aromatic cluster was found in each of the small domains [4] and mutation of Phe205Met in TmIDH (Phe207 in CtIDH) resulted in a decrease in Tm by −3.5° C [9]. This aromatic cluster in CtIDH has the same size as in DpIDH with 11 aromatic residues (residues 190, 195, 207, 217, 221, 225, 229, 233, 237, 244, 246) which is bigger compared to TmIDH with 7 aromatic residues.
In the clasp domain of the CtIDH–NADP structure there are some ion-pair interactions which include Glu170-Lys151, Lys151-Glu153, Glu153-Arg165 where only the latter is <4 Å and is therefore considered to be the strongest. Compared to DpIDH–NADP the clasp domain in CtIDH–NADP is less stabilized by ionic interactions, but slightly better bounded to the small domain by hydrogen bonds (Table 4).
Stabilization of the N and C termini has been found to be important for thermal stability in hyperthermophilic IDHs [8,9,18]. The first prominent difference between CtIDH and the other IDH homologs was the disruption of a conserved ion-pair due to a mutation of a lysine to aspargine. In TmIDH one ion-pair Lys29-Asp389 was found to stabilize the structure substantially, since if it was disrupted the Tm decreased by -21.8 °C. This ionic interaction is also present in DpIDH, PcIDH and HcIDH [4,9], but the corresponding interaction is not found in CtIDH due to mutations corresponding to Asn29 and Glu393. Still, in the same region of CtIDH–NADP there are three locally stabilizing interactions involving Lys383-Glu393-Lys396, Glu388-Lys392-Asp389 and Arg395-Glu399 and a hydrogen bond from Glu399 to Tyr34 (Fig. 6a). In addition there are ionic interactions connecting Glu12-Lys27-Glu32 (Fig. 6a). Finally, two short strong ionic interactions involving Arg340-Asp344 are found in CtIDH–NADP and also extended by two hydrogen bonds (Asp344-Val8, Arg340-Tyr67) in the same region (Fig. 6a). The N and C termini of CtIDH–NADP seem to be stabilized by other ionic interactions than in DpIDH and TmIDH, but still the number of ionic interactions is high and several hydrogen bonds are well defined, which probably all together stabilize the CtIDH structure.
Fig. 6.

(a) Ionic interactions in the N and C termini of CtIDH–NADP. (b) Two four member Met-clusters at the dimer interface of CtIDH–NADP and two adjacent Met residues (327, 288). More details are described in the text.
The number of methionine residues in CtIDH, DpIDH and TmIDH are 16 (4.0%), 14 (3.5%) and 13 (3.3%) respectively, and their locations in the structure have been analyzed. In DpIDH a large methionine cluster has been described at the dimer interface with six adjacent Met-residues [4]. At the dimer interface of CtIDH the cluster is smaller with Met275, Met278, Met275′ and Met278′ adjacently positioned, and interestingly CtIDH also has another cluster in the clasp domain with Met178, Met180, Met178′ and Met180′ (Fig. 6b). Both these clusters are also found in TmIDH. For the remaining Met-residues most of them are distributed throughout in the structures, but unique to CtIDH are Met327 and Met288 in the large domain which are facing each other. The role of the Met-clusters in CtIDH could be to maintain the flexibility of the enzyme during catalysis and thus affect the catalytic rate.
The detailed structural investigations of CtIDH display typical thermophilic structural characteristics with a higher number of hydrogen and ionic interactions per residue than the mesophilic homologs PcIDH and HcIDH [1,16]. Moreover, fewer ionic interactions and only three-member ion-pair networks and not the enlarged ionic networks were found in CtIDH as compared to the hyperthermophilic TmIDH and ApIDH [8,9].
3.4. Active site
The amino acids involved in the binding of cofactor and substrate are highly conserved in all IDH enzymes. In the active site of CtIDH–NADP the isocitrate ion is bound to the conserved positively charged residues Arg100, Arg132, Arg109, and Lys210′, and hydrogen bonds are made to Ser94, Asn96, Thr77, Tyr139, Asp250′ and Asp273 (Fig. 7b). The orientation of the isocitrate and the interacting residues in CtIDH–NADP are very similar to DpIDH-iso (PDB 2UXR) and DpIDH–NADP (Fig. 7e). The metal is interpreted as Mg2+ and is penta-coordinated by a water molecule (W2), Asp250′, Asp273 and by two oxygen atoms in the isocitrate in a bidentate mode. Additionally W2 is stabilized by binding to Asp277.
Fig. 7.

The NADP and isocitrate binding clefts (left panels) in (a) CtIDH-NAPD, (d) DpIDH–NADP and (g) CtIDH-Open. Isocitrate binding residues (middle panels) in the (b) CtIDH–NADP and (e) DpIDH–NADP crystal structures. The NADP+ interacting residues in (c) CtIDH–NADP and (f) DpIDH–NADP not including the hydrogen bonded water molecules. For NADP+ the non-defined NADP nicotinamide groups are also including.
In DpIDH–NADP the isocitrate interactions residues are found at almost identical positions as in DpIDH-iso, but one difference is the octahedral-geometry of the magnesium by an additional water molecule (not labeled but shown in Fig. 7e). Further details on differences in bonds lengths and the number of hydrogen bonded water molecules have not been examined due to the different resolutions (2.5 Å versus 1.93 Å) of the compared structures.
In both complex structures presented, CtIDH–NADP and DpIDH–NADP, one metal, one isocitrate and one apo-NADP (lacking the nicotinamide group) are found in the active sites. For CtIDH–NADP the mean B-value of the cofactor is 52.1 Å 2 which is quite high. Still the observed electron density map clearly indicated the presence of the cofactor except for 8 atoms in the nicotinamide group which was refined with zero occupancy. For the rest of the cofactor, the three phosphate groups and the adenine moiety are nicely defined in the electron density (Fig. 3a and c). Due to the lack of electron density of the C4 of the nicotinamide moiety, which has been shown to be the involved in the initial hydride transfer, we could not perform a more thorough investigation of the acid–base chemistry.
Both quaternary complexes reveal that the 2′-phosphomonoester group forms ion pairs to His312 (CtIDH) and Arg255′ (CtIDH)(Fig. 7a and c). The tight interaction found for His312 to NADP+ in the presented crystal structure (Fig. 7c), further supports the importance of this residue in the binding of NADP+. Similar interactions are observed within all known structures of subfamily II IDH [16,32], also mutational studies in PcIDH confirm the importance of this histidine [46]. The ionic interactions between the Arg255′ (CtIDH) or Arg254′ (DpIDH) to the 2′-phosphomonoester group of NADP+ are unique for CtIDH and DpIDH, therefore this arginine residue may represent a different cofactor discriminating interaction. In the other known structures there is a Glutamine (Gln) in this position [9,16,32], making a hydrogen bond to the 2′-phosphomonoester in ScIDH (Gln259′). The conformation of Arg255′ is stabilized by the ion pair formation to Asp251′. In addition, a more basic/neutral environment in the binding pocket in the vicinity of the 2′-phosphomonoester, seems more complementary to the negatively charged phosphate-group in CtIDH compared to DpIDH (Fig. 2c and e).
In DpIDH–NADP the cofactor to protein interactions are similar as described in CtIDH–NADP, with His311 and Arg254′ forming ionic interactions to the 2′-phosphomonoester group. In addition, Arg374 is pointing towards the 2′-phosphomonoester group and is 4.5 Å away from one of the oxygen atoms in the cofactor (Fig. 7d), thus forming a weak ionic interaction. For DpIDH–NADP the adenine moiety has both hydrophobic and hydrophilic interactions with the side-chains of Leu285, Val308, His305, Asn324 and His311. In particular there are two short hydrogen bonds from the main chain of Asn324 N and O to the N1 and N6 atoms of the adenine, respectively (Fig. 7f). In CtIDH Asn325 makes the same interactions. The nature and total number of interactions may explain the high affinity found for NADP+ for both DpIDH and CtIDH.
However, by comparing the NADP+ interactions in CtIDH–NADP and DpIDH–NADP to those found in HcIDH (PDB 1T0L) and ScIDH (PDB 2QFX) one of the largest differences is the interactions of the 3′OH group of the adenosine ribose moiety. In CtIDH–NADP there is a short interaction (3.3 Å) from Arg255′ to the 3′OH group of the adenosine ribose moiety and a similar interaction is found in DpIDH–NADP (3.4 Å; Fig. 7c and f). Whereas in the quaternary ScIDH complex [32], it is the Arg316 in the large domain (Arg311 in CtIDH) that is stabilizing the same 3′OH group with a distance of only 2.8 Å. In the Arg311 equivalent in HcIDH (Arg314) this distance is only 3.2 Å. Hence, the interactions to the 3′OH group of the adenosine ribose are from the small domain in the CtIDH and DpIDH, whereas for ScIDH and HcIDH the interactions are from the large domain.
The number of water molecules hydrogen bonded to the NADP+ molecules in the two presented structure are different, probably reflected by the difference in resolution (2.50 Å for CtIDH–NADP and 1.93 Å for DpIDH–NADP), and these interactions are therefore not investigated in more detail.
3.5. Opening and closure of the overall structures: a unique active site locking mechanism
A profound difference between subfamily I and subfamily II IDHs is the insertion of two extra α-helices, α4 and α11, in the subfamily II enzymes. For the eukaryotic ScIDH [32] these helices were proposed to be important for a more fine-tuned and complex catalytic mechanism compared to EcIDH. The accepted catalytic mechanism for EcIDH which is from subfamily I, is a random binding of substrate and cofactor leading to the ordered enzyme-substrate complex [2,7,20–24]. In ScIDH these α-helices act as a lid by covering the top of the NADP+- and isocitrate-binding site and as a result the entrance of the active site is significantly narrowed. In the comparison of the closed CtIDH–NADP and the CtIDH-Open structure (Fig. 5) the movement of the α4 helix is evident. The fact that the electron density is not defined for α11 in any of the four molecules in CtIDH–NADP, indicates high flexibility in this structural element and leads to the perception that the α11 helix is moving and is adaptable upon opening of the NADP+ binding site. From our earlier work we observed that helix α11 and Arg310 also moved comparing the native DpIDH to the DpIDH-Iso (PDB 2UXR) structure [4]. In the DpIDH-Iso structure Arg310 is defined in the electron density map and α11 moved closer to the small domain forming a narrower NADP+ binding site. In the new DpIDH–NADP structure presented here, the width of the NADP+ site is even shorter (Table 5) and the helix α11 (see Fig. 7d) closes the NADP+ site more tightly than in DpIDH-Iso. We do not suspect the crystal packings are responsible for the observed movements and changes. The conformation of α11 and Arg311 (Arg310 in DpIDH) in CtIDH–NADP is very similar to DpIDH–NADP (Fig. 7a and d). Therefore we suggest a new model of the active site locking mechanism (Fig. 8); where we present one open-CtIDH and one locked CtIDH–NADP structure, an open TmIDH structure, a closing structure of the apo native DpIDH and a closed complex structure with isocitrate of DpIDH-Iso reported earlier [4], and a locked structure of DpIDH–NADP; which together provides a detailed overview of the important domain movements for efficient catalysis.
Table 5.
Defined widths of the NADP+ and isocitrate binding sites as also shown in Figure 7.
| CtIDH-Open | TmIDH (Open) | DpIDH | DpIDH-Iso | CtIDH–NADP | DpIDH–NADP | |
|---|---|---|---|---|---|---|
| PDB entry | 4AOY | 1ZOR | 2UXQ | 2UXR | 4AOU | 4AOV |
| Width of NADP site (Å)a | 18.7 | 15.6 | 13.0 | 11.8 | 11.8 | |
| Width of isocitrate (Å) siteb | 19.6 | 18.5 | 11.7 | 10.6 | 10.5 | 10.4 |
| RMSD for CtIDH-Open (Å)c (no of CA atoms) | 1.22 (364) | 2.65 (363) | 2.94 (345) | 2.80 (334) | 3.17 (350) | |
| RMSD for CtIDH-NADH (Å)c (no of CA atoms) | 2.44 (348) | 1.31 (400) | 0.93 (398) | 0.85 (392) |
Distance corresponding to Arg311 CA – Asp251′ CA in CtIDH, Arg310 CA – Asp250′ CA in DpIDH,
Distance corresponding to Thr77 CA – Lys210′ CA in CtIDH, Thr76 CA – Lys209′ CA in DpIDH.
RMSD for the most similar monomers.
Fig. 8.
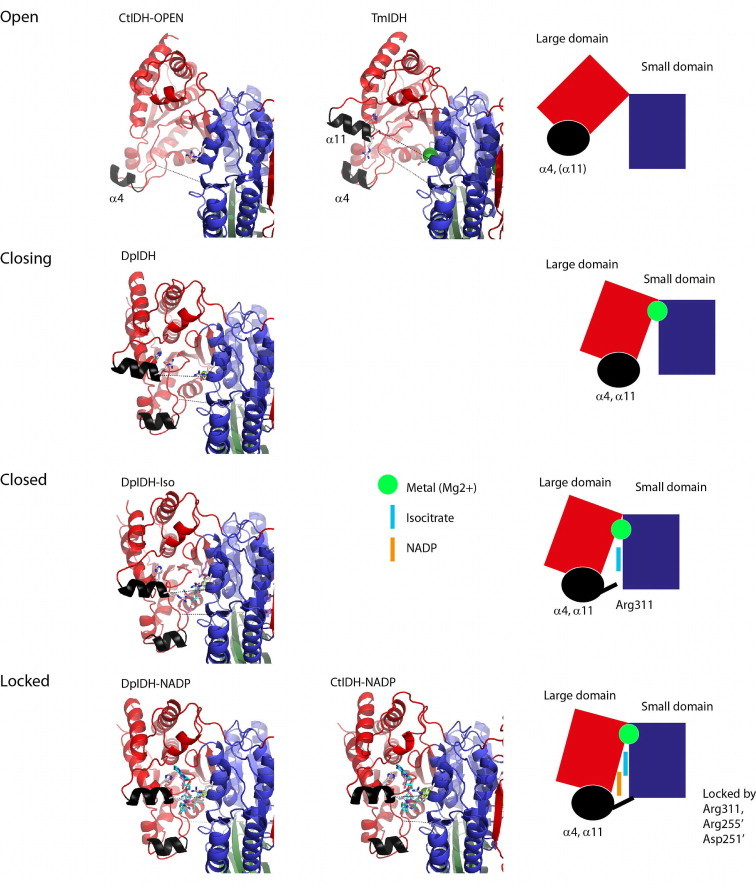
The locking of the large domain onto the small domain seems unique in CtIDH and DpIDH starting from open (CtIDH-Open, TmOpen), to the closing (DpIDH), the closed (DpIDH-Iso) to finally the locked state (CtIDH–NADP, DpIDH–NADP). Here helix α4 comes in place, Arg311 is important for binding the large to the small domain and finally Arg311, Asp251′ and Arg255′ make unique interactions and binding properties for NADP+.
In our new model (Fig. 8) the most open structure is CtIDH-Open with a isocitrate binding site width of 19.6 Å (Table 5) and the α11 not defined in the electron density. Then in TmIDH, α11 is defined and the NADP+ and isocitrate sites are still wide (18.7 Å and 18.5 Å) but smaller than the former. The apo native DpIDH is defined as a closing state, because even if the complete large domain is clearly shifted towards the small domain (compared to the open state), the main-chain is still around 2 Å more open than the closed DpIDH-Iso structure with bound isocitrate. Finally the most locked states are represented by the structures of the quaternary complexes with NADP+, isocitrate and Mg2+, (CtIDH-NAPD, DpIDH-NAPD) as reflected by the most narrow NADP+ and isocitrate sites. These quaternary structures could represent a productive conformation of the protein, with substrate and cofactor ready for catalysis. All the domain movements are also reflected by the RMSD towards CtIDH-Open or CtIDH-NAPD (Table 5).
The importance of the α4 and α11 helices is also seen in our structures. Firstly, α11 is disordered (CtIDH-Open) and then it becomes ordered and moves closer to the active site, as displayed by the closing state of the apo native DpIDH (2UXQ), but the Arg311 equivalent (Arg310) makes no interactions and is not defined. Further movement of α11 closes the active site, as displayed in DpIDH-iso, and then finally locking of α11 onto the active site cleft by the unique interactions from Arg311 to Asp250′ and also Arg255′ (CtIDH–NADP, DpIDH–NADP). Asp250′ enables and helps Arg255′ to interact with the NADP+ cofactor and stabilize the 3′OH group of the adenosine ribose moiety (Fig. 7c and f). Secondly, α4 is first positioned far away (19.6 Å in CtIDH-Open; Table 5), and becomes closer when α11 is defined (11.7 Å, apo native DpIDH) and even tighter when isocitrate and/or NADP+ is bound (10.4–10.6 Å; CtIDH–NADP, DpIDH–NADP, DpIDH-Iso). The closing of large domain and the movement of α11 and the locking by Arg311 to the small domain, together with residue determinants from both the large (His312) and the small domain (Arg255′) for the NADP+ selectivity are important subtle movements in CtIDH and DpIDH, regulating their catalytic efficiency and selectivity.
The crystal structures of the thermophilic IDH from C. thermocellum fill the gap in the temperature scale within subfamiliy II. The bacterial homologs in this study (DpIDH, DhIDH, CtIDH and TmIDH) share a high level of primary sequence identity of about 60%, and a moderate identity to eukaryotic subfamily II homologs of about 50% (HcIDH, PcIDH and ScIDH). Even though, the secondary structure elements are however highly conserved together with the active site given by the cofactor and substrate binding residues (Fig. 4), the three dimensional structures provide the distinct differences that reveal the structural explanations to the activity-stability-flexibility relationship.
The prokaryotic CtIDH and DpIDH IDHs are structurally very similar to the eukaryotic subfamily II IDHs, however they display differences in cofactor interaction and locking of the active site. For that reason, we suggest a new active site locking mechanism (Fig. 8), which consists of an open and locked CtIDH structure and a closing, closed and locked structure of DpIDH. These organized subtle movements in CtIDH and DpIDH regulate the catalytic efficiency at their operational temperature.
Acknowledgements
This work was supported by the Norwegian Research Council (Project No. 153774/420). The Norwegian Structural Biology Centre (NorStruct) is supported by the National Functional Genomics Program (FUGE), Research Council of Norway (RCN). Provision of beamtime at ID29 and SNBL at the European Synchrotron Radiation Facility (ESRF) and at BESSY, Berlin BL14.2 is gratefully acknowledged. We thank Marit Steine Madsen for skillful technical help and Thomas Jeffrey Keen for language improvements.
References
- 1.Ceccarelli C., Grodsky N.B., Ariyaratne N., Colman R.F., Bahnson B.J. Crystal structure of porcine mitochondrial NADP+-dependent isocitrate dehydrogenase complexed with Mn2+ and isocitrate. Insights into the enzyme mechanism. J. Biol. Chem. 2002;277:43454–43462. doi: 10.1074/jbc.M207306200. [DOI] [PubMed] [Google Scholar]
- 2.Dean A.M., Koshland D.E., Jr. Kinetic mechanism of Escherichia coli isocitrate dehydrogenase. Biochemistry. 1993;32:9302–9309. doi: 10.1021/bi00087a007. [DOI] [PubMed] [Google Scholar]
- 3.Doyle S.A., Fung S.Y., Koshland D.E., Jr. Redesigning the substrate specificity of an enzyme: isocitrate dehydrogenase. Biochemistry. 2000;39:14348–14355. doi: 10.1021/bi001458g. [DOI] [PubMed] [Google Scholar]
- 4.Fedøy A.E., Yang N., Martinez A., Leiros H.K., Steen I.H. Structural and functional properties of isocitrate dehydrogenase from the psychrophilic bacterium Desulfotalea psychrophila reveal a cold-active enzyme with an unusual high thermal stability. J. Mol. Biol. 2007;372:130–149. doi: 10.1016/j.jmb.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 5.Huang Y.C., Grodsky N.B., Kim T.K., Colman R.F. Ligands of the Mn2+ bound to porcine mitochondrial NADP-dependent isocitrate dehydrogenase, as assessed by mutagenesis. Biochemistry. 2004;43:2821–2828. doi: 10.1021/bi030253f. [DOI] [PubMed] [Google Scholar]
- 6.Huang Y.C., Colman R.F. Location of the coenzyme binding site in the porcine mitochondrial NADP-dependent isocitrate dehydrogenase. J. Biol. Chem. 2005;280:30349–30353. doi: 10.1074/jbc.M505828200. [DOI] [PubMed] [Google Scholar]
- 7.Hurley J.H., Dean A.M., Koshland D.E., Jr., Stroud R.M. Catalytic mechanism of NADP(+)-dependent isocitrate dehydrogenase: implications from the structures of magnesium-isocitrate and NADP+ complexes. Biochemistry. 1991;30:8671–8678. doi: 10.1021/bi00099a026. [DOI] [PubMed] [Google Scholar]
- 8.Karlstrom M., Stokke R., Steen I.H., Birkeland N.K., Ladenstein R. Isocitrate dehydrogenase from the hyperthermophile Aeropyrum pernix: X-ray structure analysis of a ternary enzyme-substrate complex and thermal stability. J. Mol. Biol. 2005;345:559–577. doi: 10.1016/j.jmb.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 9.Karlstrom M., Steen I.H., Madern D., Fedoy A.E., Birkeland N.K., Ladenstein R. The crystal structure of a hyperthermostable subfamily II isocitrate dehydrogenase from Thermotoga maritima. FEBS J. 2006;273:2851–2868. doi: 10.1111/j.1742-4658.2006.05298.x. [DOI] [PubMed] [Google Scholar]
- 10.Kim S.Y., Hwang K.Y., Kim S.H., Sung H.C., Han Y.S., Cho Y. Structural basis for cold adaptation. Sequence, biochemical properties, and crystal structure of malate dehydrogenase from a psychrophile Aquaspirillium arcticum. J. Biol. Chem. 1999;274:11761–11767. doi: 10.1074/jbc.274.17.11761. [DOI] [PubMed] [Google Scholar]
- 11.Kim T.K., Lee P., Colman R.F. Critical role of Lys212 and Tyr140 in porcine NADP-dependent isocitrate dehydrogenase. J. Biol. Chem. 2003;278:49323–49331. doi: 10.1074/jbc.M303781200. [DOI] [PubMed] [Google Scholar]
- 12.Kim T.K., Colman R.F. Ser95, Asn97, and Thr78 are important for the catalytic function of porcine NADP-dependent isocitrate dehydrogenase. Protein Sci. 2005;14:140–147. doi: 10.1110/ps.041091805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee P., Colman R.F. Thr(373), Asp(375), and Lys(260) are in the coenzyme site of porcine NADP-dependent isocitrate dehydrogenase. Arch. Biochem. Biophys. 2006;450:183–190. doi: 10.1016/j.abb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Singh S.K., Matsuno K., LaPorte D.C., Banaszak L.J. Crystal structure of Bacillus subtilis isocitrate dehydrogenase at 1.55 Å. Insights into the nature of substrate specificity exhibited by Escherichia coli isocitrate dehydrogenase kinase/phosphatase. J. Biol. Chem. 2001;276:26154–26163. doi: 10.1074/jbc.M101191200. [DOI] [PubMed] [Google Scholar]
- 15.Steen I.H., Madern D., Karlstrom M., Lien T., Ladenstein R., Birkeland N.K. Comparison of isocitrate dehydrogenase from three hyperthermophiles reveals differences in thermostability, cofactor specificity, oligomeric state, and phylogenetic affiliation. J. Biol. Chem. 2001;276:43924–43931. doi: 10.1074/jbc.M105999200. [DOI] [PubMed] [Google Scholar]
- 16.Xu X., Zhao J., Xu Z., Peng B., Huang Q., Arnold E., Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004;279:33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- 17.Stokke R., Birkeland N.K., Steen I.H. Thermal stability and biochemical properties of isocitrate dehydrogenase from the thermoacidophilic archaeon Thermoplasma acidophilum. Extremophiles. 2007;11:397–402. doi: 10.1007/s00792-006-0045-y. [DOI] [PubMed] [Google Scholar]
- 18.Stokke R., Karlstrom M., Yang N., Leiros I., Ladenstein R., Birkeland N.K., Steen I.H. Thermal stability of isocitrate dehydrogenase from Archaeoglobus fulgidus studied by crystal structure analysis and engineering of chimers. Extremophiles. 2007;11:481–493. doi: 10.1007/s00792-006-0060-z. [DOI] [PubMed] [Google Scholar]
- 19.Stokke R., Madern D., Fedoy A.E., Karlsen S., Birkeland N.K., Steen I.H. Biochemical characterization of isocitrate dehydrogenase from Methylococcus capsulatus reveals a unique NAD+-dependent homotetrameric enzyme. Arch. Microbiol. 2007;187:361–370. doi: 10.1007/s00203-006-0200-y. [DOI] [PubMed] [Google Scholar]
- 20.Bolduc J.M., Dyer D.H., Scott W.G., Singer P., Sweet R.M., Koshland D.E., Jr., Stoddard B.L. Mutagenesis and Laue structures of enzyme intermediates: isocitrate dehydrogenase. Science. 1995;268:1312–1318. doi: 10.1126/science.7761851. [DOI] [PubMed] [Google Scholar]
- 21.Dean A.M., Golding G.B. Protein engineering reveals ancient adaptive replacements in isocitrate dehydrogenase. Proc Natl Acad Sci U S A. 1997;94:3104–3109. doi: 10.1073/pnas.94.7.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurley J.H., Chen R., Dean A.M. Determinants of cofactor specificity in isocitrate dehydrogenase: structure of an engineered NADP+ → NAD+ specificity-reversal mutant. Biochemistry. 1996;35:5670–5678. doi: 10.1021/bi953001q. [DOI] [PubMed] [Google Scholar]
- 23.Stoddard B.L. New results using Laue diffraction and time-resolved crystallography. Curr. Opin. Struct. Biol. 1998;8:612–618. doi: 10.1016/s0959-440x(98)80153-x. [DOI] [PubMed] [Google Scholar]
- 24.Stoddard B.L., Cohen B.E., Brubaker M., Mesecar A.D., Koshland D.E., Jr. Millisecond Laue structures of an enzyme-product complex using photocaged substrate analogs. Nat. Struct. Biol. 1998;5:891–897. doi: 10.1038/2331. [DOI] [PubMed] [Google Scholar]
- 25.Aktas D.F., Cook P.F. A lysine-tyrosine pair carries out acid-base chemistry in the metal ion-dependent pyridine dinucleotide-linked β-hydroxyacid oxidative decarboxylases. Biochemistry. 2009;48:3565–3577. doi: 10.1021/bi8022976. [DOI] [PubMed] [Google Scholar]
- 26.Karshikoff A., Ladenstein R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: a “traffic rule” for hot roads. Trends Biochem. Sci. 2001;26:550–556. doi: 10.1016/s0968-0004(01)01918-1. [DOI] [PubMed] [Google Scholar]
- 27.Vieille C., Zeikus G.J. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szilagyi A., Zavodszky P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure. 2000;8:493–504. doi: 10.1016/s0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 29.Jaenicke R., Bohm G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 30.Smalås A.O., Leiros H.-K.S., Os V., Willassen N.P. Cold adapted enzymes. Biotechnol Annu Rev. 2000;6:1–57. doi: 10.1016/s1387-2656(00)06018-x. [DOI] [PubMed] [Google Scholar]
- 31.Siddiqui K.S., Cavicchioli R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006;75:403–433. doi: 10.1146/annurev.biochem.75.103004.142723. [DOI] [PubMed] [Google Scholar]
- 32.Peng Y., Zhong C., Huang W., Ding J. Structural studies of Saccharomyces cerevesiae mitochondrial NADP-dependent isocitrate dehydrogenase in different enzymatic states reveal substantial conformational changes during the catalytic reaction. Protein Sci. 2008;17:1542–1554. doi: 10.1110/ps.035675.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 34.Eisenthal R., Cornish-Bowden A. The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. Biochem J. 1974;139:715–720. doi: 10.1042/bj1390715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lonhienne T., Gerday C., Feller G. Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim. Biophys. Acta. 2000;1543:1–10. doi: 10.1016/s0167-4838(00)00210-7. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallog. 1993;24:795–800. [Google Scholar]
- 37.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.Murshudov G.N., Vagin A.A., Lebedev A., Wilson K.S., Dodson E.J. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 39.Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.McDonald I.K., Thornton J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 41.Rocchia W., Sridharan S., Nicholls A., Alexov E., Chiabrera A., Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J. Comput. Chem. 2002;23:128–137. doi: 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- 42.Herrero A.A., Gomez R.F. Development of ethanol tolerance in Clostridium thermocellum: effect of growth temperature. Appl. Environ. Microbiol. 1980;40:571–577. doi: 10.1128/aem.40.3.571-577.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feller G. Protein stability and enzyme activity at extreme biological temperatures. Journal of Physics-Condensed Matter. 2010;22:323101. doi: 10.1088/0953-8984/22/32/323101. [DOI] [PubMed] [Google Scholar]
- 44.Marx JC, Collins T, D’Amico S, Feller G & Gerday C (2007) Cold-Adapted Enzymes from Marine Antarctic Microorganisms. Mar Biotechnol (NY). [DOI] [PubMed]
- 45.Valencia E., Larroy C., Ochoa W.F., Pares X., Fita I., Biosca J.A. Apo and Holo structures of an NADPH-dependent cinnamyl alcohol dehydrogenase from Saccharomyces cerevisiae. J. Mol. Biol. 2004;341:1049–1062. doi: 10.1016/j.jmb.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 46.Huang Y.C., Colman R.F. Evaluation by mutagenesis of the roles of His309, His315, and His319 in the coenzyme site of pig heart NADP-dependent isocitrate dehydrogenase. Biochemistry. 2002;41:5637–5643. doi: 10.1021/bi0200716. [DOI] [PubMed] [Google Scholar]