

Abstract
Human type 2 diabetes is associated with β-cell apoptosis, and human islets from diabetic donors are ∼80% deficient in PAK1 protein. Toward addressing linkage of PAK1 to β-cell survival, PAK1–siRNA targeted MIN6 pancreatic β-cells were found to exhibit increased caspase-3 cleavage, cytosolic cytochrome-C and the pro-apoptotic protein Bad. PAK1+/− heterozygous mouse islets recapitulated the upregulation of Bad protein expression, as did hyperglycemic treatment of human or mouse islets; Bad levels were exacerbated most in PAK1+/− islets subjected to hyperglycemic stress. These data implicate PAK1 in β-cell survival via quenching of Bad protein expression, and suggest PAK1 as potential molecular target to preserve β-cell mass.
Keywords: PAK1, Bad, Pancreatic β-cell, Mouse islet, Human islet, Apoptosis
Highlights
▸ Bad expression in islets was increased by chronic exposure to hyperglycemic conditions. ▸ Depletion of PAK1 in the absence of glucose induced Bad expression. ▸ Bad expression increased most in PAK1+/− heterozygous islets subjected to hyperglycemia. ▸ PAK1 is required to protect against β-cell apoptosis under hyperglycemic conditions.
1. Introduction
Progressive deterioration in β-cell function and mass occurs in the disease progression of type 2 diabetes (T2D). Reports suggest that ∼50% of islets are in state of dysfunction or outright failure at the time of diagnosis [1–3]. The serine/threonine p21-activated kinase 1 (PAK1) is a ubiquitously expressed protein that is implicated as a positive regulator of β-cell function; a paucity of PAK1 protein in islets is correlated with human T2D [4–6]. With regards to this, PAK1 is implicated in the promotion of cell survival in neuronal cells [7] and protective against ischemia/reperfusion injury in cardiac cells [8,9]. However the role of PAK1 in β-cell survival has never been assessed.
During the process of apoptosis, two dominant endogenous cell fate regulators that target and block different steps from proceeding are Inhibitor of Apoptosis Proteins and the Bcl-2 family [10]. The pro-apoptotic protein Bad is a member of the Bcl-2 protein family, and plays an important role in glucolipotoxicity induced β-cell death in cultured human islets [11,12]. PAK1 has been shown to be an important regulator of Bad proteins in vitro, phosphorylating Bad to inhibit its pro-apoptotic effects [13,14]. Pharmacological inhibition of PAK1 activation results in reduced phosphorylation of Bad, concurrent with increased apoptosis in the H358 lung cancer and MPNST cell lines [13]. Whether PAK1 regulates Bad function in islet β-cells remains untested.
Herein, we examined a potential role of PAK1 in β-cell survival, using multiple model systems: human islets, PAK1+/− heterozygous mouse islets, and PAK1-depleted MIN6 β-cells. Our findings reveal a role for PAK1 in maintaining Bad protein expression at low levels in islet β-cells. Bad expression in islets was increased by chronic exposure to hyperglycemic conditions, and could be induced by depletion of PAK1 in the absence of glucose; PAK1+/− heterozygous islets subjected to hyperglycemia showed the largest increase in Bad expression. Given that restoration of PAK1 to normal levels corresponded to reduced Bad expression, this suggests that PAK1 is required to protect against β-cell apoptosis, particularly under glucotoxic/hyperglycemic stress conditions, and that attenuated PAK1 abundance could render islet β-cells more susceptible to apoptosis.
2. Materials and methods
2.1. Materials
The rabbit antibodies of PAK1, Bad, phospho-Bad(Ser112), Caspase-3, Cytochrome-C were purchased from Cell Signaling (Danvers, MA). Rabbit anti-actin was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-rabbit horseradish peroxidase and anti-mouse horseradish peroxidase secondary antibodies were acquired from Bio-Rad. Mouse PAK1–siRNA oligonucleotide duplexes and siRNA non-targeting control oligonucleotides were purchased from Ambion (Austin, TX) as described previously [4]. Lipofectamine 2000, the SuperScript First Strand cDNA Synthesis kit and Platinum SYBR Green qPCR SuperMix-UDG kit were purchased from Invitrogen. Enhanced chemiluminescence (ECL) reagent was obtained from Amersham Biosciences. d-glucose was obtained from Sigma. The pCMV-myc-PAK1 and pCMV vector plasmids were a gift from Dr. Lawrence Quilliam (Indiana University School of Medicine, Indianapolis, IN).
2.2. Methods
2.2.1. Cell culture, transient transfection, and immunoblotting
MIN6 mouse pancreatic β-cells were cultured in DMEM (25 mM glucose) supplemented with 15% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml l-glutamine, and 50 μM β-mercaptoethanol as described previously [4]. For PAK1 depletion, MIN6 cells were transiently transfected with PAK1–siRNA oligonucleotides duplexes along with pCMV6 vector plasmid or with pCMV6-myc-PAK1 plasmid as previously reported [4] and incubated for 48 h. Cells were harvested in 1% Nonidet P-40 lysis buffer and lysates cleared by centrifugation at 14,000 g for 10 min at 4 °C. Proteins present in lysates were resolved by 12% or 15% SDS–PAGE, transferred to PVDF membrane and subjected to immunoblotting. Membranes were incubated with primary antibody at 4 °C overnight, with secondary antibodies conjugated to horseradish peroxidase used for 1 h at room temperature. Bands were visualized by enhanced chemiluminescence.
2.2.2. Mouse and human islets
The PAK1+/− heterozygous mouse is a classic whole-body gene-ablation model on the C57Bl6J strain background, generated as previously described [15]. All islets were obtained using paired littermate mice as controls. Mouse pancreatic islets were isolated as previously described [4]. Briefly, pancreata from 10- to 16-week-old male mice were batch-digested with collagenase, purified using a Ficoll density gradient, and incubated in CRML at 37 °C, 5% CO2 for further experiments. All studies involving mice followed the guidelines for the use and care of laboratory animals at the Indiana University School of Medicine. Human islets were obtained through the Integrated Islet Distribution Program (IIDP), accepted under the following criteria: 75% or better viability and purity, normal BMI non-diabetic donor, obtained within 2 days of isolation. Upon arrival islets were immediately hand-picked to exclude non-islet material and placed in low or high-glucose CMRL for 5 days as described in the figure legend.
2.2.3. Subcellular fractionation
Cytosolic fractions were prepared from MIN6 β-cells as previously described [4]. Briefly, MIN6 cells were washed with cold phosphate-buffered saline and harvested into 1 ml of homogenization buffer (20 mM Tris–HCl, pH 7.4, 0.5 mM EDTA, 0.5 mM EGTA, 250 mM sucrose, and 1 mM dithiothreitol containing the following protease inhibitors: 10 μg/ml leupeptin, 4 μg/ml aprotinin, 2 μg/ml pepstatin, and 100 μM phenylmethylsulfonyl fluoride). Cells were disrupted by 10 strokes through a 27-gauge needle, and homogenates were centrifuged at 900g for 10 min. Postnuclear supernatants were centrifuged at 5500g for 15 min, and the subsequent supernatant was centrifuged at 25,000g for 20 min to obtain the secretory granule fraction in the pellet. The supernatant was further centrifuged at 100,000g for 1 h to obtain the cytosolic fraction.
2.2.4. Islet RNA isolation and quantitative PCR (Q-PCR)
Total RNA from mouse islets was obtained using the RNeasy mini kit (Qiagen). RNA (0.5 mg) was reverse transcribed with the SuperScript First Strand cDNA Synthesis kit (Invitrogen), and 10% of the product was used for Q-PCR. The primers used were as follows: Bad primers, forward 5′-agagtatgttccagatcccag-3′ and reverse 5′-gtc ctcgaaaagggctaagc-3′; GAPDH primers, forward 5′-atggtgaa ggtcggtgtgaacg-3′ and reverse 5′-gttgtcatggatgaccttggcc-3′. The Q-PCR conditions were as follows: 50 °C for 2-min hold (UDG incubation), 95 °C for 2-min hold, 40 cycles of 95 °C for 15 s, and 60 °C for 30 s.
2.2.5. Statistical analysis
Student's t-test was used to evaluate statistical significance.
3. Results
3.1. PAK1 regulates expression of apoptotic factors in β-cells
To initiate study into the requirement for PAK1 in β-cell survival, we first employed MIN6 cells depleted of PAK1 using siRNA. Depletion of PAK1 levels by ∼50% significantly increased caspase-3 cleavage levels by 2-fold compared with control siRNA-expressing cells (siCon) (Fig. 1A). Moreover, we assessed cytosolic levels of cytochrome-C as a measure of the ‘intrinsic’ signaling pathway of apoptosis, wherein mitochondria release cytochrome-C from the mitochondrial intermembrane space [16]. Indeed, MIN6 cells depleted of ∼50% endogenous PAK1 released substantially elevated levels of cytochrome-C into the cytosolic fraction (Fig. 1B). These data implicate PAK1 expression in a regulatory role to restrict expression of apoptotic factors and the intrinsic apoptotic pathway in β-cells.
Fig. 1.
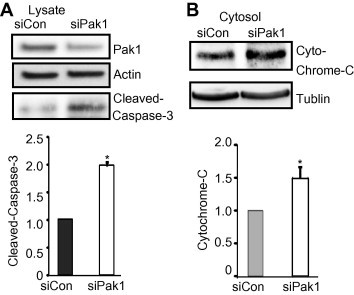
Depletion of PAK1 increases expression of apoptotic factors in β-cells. MIN6 cells were transfected with PAK1–siRNA (siPAK1) or control (siCon) siRNA oligonucleotides. After 48 h incubation, cells were washed twice and incubated with glucose-free MKRBB for 2 h before harvesting. (A) Whole cell detergent lysates were prepared and subjected to 12% SDS–PAGE for immunoblotting with antibodies indicated. Data represent the average ± SE for 3 independent experiments; *p < 0.05, versus siCon. (B) Cytosolic fractions were prepared as described in Section 2.2. Cytosolic fraction protein was subjected to 15% SDS–PAGE for immunoblotting (IB). Ponceau S staining was used as a protein loading control. Data represent the average ± SE for 3 independent experiments; *p < 0.05, versus siCon.
3.2. Hyperglycemia/glucotoxic stress increases Bad protein expression in human and mouse pancreatic islets
Elevated fasting blood glucose is a hallmark characteristic of individuals with frank T2D [3,17,18], wherein hyperglycemia is presumed to ultimately enhance β-cell death [19,20]. Relatedly, Bad protein has been implicated in glucolipotoxicity-induced β-cell death [11,12]. To determine if hyperglycemia/glucotoxic stress was sufficient to alter Bad protein expression, human or mouse islets were incubated in medium containing 5.8 or 25 mM glucose for 5 days, a model system used by others [12]. As shown in Fig. 2, glucotoxicity was sufficient to substantially elevate Bad protein levels in both human and mouse islets.
Fig. 2.
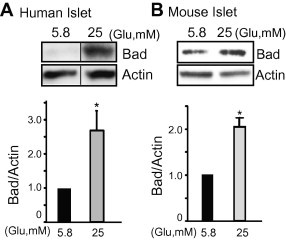
Glucotoxic/hyperglycemic stress induced Bad protein levels in pancreatic β-cells. (A) Human islets were incubated in CMRL in the presence of 5.8 or 25 mM glucose for 5 days. Islets were then washed twice before harvesting and whole cell lysates were subjected to immunoblotting with antibodies indicated. Data represent the average ± SE for 3 independent donor batches of human islets; *p < 0.05, versus 5.8 mM glucose-treated islets. Vertical lines indicate the splicing of lanes from within the same gel image. (B) Islets from wild type mice were incubated in CMRL media in the presence of 5.8 or 25 mM glucose for 5 days. Islets were then washed twice before harvesting. Whole cell detergent lysates were subjected to immunoblotting with antibodies indicated. Data represent the average ± SE for 5 sets of mouse islets; *p < 0.05, versus 5.8 mM glucose.
3.3. PAK1 depletion results in enhanced Bad protein expression in islet β-cells
Since PAK1 in known to phosphorylate Bad, which leads to Bad degradation [13,14], we evaluated Bad and phosphorylated Bad (pBad) levels in PAK1+/− (Het) mouse islets. PAK1+/− heterozygous mouse islets contained ∼2-fold more Bad protein when compared with PAK1+/+ wild-type (WT) islets (Fig. 3A). Bad expression was also significantly elevated in islets from PAK1−/− KO mice (2.1 ± 0.2-fold increase versus WT, p < 0.05). Coincident with this, the level of pBadSer112 was diminished in PAK1+/− islets, and when normalized to total Bad levels, the relative amount of pBadSer112 in these islets was reduced to 41 ± 8% ( n = 3, p < 0.0002). In addition, quantitative real-time PCR revealed an increase of Bad mRNA in PAK1+/− Het islets (Fig. 3B), suggesting that in the islet, at least in part, PAK1 regulates Bad protein expression via a transcriptional step.
Fig. 3.
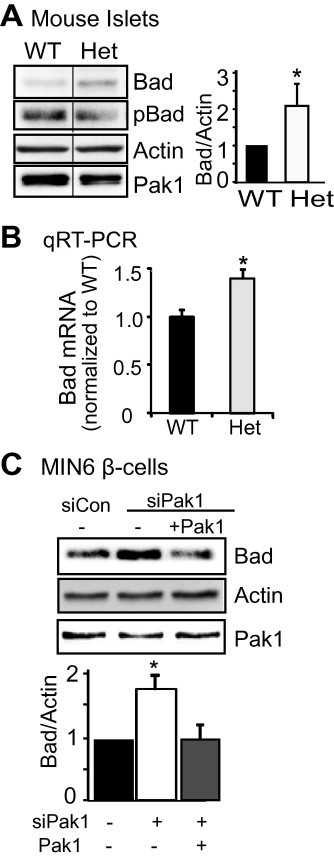
Depletion of PAK1 increased Bad expression. (A) Bad expression increased in PAK1+/− heterozygous (Het) islets. Islets were isolated from PAK1+/− Het and wild type (WT) littermate mice and homogenized. Proteins were resolved by 12% SDS–PAGE for immunoblotting with antibodies as indicated. Data represent the average ± SE for 5 pairs of mice; *p < 0.05, versus WT. Vertical lines indicate the splicing of lanes from within the same gel image. (B) Bad mRNA expression in PAK1+/− mice. Islets were isolated from PAK1+/− Het and littermate PAK1+/+ WT mice for use in Q-PCR analysis (quantified relative to GAPDH from 3 batches of islets, *p < 0.05 versus WT). (C) MIN6 cells were transfected with PAK1 siRNA (+) or control (−) oligonucleotides with pCMV6 vector or pCMV6-myc-Pak1 plasmid. After 48 h incubation, cells were washed twice and incubated with glucose-free MKRBB for 2 h before harvesting. Whole cell detergent lysates were prepared and subjected to 12% SDS–PAGE for immunoblotting with antibodies indicated. Data represent the average ± SE for 3 independent experiments; *p < 0.05, versus siCon.
Since islets contain a population of ∼20% of cells that are non-β-cells, we confirmed that this Bad elevation was in the β-cells by evaluating PAK1-depleted clonal MIN6 β-cells, which showed significantly increased Bad protein levels as well. Restoration of PAK1 protein level by co-transfection with exogenous PAK1 correspondingly normalized the aberrant increase in Bad protein induced by PAK1-depletion (Fig. 3C). Overall these data suggest that the abundance of PAK1 specifically in the β-cells of the islet is important for maintaining a low level of Bad expression.
3.4. Hyperglycemia induced Bad up-regulation is exacerbated in PAK1+/− heterozygous islets
We have shown that Bad expression in islets was increased by chronic exposure to hyperglycemic conditions, or could be induced by depletion of PAK1 in the absence of glucose. As shown in Fig. 4, PAK1+/− Het islets subjected to hyperglycemia showed the largest increase in Bad expression, suggesting that attenuated PAK1 abundance could render islet β-cells more susceptible to apoptosis.
Fig. 4.
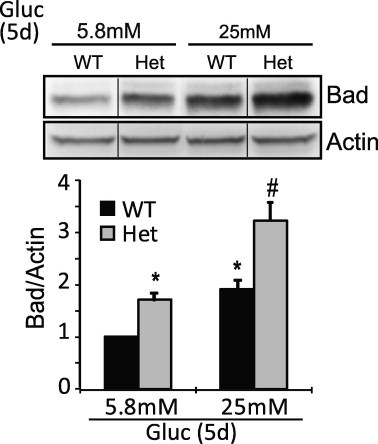
Hyperglycemia/glucotoxic stress induced Bad expression is exacerbated in islets from PAK1+/− heterozygous (Het) mouse. Islets from WT or PAK1+/− Het mice were incubated in the presence of 5.8 or 25 mM glucose for 5 days (Gluc, 5d). Islets were then washed twice before harvesting. Detergent lysates were subjected to immunoblotting with antibodies indicated. Data represent the average ± SD from 3 independent batches of islets (*p < 0.05 versus WT islets in 5.8 mM glucose;#p < 0.05 versus WT islets in 25 mM glucose). Vertical lines indicate the splicing of lanes from within the same gel image.
4. Discussion
Type 2 diabetes (T2D) occurs as a result of pancreatic β-cell failure in combination with insulin resistance in peripheral tissues. It has been reported that at the onset of clinical diagnosis, there is a ∼50% reduction in β-cell mass in T2D, which has been attributed to a 3- to10-fold increase in β-cell apoptosis [2,3]. Implicated in the progression to T2D is the upward rise in blood glucose levels [3,17,18], and in the later stages of T2D, pro-inflammatory cytokines on a base of chronically elevated blood glucose will ultimately enhance β-cell death [19,20]. Thus, understanding the details of how hyperglycemia, which begins prior to the onset of frank T2D, leads to β-cell apoptosis will be essential for future development of therapies that preserve β-cell mass and avoid the otherwise inevitable progression toward β-cell failure and T2D onset. Here we showed that hyperglycemic conditions or PAK1 loss is sufficient to trigger the up-regulation of Bad protein in islet β-cells, while up-regulation of Bad protein level induced by glucotoxic stress was further exacerbated in PAK1+/− mouse islets. This suggests that PAK1 plays a role in restricting Bad protein abundance in islet β-cells, and could confer protection against apoptosis under stress.
We previously demonstrated that PAK1−/− knockout mice show normal β-cell mass and insulin content in islets [5], despite the observations that Bad expression is elevated in islets from PAK1−/− knockout mice. One explanation for this discrepancy may be that the original β-cell mass determinations were made in younger mice (10 weeks) wherein β-cell mass loss was not yet or fully developed. Additionally or alternatively, it is also possible that prolonged hyperglycemic stress is required to fully induce Bad expression in vivo and hence drive β-cell apoptosis to yield measurable loss of β-cell mass; PAK1−/− mice at 10 weeks of age did not show fasting hyperglycemia [5]. Considering this new information showing the potential for PAK1 involvement in Bad expression and apoptosis regulation, we plan to pursue longitudinal studies of β-cell mass and apoptosis of the PAK1−/− and PAK+/− mouse models, with inclusion of diet-induced stress to drive hyperglycemia.
Our data suggest that PAK1 is required to control the level of Bad in the β-cell, loss of PAK1 in islets from PAK+/− mouse leading to a 2-fold increase of protein expression. Q-PCR suggests that PAK1 may regulate Bad protein expression at the transcriptional level. However, our data suggest that PAK1 may also regulate post-translationally, via phosphorylation of Bad, as supported by our data showing reduced relative levels of pBad in PAK1+/− islets as compared with WT. It has been shown that phosphorylation of BadSer112 by several kinases control its stability [21,22]. PAK1 has been shown to phosphorylate Bad in vitro and in vivo on Ser112 [13,14]. Thus, our data suggest that PAK1 may regulate Bad expression via phosphorylation-mediated degradation. Detailed mechanisms of the regulation of Bad protein by PAK1 as well as the potential regulation of additional members of the Bcl-2 family by PAK1 are currently under investigation. In addition, it remains to be tested whether PAK1 protein activation/signaling and/or scaffolding destabilize Bad protein.
In summary, these data implicate PAK1 as a new hub of susceptibility for β-cell apoptosis, and we speculate that efforts to minimize loss of PAK1 could assist to regain control over Bad levels. Efforts to determine why PAK1 levels are attenuated in islets of T2D patients are underway, to determine whether in response to chronic hyperglycemia or otherwise, PAK1 protein is selectively reduced, ultimately rendering the islet more susceptible to apoptosis. Combined with the known positive role for PAK1 signaling in glucose-stimulated insulin secretion and in peripheral insulin signaling in skeletal muscle [5], PAK1 has potential as a therapeutic target in diabetes.
Acknowledgments
We are grateful to Dr. Lawrence Quilliam (Indiana University School of Medicine) and Dr. John Hutton (University of Colorado Health Sciences Center) for their gifts of the pCMV-myc-PAK1 plasmid and MIN6 cells, respectively. This study was supported by CTSI-KL2 RR025760 (to Z.W.) and by Grants DK067912 and DK076614 (to D.C.T.). We thank Dr. Stephanie Yoder and Michael Kalwat for technical assistance, as well as Natalie Stull for assistance with islet isolation. Human islets were obtained from the Integrated Islet Distribution Program (IIDP).
References
- 1.Wajchenberg B.L. Clinical approaches to preserve beta-cell function in diabetes. Adv. Exp. Med. Biol. 2010;654:515–535. doi: 10.1007/978-90-481-3271-3_23. [DOI] [PubMed] [Google Scholar]
- 2.Meier J.J., Breuer T.G., Bonadonna R.C., Tannapfel A., Uhl W., Schmidt W.E., Schrader H., Menge B.A. Pancreatic diabetes manifests when beta cell area declines by approximately 65% in humans. Diabetologia. 2012;55:1346–1354. doi: 10.1007/s00125-012-2466-8. [DOI] [PubMed] [Google Scholar]
- 3.Butler A.E., Janson J., Bonner-Weir S., Ritzel R., Rizza R.A., Butler P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z., Oh E., Thurmond D.C. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J. Biol. Chem. 2007;282:9536–9546. doi: 10.1074/jbc.M610553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z., Oh E., Clapp D.W., Chernoff J., Thurmond D.C. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J. Biol. Chem. 2011;286:41359–41367. doi: 10.1074/jbc.M111.291500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z., Thurmond D.C. Differential phosphorylation of RhoGDI mediates the distinct cycling of Cdc42 and Rac1 to regulate second-phase insulin secretion. J. Biol. Chem. 2010;285:6186–6197. doi: 10.1074/jbc.M109.072421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson K., D’Mello S.R. p21-Activated kinase-1 is necessary for depolarization-mediated neuronal survival. J. Neurosci. Res. 2005;79:809–815. doi: 10.1002/jnr.20415. [DOI] [PubMed] [Google Scholar]
- 8.Egom E.E., Ke Y., Musa H., Mohamed T.M., Wang T., Cartwright E., Solaro R.J., Lei M. FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J. Mol. Cell. Cardiol. 2010;48:406–414. doi: 10.1016/j.yjmcc.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egom E.E. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1487–H1495. doi: 10.1152/ajpheart.01003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi D., Woo M. Executioners of apoptosis in pancreatic {beta}-cells: not just for cell death. Am. J. Physiol. Endocrinol. Metab. 2010;298:E735–E741. doi: 10.1152/ajpendo.00696.2009. [DOI] [PubMed] [Google Scholar]
- 11.McKenzie M.D. Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins Bim and Puma and multi-BH domain protein Bax. Diabetes. 2010;59:644–652. doi: 10.2337/db09-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Federici M. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: a potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes. 2001;50:1290–1301. doi: 10.2337/diabetes.50.6.1290. [DOI] [PubMed] [Google Scholar]
- 13.Ye D.Z., Jin S., Zhuo Y., Field J. p21-Activated Kinase 1 (Pak1) phosphorylates BAD directly at Serine 111 in vitro and indirectly through Raf-1 at Serine 112. PLoS One. 2011;6:e27637. doi: 10.1371/journal.pone.0027637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schurmann A., Mooney A.F., Sanders L.C., Sells M.A., Wang H.G., Reed J.C., Bokoch G.M. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol. Cell. Biol. 2000;20:453–461. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen J.D. p21-Activated kinase regulates mast cell degranulation via effects on calcium mobilization and cytoskeletal dynamics. Blood. 2009;113:2695–2705. doi: 10.1182/blood-2008-06-160861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caroppi P., Sinibaldi F., Fiorucci L., Santucci R. Apoptosis and human diseases: mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Curr. Med. Chem. 2009;16:4058–4065. doi: 10.2174/092986709789378206. [DOI] [PubMed] [Google Scholar]
- 17.Leonardi O., Mints G., Hussain M.A. Beta-cell apoptosis in the pathogenesis of human type 2 diabetes mellitus. Eur. J. Endocrinol. 2003;149:99–102. doi: 10.1530/eje.0.1490099. [DOI] [PubMed] [Google Scholar]
- 18.Robertson R.P., Harmon J., Tran P.O., Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl. 1):S119–S124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 19.Mathis D., Vence L., Benoist C. Beta-cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 20.Robertson R.P., Harmon J., Tran P.O., Tanaka Y., Takahashi H. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52:581–587. doi: 10.2337/diabetes.52.3.581. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y., Sun S.Y., Owonikoko T.K., Sica G.L., Curran W.J., Khuri F.R., Deng X. Rapamycin induces bad phosphorylation in association with its resistance to human lung cancer cells. Mol. Cancer Ther. 2012;11:45–56. doi: 10.1158/1535-7163.MCT-11-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fueller J., Becker M., Sienerth A.R., Fischer A., Hotz C., Galmiche A. C-RAF activation promotes BAD poly-ubiquitylation and turn-over by the proteasome. Biochem. Biophys. Res. Commun. 2008;370:552–556. doi: 10.1016/j.bbrc.2008.03.141. [DOI] [PubMed] [Google Scholar]