

Abstract
Background
Promoter hypermethylation is a well documented mechanism for tumor-specific alteration of suppressor gene activity in human malignancy including Head and Neck Cancer (HNC). The abrogation of specific suppressor gene activity may influence tumor behavior and clinical outcome. In this study we examined methylation of DCC, KIF1A, EDNRB, and p16INK4a in a large cohort of HNC patients from ECOG 4393/RTOG 9614 to identify clinical correlates of methylation of these genes.
Methods
Methylation was assessed by quantitative methylation-specific polymerase chain reaction in DNA from tumor specimens and was considered as a continuous and a binary variable. Clinical data including demographics, stage, risk factor exposure, treatment, and outcome were collected by ECOG and RTOG. Methylation status was also correlated with mutation of TP53 (previously reported) and HPV status.
Results
Methylation results were available for 368 cases, 353 of which also have p53 mutation status. At least one methylation event was present in all tumors. In multivariate analysis of the entire cohort, methylation of p16 was associated with decreased survival (HR=1.008, p=0.045). However, in tumors with disruptive TP53 mutation (poor prognostic group), the additional presence of methylation of p16 was protective (p=0.019 considering p16 methylation as a continuous variable).
Conclusion
Methylation of tumor-related genes contributes to the biological behavior of HNC and influences overall survival in conjunction with other known prognostic molecular events.
Keywords: head and neck cancer, methylation, survival
INTRODUCTION
Head and Neck Squamous Cell Carcinoma (HNC) arises in the mucosa of the upper aerodigestive tract as the result of an accumulation of molecular alterations that affect cellular behavior resulting in a malignant phenotype. These alterations may involve the DNA coding sequence directly (genetic) or indirectly (epigenetic) and result in variation in expression and/or function of genes with tumor suppressive or oncogenic activity. Primary sequencing of the cancer genome of HNC has identified numerous target genes (1–2), however, the scope and number of identified gene mutations is insufficiently robust to account for the entire cancer phenotype. Epigenetic alterations, specifically promoter hypermethylation, are well documented to contribute to tumorigenesis through the abrogation of expression of tumor suppressor genes (3). Many candidate genes have been found to display promoter methylation in HNC (4). These genetic and epigenetic alterations interact in complex and integrated fashion affecting key cellular functional pathways that control cell cycle and proliferation, response to genotoxic stress and apoptosis, metastasis and invasion, and terminal differentiation. It is reasonable to hypothesize that analysis of molecular alteration in cohorts of cancer patients may provide valuable insight into their individual and cumulative effect on tumor aggressiveness and prognosis.
Aberrant DNA methylation of a particular gene or a set of selected genes may be used as a cancer biomarker for clinical assessment. Similar to genetic mutations, promoter-specific DNA hypermethylation results in silencing tumor suppressor genes, mediating tumorigenesis, invasiveness and metastasis (5–6). As a stable, clonally propagated event, aberrant DNA methylation has significant tumor type specificity and variability between patients, which may indicate clinical usefulness (7). Single or panels of DNA methylation markers have been proposed as suitable prognostic biomarkers for cancer progression and outcomes. Methylated PITX2 promoter has been associated with a poor outcome in breast and prostate cancer (8–9). The promoter hypermethylation of CDKN2A, CDH13, RASSF1A and APC were associated with early recurrence in stage I non-small-cell lung carcinoma (10). Other prognostic hyperemethylated genes in HNC have been reported: DCC, hMLH1, MGMT, HMT, ATM, GSTP1, MINT, etc (11). DNA methylation biomarkers show promise for clinical implementation, however, the identification of reliable prognostic biomarkers await confirmation from large, well defined sample cohorts (12).
The immense complexity of both clinical and biological factors that serve as potential prognostic factors in any cohort of HNC dictates that the clinical data used to evaluate the effect of individual molecular events be very robust. Large, well defined cohorts followed for adequate periods of time with rigorous quality control for data collection are necessary in order to provide the statistical power needed to assess the relative contribution of various molecular factors and their independence with respect to clinical parameters that are more readily and economically derived. ECOG 4393/RTOG 9614 is a cooperative group, multi-institutional study involving the collection of tumor, and resection margin tissue samples along with clinical and treatment outcome data from a large cohort of patients with HNC. Molecular analysis of tissue samples for genetic and epigenetic alterations was performed at a centralized location (Johns Hopkins Head and Neck Tumor Laboratory) while clinical and outcome data was collected by the cooperative groups and statistical analysis performed by the Eastern Cooperative Group (ECOG). With 480 evaluable cases treated with primary surgery with curative intent and followed for over 10 years after surgery, this cohort provides one of the best resources for exploring possible associations between molecular alterations and clinical tumor behavior available in the world. We have been examining the potential utility of candidate tumor promoter hypermethylation events in this cohort for molecular detection of rare cancer cells in the margin samples (the primary goal of the protocol) in order to improve the sensitivity and specificity of molecular margin analysis. As a result, the presence of methylation of four genes (p16, DCC, KIF1A, and EDNRB) in tumor samples has been determined providing opportunity to test these events individually and in combination for associations with survival. This strategy has already been performed to examine the prognostic significance of TP53 gene mutation in the cohort. The value of the ECOG 4393/RTOG 9614 cohort was demonstrated by our ability to display a prognostic association between those TP53 mutations which disrupted the DNA binding capacity of the p53 protein and decreased overall survival (13). The current report expands on and refines those earlier findings by adding the information about the spectrum of tumor-specific methylation of the four target genes to that of TP53 mutation.
METHODS
STUDY DESIGN
The study cohort was from a prospective, multicenter study involving 18 member institutions of ECOG and Radiation Therapy Oncology Group (RTOG) between 1996 and 2002 (study no. E4393/R9614). The first objective of the study protocol was to determine the clinical utility of molecular detection of residual cancer cells in tumor margins and the second independent objective was to seek association between molecular alterations in HNC and survival. The protocol was approved by the cooperative groups and the investigational review board of each participating institution. Written informed consent was obtained from each patient. The study enrolled 560 eligible patients with HNC who underwent primary surgical extirpation of tumor with curative intent. The cooperative group data managers collected and reviewed demographic, clinical, and pathologic information for each patient perioperatively and follow-up patient information at scheduled intervals (at every 6 months for the first 3 years and annually thereafter).
TUMOR SPECIMENS
Tumor Sample Collection, DNA Extraction, and Bisulfite Treatment
Tumor and margin samples were collected during operation and rapidly frozen in −80°C. The samples were shipped on dry ice by overnight courier to the Johns Hopkins Head and Neck Cancer Research Laboratory, and kept frozen in liquid nitrogen until DNA extraction. Frozen tumor specimens were microdissected in series of 5-µm sections and processed with hematoxylin and eosin (H&E) staining for light microscopic examination to ensure the presence of HNC. Tumor samples with at least 70% cancer cells were candidates for molecular studies. Frozen tissues with less than 70% tumor cells were microdissected to enrich the tumor-cell content. Additional 12-µm sections were then cut and digested with DNA elution buffer containing 1% sodium dodecyl sulfate and 50 µg/mL proteinase K (Boehringer, Mannheim, Germany) at 48°C for 2–3 days. Initial, middle, and last sections cut for DNA extraction were also stained with H&E to ensure the presence of > 70% HNC. Genomic DNA was extracted with phenol-chloroform extraction and ethanol precipitation, eluted in low-salt 10 mM Trix-HCl and 2.5 mM EDTA (LoTE) buffer.
DNA (2 µg) extracted from tissue samples was subjected to bisulfite treatment, which modifies CpG islands, using the EpiTect® Bisulfite Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions.(14–15) The bisulfite-treated DNA was resuspended in 50 uL of elution butter and stored at −80°C.
Quantitative Methylation-Specific PCR
The bisulfite-treated DNA was used as a template for fluorescence-based real-time polymerase chain reaction (PCR) and was amplified using quantitative methylation-specific PCR (Q-MSP) as described previously.(16) Genes selected for this study came from the studies previously performed by the authors chosen for utility (specificity and sensitivity) for detection of HNC in saliva: p16, DCC, KIF1A, and EDNRB.(17–20) Primers and probes were designed to amplify the bisulfate-modified DNA for four gene promoters and internal reference gene (ACTB). The sequences of primers and probes are available in previous publication.(21) For all Q-MSP reactions, 3 µL of bisulfite-treated DNA were added to a final volume of 20 µL consisting of each 600 nM primer and 200 µM probe, 0.75 U platinum Taq polymerase (Invitrogen, Carlsbad, CA), 200 nM ROX reference dye (Invitrogen), 200 µM dNTP each, 16.6 mM ammonium sulfate, 67 mM Trizma (Sigma, St Louis, MO), 6.7 mM magnesium chloride, 10 mM mercaptoethanol and 0.1% dimethylsulfoxide. All reactions were carried out in triplicate to ensure consistent results in 384-well plates using a 7900 Sequence Detector System (Perkin-Elmer Applied Biosystems, Norwalk, CT). Thermal cycling included a first denaturation step at 95°C for 10 min, followed by 50 cycles of 95°C for 15 sec and 60°C for 1 min. As a standard reference for Q-MSP, leukocyte DNA from a healthy individual was completely methylated using excess SssI methyltransferase (New England Biolab Inc, Beverly, MA) and methylated DNA was serially diluted 45 to 0.00045 ng to construct a calibration curve for each plate.(22) Each plate contained wells with only DNA- and RNA-free water and the reaction mix for negative control to ensure no contamination. Each reaction was performed in triplicate and the average of the triplicate was considered for analysis. The methylation level of each gene in each DNA sample was normalized to ACTB and calculated as an equation of (gene of interest/ACTB) ×100. The normalized value is set to 100 if the ratio (gene of raterest/ACTB) is greater than 1.
TP53 Mutational Analysis
TP53 mutation data has been previously published and was derived with the use of the GeneChip p53 assay (Affymetrix) and the Surveyor DNA endonuclease–denaturing high-performance liquid chromatography (DHPLC) for high-throughput detection of mutations in exons 2 to 11 in head and neck primary tumors.(13) The TP53 mutations were grouped as “disruptive” and “nondisruptive”, as previously defined. Disruptive mutations included nonconservative mutations occurring within key DNA binding domains (L2–L3 regions) or stop codons in any region, and all other mutations were defined as nondisruptive mutations.
HPV detection
The presence or absence of HPV DNA has been determined using in-situ hybridization for a subset of the ECOG cohort and previously published (23). Additional samples have been screened using quantitative PCR. To quantify the viral load of HPV-16, genomic DNA from patients was used for real-time PCR. Specific primers and probes sets are reported in a previous publication (24). PCR for housekeeping gene β-actin was performed in duplicate and parallel to normalize the input DNA. The final primer and probe concentrations were 0.3 and 0.1 mmol/L, respectively, in a total volume of 10 mL. Each reaction was run for 50 cycles. All experiments were performed at least twice in duplicate. Four water negative controls were included in each run. Samples in which 4 results were not concordant were repeated twice more in duplicate as they were usually due to failed PCR in one of the initial reactions. By using serial dilutions, standard curves were developed for the HPV 16 viral load using CaSki (American Type Culture Collection, Manassas, VA) cell line genomic DNA, as it has been previously characterized to harbor 600 copies of HPV-16 DNA per genome equivalent (6.6 pg of DNA/genome). Standard curves were developed for HPV 16 E6 and E7, using serial dilutions of genomic DNA extracted from CaSki cells with following concentrations: 25, 2.5, 0.25, 0.025, 0.0025, and 0.00025 ng of genomic DNA. A standard curve was developed as well for the β-actin housekeeping gene which has 2 copies per genome equivalent, using the same serial dilutions of the CaSki genomic DNA. For all samples, duplicate control β-actin amplifications of 10 ng of total DNA were positive. Only tumors arising in the oropharynx were included in PCR HPV testing. HPV status was available for 69 oropharynx samples (23 negative, 46 positive) and one sample from the oral cavity (negative). Cases of oropharyngeal tumors for which HPV status could not be determined were coded as unknown. HPV status was imputed to be negative for samples with missing HPV status from sites other than the oropharynx.
STATISTICAL ANALYSIS
Laboratory and clinical data were submitted to the ECOG central office and analyzed at the ECOG statistical center, including patient demographics, staging, risk factor exposure, pathology, treatment, and outcome. Descriptive statistics were used to summarize the methylation levels by baseline characteristics of the patients. Wilcoxon or Kruskal-Wallis rank-sum test was used to test for differences in methylation levels among baseline categories. Overall survival was defined as the time between study entry (surgery) and death from all causes or last follow-up. Patients who were alive at their last follow-up were censored. The Kaplan-Meier method (25) was used to estimate survival curves. Multivariate Cox proportional hazards models (26) were used to assess the significance of variable on overall survival. Multivariate models considered TP53 status, marker methylation levels, HPV status, tumor site and stage, nodal stage, smoking history, average alcohol use, treatment type, interaction between TP53 status and HPV status, and interaction between TP53 status and marker methylation levels. Models considering the effect of accumulation of methylation did not include individual marker methylation levels. Methylation levels were included as continuous or categorical (high/low defined by the k-means algorithm [35] with k=2) variables, and all other variables except for age were categorical. The k-means algorithm defines two groups where the within-group sum of squares is minimized. P-values for hazard ratios were calculated using the likelihood-ratio test, and values of less than 0.05 were considered to indicate statistical significance. Fisher’s exact test (categorical variable with two categories), chi-square test (categorical variable with three or more categories) and Wilcoxon test (continuous variable) were used to test for differences in characteristics between included and excluded patients. Linear models were used to assess the association between methylation levels and smoking history while controlling for age, gender, site and HPV status.
RESULTS
The ECOG4393/RTOG 9614 population includes 560 subjects with HNC. Sixteen subjects did not meet entry criteria, DNA was not available for 176 others, and 15 did not have p53 mutation status, leaving 353 tumor samples/cases that form the basis of this analysis. A comparison of clinical characteristics between included and excluded patients is shown in Table I The quantity of methylation for each of the four target genes compared to β actin is shown in Figure 1. The relative quantity of methylation by QMSP was considered both as a continuous and as dichotomous (high and low) values. Tumors from 48 cases displayed below-median methylation in all four target genes, while 50 cases displayed above-median levels of methylation of all four genes. Associations of promoter methylation with clinical features including tumor site, T and N stage, gender, treatment modality, and use of alcohol and tobacco as well as with the molecular factor, status of p53 mutation and HPV status, were examined (Table II). There was some variability of methylation by tumor site with higher methylation of p16 and KIF1A in oral cavity tumors while p16 methylation was higher in lower T stage tumors. Of note, methylation of p16, DCC, EDNRB and KIF1A varies across the amount of tobacco used (Figure 2). Methylation level for these markers is negatively associated with tobacco use when explored in a multivariate linear model (Table III).
Table I.
Characteristics of patients included and excluded from analysis. Numbers in parentheses are percentages of included or excluded patients in that category, unless indicated otherwise. Fisher’s exact test (categorical variable with two categories), chi-square test (categorical variable with three or more categories) and Wilcoxon test (continuous variable) were used to test for differences in patient characteristics between included and excluded.
| Variable | Category | Excluded | Included | Total | p-value |
|---|---|---|---|---|---|
| Total | 207 | 353 | 560 | – | |
| Age | Mean (SD) | 60.0(13.0) | 60.6(13.3) | 60.3(13.2) | 0.665 |
| Median (Q1,Q3) | 60 (51,70) | 61 (52,69) | 61 (52,69) | ||
| [Min, Max] | [1,91] | [17,98] | [1,98] | ||
| Age Category | <=64 | 124(59.9) | 205(58.1) | 329(58.8) | 0.063 |
| >64 | 80(38.6) | 148(41.9) | 228(40.7) | ||
| Unknown | 3(1.4) | 0(0.0) | 3(0.5) | ||
| Gender | Male | 156(75.4) | 252(71.4) | 408(72.9) | 0.326 |
| Female | 51(24.6) | 101(28.6) | 152(27.1) | ||
| Race | White | 162(78.3) | 301(85.3) | 463(82.7) | 0.053 |
| Hispanic | 11(5.3) | 19(5.4) | 30(5.4) | ||
| Black | 29(14.0) | 31(8.8) | 60(10.7) | ||
| Other | 5(2.4) | 2(0.6) | 7(1.2) | ||
| Site | Oral cavity | 85(41.1) | 156(44.2) | 241(43.0) | 0.353 |
| Oropharynx | 47(22.7) | 80(22.7) | 127(22.7) | ||
| Hypopharynx | 16(7.7) | 23(6.5) | 39(7.0) | ||
| Larynx | 43(20.8) | 73(20.7) | 116(20.7) | ||
| Salivary glands | 1(0.5) | 0(0.0) | 1(0.2) | ||
| Other | 13(6.3) | 18(5.1) | 31(5.5) | ||
| Multiple | 0(0.0) | 3(0.8) | 3(0.5) | ||
| Unknown | 2(1.0) | 0(0.0) | 2(0.4) | ||
| PT Stage | T1 | 55(26.6) | 76(21.5) | 131(23.4) | 0.000 |
| T2 | 38(18.4) | 127(36.0) | 165(29.5) | ||
| T3 | 26(12.6) | 61(17.3) | 87(15.5) | ||
| T4 | 24(11.6) | 79(22.4) | 103(18.4) | ||
| TX or Tis | 4(1.9) | 9(2.5) | 13(2.3) | ||
| Unknown | 60(29.0) | 1(0.3) | 61(10.9) | ||
| PN Cat | N0 | 41(19.8) | 130(36.8) | 171(30.5) | 0.000 |
| N1 | 20(9.7) | 48(13.6) | 68(12.1) | ||
| N2 | 51(24.6) | 121(34.3) | 172(30.7) | ||
| N3 | 0(0.0) | 3(0.8) | 3(0.5) | ||
| NX | 39(18.8) | 50(14.2) | 89(15.9) | ||
| Unknown | 56(27.1) | 1(0.3) | 57(10.2) | ||
| Treatment | Surgery Only | 21(10.1) | 110(31.2) | 131(23.4) | 0.000 |
| Surgery + postoperative therapy | 32(15.5) | 172(48.7) | 204(36.4) | ||
| Salvage surgery | 14(6.8) | 65(18.4) | 79(14.1) | ||
| Unknown | 140(67.6) | 6(1.7) | 146(26.1) | ||
| Smoking | Never smoked | 36(17.4) | 66(18.7) | 102(18.2) | 0.377 |
| Pipe or cigar only | 7(3.4) | 15(4.2) | 22(3.9) | ||
| Cigarette: <20 pack-yrs | 31(15.0) | 38(10.8) | 69(12.3) | ||
| Cigarette: 20–40 pack-yrs | 44(21.3) | 96(27.2) | 140(25.0) | ||
| Cigarette: >40 pack-yrs | 82(39.6) | 131(37.1) | 213(38.0) | ||
| Unknown | 7(3.4) | 7(2.0) | 14(2.5) | ||
| Alcohol | <10 oz/wk | 108(52.2) | 186(52.7) | 294(52.5) | 0.531 |
| 10–32 oz/wk | 34(16.4) | 70(19.8) | 104(18.6) | ||
| >32 oz/wk | 50(24.2) | 69(19.5) | 119(21.2) | ||
| Unknown | 15(7.2) | 28(7.9) | 43(7.7) | ||
Figure 1.
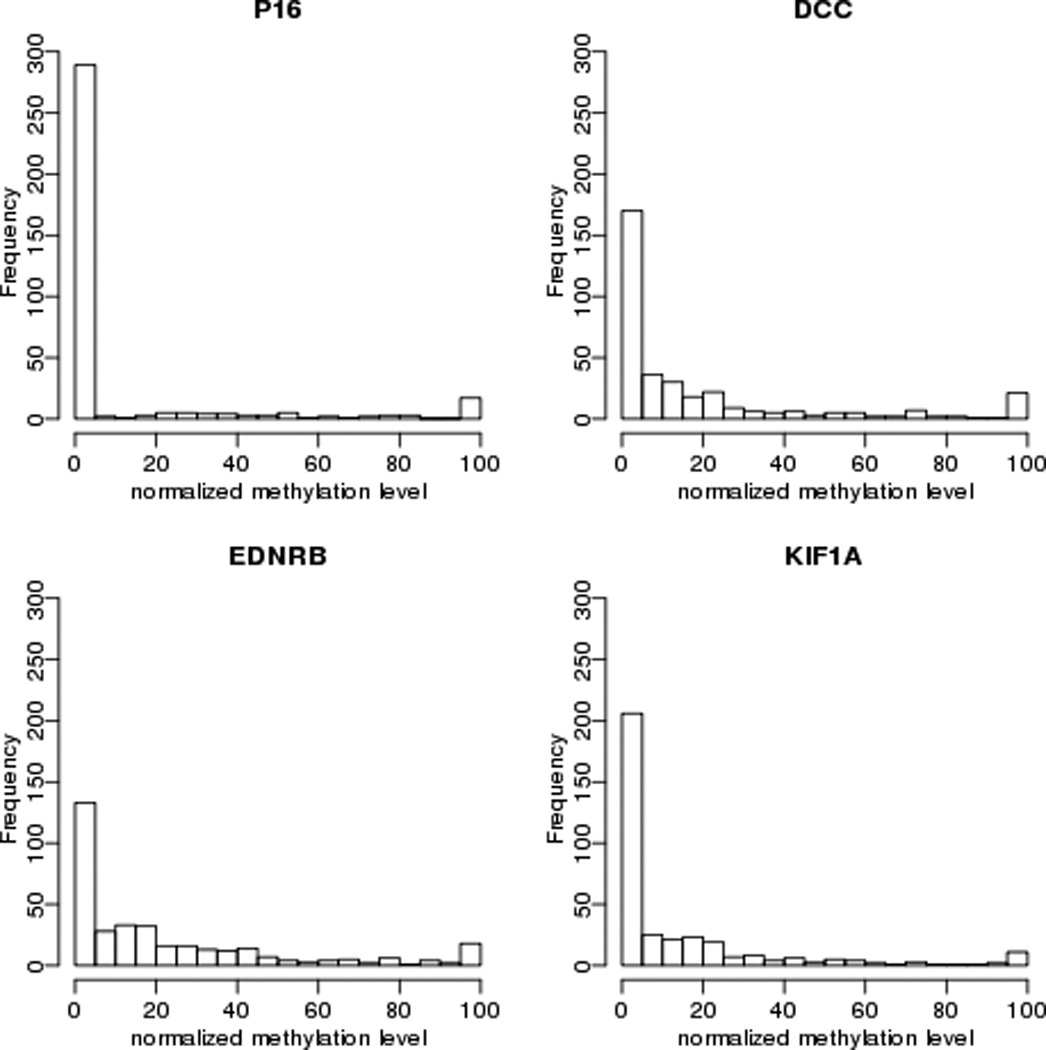
Relative level of promoter hypermethylation of four target genes in tumors of ECOG 4393/RTOG9614 cohort included in this analysis (n=353). X-axis indicates methylation level of target genes normalized to β-Actin.
Table II.
Methylation levels by baseline characteristics (median, [1st quartile,3rd quartile]). Wilcoxon or Kruskal-Wallis rank sum test is used to test for differences in methylation levels among baseline categories.
| Number of patients | % patients | P16 | DCC | EDNRB | KIF1A | |
|---|---|---|---|---|---|---|
| Sex | ||||||
| Female | 101 | 29 | 0 [0,1] | 7 [1,15] | 14 [2,37] | 4 [0,17] |
| Male | 252 | 71 | 0 [0,0] | 5 [0,24] | 12 [2,31] | 2 [0,16] |
| pval | 0.59 | 0.87 | 0.38 | 0.57 | ||
| Race or ethnic group | ||||||
| White | 301 | 85 | 0 [0,0] | 6 [0,21] | 14 [2,36] | 2 [0,18] |
| Hispanic | 19 | 5 | 0 [0,0] | 4 [1,19] | 12 [2,27] | 2 [0,8] |
| Black | 31 | 9 | 0 [0,0] | 2 [0,25] | 3 [2,12] | 1 [0,11] |
| Other | 2 | 1 | 0 [0,0] | 38 [20,57] | 8 [5,12] | 15 [10,20] |
| pval | 0.21 | 0.82 | 0.12 | 0.49 | ||
| Age at study entry | ||||||
| <55 yr | 114 | 32 | 0 [0,0] | 4 [1,21] | 9 [2,32] | 2 [0,14] |
| 55–64 yr | 91 | 26 | 0 [0,0] | 6 [0,23] | 13 [1,38] | 2 [0,14] |
| >64 yr | 148 | 42 | 0 [0,1] | 7 [0,22] | 14 [2,30] | 2 [0,23] |
| pval | 0.98 | 0.89 | 0.62 | 0.85 | ||
| Cell differentiation | ||||||
| Well differentiated | 64 | 18 | 0 [0,1] | 10 [1,35] | 13 [3,32] | 9 [1,34] |
| Moderately differentiated | 201 | 57 | 0 [0,1] | 5 [0,17] | 13 [1,31] | 2 [0,16] |
| Poorly differentiated | 66 | 19 | 0 [0,0] | 4 [0,26] | 12 [2,35] | 1 [0,6] |
| Undifferentiated | 2 | 1 | 0 [0,0] | 30 [15,44] | 1 [1,1] | 1 [1,2] |
| Unknown | 20 | 6 | 0 [0,0] | 19 [2,36] | 19 [2,43] | 1 [1,14] |
| pval | 0.01 | 0.05 | 0.26 | 0 | ||
| Primary tumor site | ||||||
| Oral cavity | 156 | 44 | 0 [0,25] | 9 [1,23] | 14 [3,40] | 8 [0,28] |
| Oropharynx | 80 | 23 | 0 [0,0] | 5 [0,22] | 13 [2,31] | 1 [0,11] |
| Hypopharynx | 23 | 7 | 0 [0,0] | 9 [0,34] | 1 [0,25] | 1 [0,2] |
| Larynx | 73 | 21 | 0 [0,0] | 2 [0,12] | 5 [1,19] | 1 [0,6] |
| Other | 18 | 5 | 0 [0,23] | 4 [0,21] | 16 [9,21] | 8 [2,15] |
| Multiple | 3 | 1 | 0 [0,50] | 4 [2,17] | 2 [1,10] | 0 [0,26] |
| pval | 0.04 | 0.14 | 0.02 | 0 | ||
| Pathological tumor stage | ||||||
| T1 | 76 | 22 | 0 [0,1] | 5 [0,16] | 8 [2,23] | 2 [0,13] |
| T2 | 127 | 36 | 0 [0,1] | 5 [0,22] | 16 [3,39] | 2 [0,17] |
| T3 | 61 | 17 | 0 [0,0] | 5 [1,24] | 10 [1,36] | 1 [0,13] |
| T4 | 79 | 22 | 0 [0,0] | 9 [1,24] | 13 [3,32] | 2 [0,17] |
| TX or Tis | 9 | 3 | 0 [0,0] | 0 [0,6] | 1 [0,27] | 1 [0,19] |
| Unknown | 1 | 0 | 8 [8,8] | 31 [31,31] | 1 [1,1] | 1 [1,1] |
| pval | 0.02 | 0.23 | 0.1 | 0.84 | ||
| Pathological nodal stage | ||||||
| N0 or NX | 180 | 51 | 0 [0,1] | 5 [0,22] | 13 [2,30] | 3 [0,19] |
| N1–N3 | 172 | 49 | 0 [0,0] | 7 [0,22] | 13 [1,37] | 2 [0,16] |
| Unknown | 1 | 0 | 8 [8,8] | 31 [31,31] | 1 [1,1] | 1 [1,1] |
| pval | 0.24 | 0.41 | 0.55 | 0.11 | ||
| Clinical TNM stage | ||||||
| I | 45 | 13 | 0 [0,6] | 1 [0,15] | 7 [1,23] | 1 [0,13] |
| II | 72 | 20 | 0 [0,2] | 8 [0,24] | 16 [3,38] | 2 [0,16] |
| III | 89 | 25 | 0 [0,0] | 9 [1,23] | 13 [3,37] | 5 [0,24] |
| IV | 136 | 39 | 0 [0,0] | 5 [0,21] | 12 [1,32] | 1 [0,15] |
| Could not be assessed | 11 | 3 | 0 [0,13] | 4 [1,19] | 2 [1,16] | 1 [1,14] |
| pval | 0.17 | 0.24 | 0.16 | 0.15 | ||
| Treatment | ||||||
| Surgery Only | 110 | 31 | 0 [0,0] | 8 [1,22] | 14 [2,32] | 4 [1,22] |
| Surgery + postoperative therapy | 172 | 49 | 0 [0,0] | 4 [0,22] | 11 [2,30] | 2 [0,16] |
| Salvage surgery | 65 | 18 | 0 [0,1] | 7 [0,18] | 13 [1,38] | 1 [0,11] |
| Unknown | 6 | 2 | 0 [0,6] | 32 [8,81] | 24 [2,52] | 2 [1,13] |
| pval | 0.6 | 0.5 | 0.84 | 0.15 | ||
| Smoking history | ||||||
| Never smoked | 66 | 19 | 0 [0,31] | 10 [1,32] | 24 [6,41] | 7 [1,29] |
| Pipe or cigar only | 15 | 4 | 0 [0,3] | 5 [0,20] | 18 [1,30] | 0 [0,20] |
| Cigarette: <20 pack-yrs | 38 | 11 | 0 [0,2] | 4 [0,17] | 15 [3,29] | 5 [0,19] |
| Cigarette: 20–40 pack-yrs | 96 | 27 | 0 [0,0] | 7 [1,23] | 13 [2,29] | 2 [0,18] |
| Cigarette: >40 pack-yrs | 131 | 37 | 0 [0,0] | 4 [0,18] | 6 [1,24] | 1 [0,11] |
| Cigarette: unknown | 7 | 2 | 0 [0,2] | 12 [1,51] | 36 [4,72] | 7 [3,10] |
| pval | 0.01 | 0.15 | 0.01 | 0.02 | ||
| Average alcohol use | ||||||
| <10 oz/wk | 186 | 53 | 0 [0,1] | 8 [1,25] | 14 [2,32] | 3 [0,16] |
| 10–32 oz/wk | 70 | 20 | 0 [0,1] | 3 [0,16] | 13 [1,38] | 2 [0,17] |
| >32 oz/wk | 69 | 20 | 0 [0,0] | 5 [0,24] | 10 [1,29] | 1 [0,17] |
| Unknown | 28 | 8 | 0 [0,0] | 5 [2,18] | 13 [1,31] | 2 [0,16] |
| pval | 0.43 | 0.24 | 0.72 | 0.75 | ||
Figure 2.

Promoter hypermethylation for p16, DCC, EDNRB and KIF1A for tumors categorized by subject smoking history. Y-axis is the square root of methylation level.
Table III.
Linear models with methylation level as the response, and smoking, age, gende, HPV status and tumor site as the covariates. Smoking is coded as continuous variable: 1=”never smoked”, 2=”pipe or cigar only”, 3=”cigarette: <20 pack-years”, 4=”cigarette: 20–40 pack-years”, 5=”cigarette: >40 pack-years”. Seven subjects had unkown smoking history and were excluded from this analysis, leaving 346 subjects in this analysis. HPV status was imputed to be negative for samples from sites other than oropharynx.
| P16 | |||
| Estimate | 95% CI | P-value | |
| Intercept | 15.514 | (−0.254, 31.282) | 0.054 |
| Smoking | −1.818 | (−3.705, 0.069) | 0.059 |
| Age | 0.076 | (−0.135, 0.286) | 0.480 |
| Gender: female | |||
| male | 2.886 | (−3.472, 9.244) | 0.373 |
| HPV: no | |||
| yes | −5.022 | (−17.960, 7.916) | 0.446 |
| unkown | −8.062 | (−26.408, 10.284) | 0.388 |
| Site: oral cavity | |||
| oropharynx | −5.574 | (−17.038, 5.890) | 0.340 |
| hypopharynx | −13.374 | (−24.780, −1.967) | 0.022 |
| larynx | −10.779 | (−18.163, −3.394) | 0.004 |
| other | 2.039 | (−10.407, 14.485) | 0.747 |
| multiple | 17.271 | (−11.786, 46.329) | 0.243 |
| DCC | |||
| Estimate | 95% CI | P-value | |
| Intercept | 4.503 | (−12.572, 21.578) | 0.604 |
| Smoking | −1.787 | (−3.830, 0.257) | 0.086 |
| Age | 0.253 | (0.025, 0.481) | 0.030 |
| Gender: female | |||
| male | 7.006 | (0.122, 13.891) | 0.046 |
| HPV: no | |||
| yes | 14.791 | (0.781, 28.801) | 0.039 |
| unkown | 9.896 | (−9.971, 29.762) | 0.328 |
| Site: oral cavity | |||
| oropharynx | −5.745 | (−18.159, 6.670) | 0.363 |
| hypopharynx | 3.556 | (−8.796, 15.908) | 0.572 |
| larynx | −6.643 | (−14.640, 1.353) | 0.103 |
| other | 0.364 | (−13.113, 13.842) | 0.958 |
| multiple | −9.684 | (−41.150, 21.783) | 0.545 |
| EDNRB | |||
| Estimate | 95% CI | P-value | |
| Intercept | 13.489 | (−3.440, 30.418) | 0.118 |
| Smoking | −2.208 | (−4.234, −0.182) | 0.033 |
| Age | 0.287 | (0.061, 0.513) | 0.013 |
| Gender: female | |||
| male | 1.321 | (−5.504, 8.147) | 0.704 |
| HPV: no | |||
| yes | 0.717 | (−13.174, 14.607) | 0.919 |
| unkown | 1.678 | (−18.019, 21.375) | 0.867 |
| Site: oral cavity | |||
| oropharynx | −0.594 | (−12.902, 11.715) | 0.924 |
| hypopharynx | −3.062 | (−15.309, 9.185) | 0.623 |
| larynx | −7.457 | (−15.385, 0.472) | 0.065 |
| other | −0.095 | (−13.458, 13.267) | 0.989 |
| multiple | −18.193 | (−49.390, 13.005) | 0.252 |
| KIF1A | |||
| Estimate | 95% CI | P-value | |
| Intercept | 11.429 | (−2.753, 25.611) | 0.114 |
| Smoking | −2.452 | (−4.149, −0.755) | 0.005 |
| Age | 0.243 | (0.053, 0.432) | 0.012 |
| Gender: female | |||
| male | 1.504 | (−4.214, 7.222) | 0.605 |
| HPV: no | |||
| yes | 4.704 | (−6.933, 16.340) | 0.427 |
| unkown | −3.572 | (−20.072, 12.928) | 0.671 |
| site: oral cavity | |||
| oropharynx | −7.934 | (−18.245, 2.377) | 0.131 |
| hypopharynx | −12.198 | (−22.457, −1.938) | 0.020 |
| larynx | −10.254 | (−16.896, −3.613) | 0.003 |
| other | −8.892 | (−20.085, 2.302) | 0.119 |
| multiple | −1.839 | (−27.973, 24.296) | 0.890 |
The effect of each factor on overall survival was analyzed. Advanced nodal status was associated with a significantly increased risk of death (HR=3.2, p<0.001) (Table IV). Disruptive p53 mutation conveyed a 1.6 fold increased risk of death compared to wild-type (HR=1.632, p=0.069). There was no difference in survival seen for methylation of KIF1a, or EDNRB taken as individual events alone. (Figure 3) While methylation of DCC appeared to be significantly associated with decreased survival in univariate analysis (p=0.038, Figure 3), this effect was not independent of other molecular factors in the multivariate model (Table IV). However, methylation of p16 was associated with decreased survival for the overall cohort in multivariate analysis (HR=1.008, p=0.045). When methylation of these genes is combined with p53 mutational status, the presence of p16 methylation conveyed a significant incremental improvement in survival for tumors with a disruptive p53 mutation (p=0.019). The log hazard ratio for death decreased for subjects with tumors with disruptive TP53 mutation by 0.017 for each unit of increase in p16 methylation level. Taking p16 as a dichotomous value, the 26 cases with disruptive p53 and positive p16 methylation had survival similar to that of cases with non-disruptive mutation with any p16 status, while those with disruptive p53 mutation and 0 p16 methylation fared more poorly (Figure 4A + B). This protective effect was independent of smoking status in multivariate analysis. It also appears to be free of interaction with HPV status. The presence of HPV DNA was not associated with risk of death or methylation status in this cohort (Tables III and IV). The observed effect of p16 methylation on survival in our cohort is independent of HPV status in our multivariate model. The protective effect was observed predominantly in cases arising outside the oropharynx which do not contain HPV. Of the 14 oropharynx cases that have disruptive TP53, only 3 have p16 methylation levels greater than 0. Due to a paucity of tumors with both HPV DNA and disruptive p53 mutation, however, it cannot be rigorously demonstrated that p16 methylation remains protective in the presence of both disruptive p53 mutation and oncogenic HPV.
Table IV.
Risk of death associated with normalized methylation level, n=353, number of deaths=227. HPV status was imputed to be negative for samples from sites other than oropharynx.
| Hazard Ratio | 95% CI | P-value | |
|---|---|---|---|
| Site: oral cavity | |||
| oropharynx | 0.723 | (0.395, 1.323) | 0.292 |
| hypopharynx | 0.887 | (0.497, 1.584) | 0.685 |
| larynx | 1.059 | (0.699, 1.604) | 0.787 |
| other | 0.666 | (0.337, 1.315) | 0.241 |
| multiple | 3.847 | (1.070, 13.835) | 0.039 |
| PNCat: N0 or NX | |||
| N1–N3 | 3.200 | (2.246, 4.561) | 0.000 |
| Treatment: surgery only | |||
| surgery+radiation | 0.622 | (0.437, 0.886) | 0.009 |
| salvage surgery | 1.524 | (0.969, 2.398) | 0.068 |
| unknown | 0.677 | (0.197, 2.327) | 0.535 |
| Gender: female | |||
| male | 0.990 | (0.680, 1.442) | 0.958 |
| age | 1.023 | (1.010, 1.036) | 0.000 |
| Smoking: never | |||
| pipe or cigar only | 0.495 | (0.219, 1.122) | 0.092 |
| cigarette: <20 pack-yrs | 0.796 | (0.429, 1.476) | 0.469 |
| cigarette: 20–40 pack-yrs | 1.036 | (0.617, 1.740) | 0.894 |
| cigarette: > 40 pack-yrs | 1.120 | (0.684, 1.836) | 0.652 |
| unknown | 0.750 | (0.218, 2.575) | 0.647 |
| Alcohol: < 10 oz/wk | |||
| 10–32 oz/wk | 1.973 | (1.326, 2.937) | 0.001 |
| >32 oz/wk | 1.280 | (0.838, 1.955) | 0.254 |
| unknown | 1.194 | (0.634, 2.248) | 0.583 |
| P53: wild-type | |||
| non-disruptive mutant | 1.622 | (1.021, 2.578) | 0.041 |
| disruptive mutant | 1.632 | (0.962, 2.770) | 0.069 |
| p16 | 1.008 | (1.000, 1.016) | 0.045 |
| dcc | 1.005 | (0.997, 1.013) | 0.249 |
| ednrb | 1.002 | (0.993, 1.011) | 0.632 |
| kif | 0.997 | (0.984, 1.010) | 0.642 |
| HPV: no | |||
| yes | 0.499 | (0.212, 1.176) | 0.112 |
| unknown | 2.312 | (0.768, 6.959) | 0.136 |
| P53 wild-type | |||
| p53 non-disruptive: p16 | 0.998 | (0.986, 1.011) | 0.813 |
| p53 disruptive: p16 | 0.983 | (0.968, 0.997) | 0.019 |
| p53 non-disruptive: dcc | 0.999 | (0.986, 1.012) | 0.875 |
| p53 disruptive: dcc | 1.003 | (0.989, 1.018) | 0.634 |
| p53n on-disruptive: ednrb | 0.987 | (0.974, 1.000) | 0.053 |
| p53 disruptive: ednrb | 0.999 | (0.979, 1.019) | 0.921 |
| p53 non-disruptive: kif | 1.009 | (0.991, 1.027) | 0.338 |
| p53 disruptive: kif | 1.005 | (0.984, 1.027) | 0.623 |
| p53 non-disruptive: hpv yes | 2.300 | (0.894, 5.919) | 0.084 |
| p53 disruptive: hpv yes | 2.196 | (0.648, 7.445) | 0.207 |
| p53 non-disruptive: hpv unknown | 0.738 | (0.074, 7.369) | 0.796 |
| p53 disruptive: hpv unknown | 0.191 | (0.021, 1.728) | 0.141 |
Figure 3.
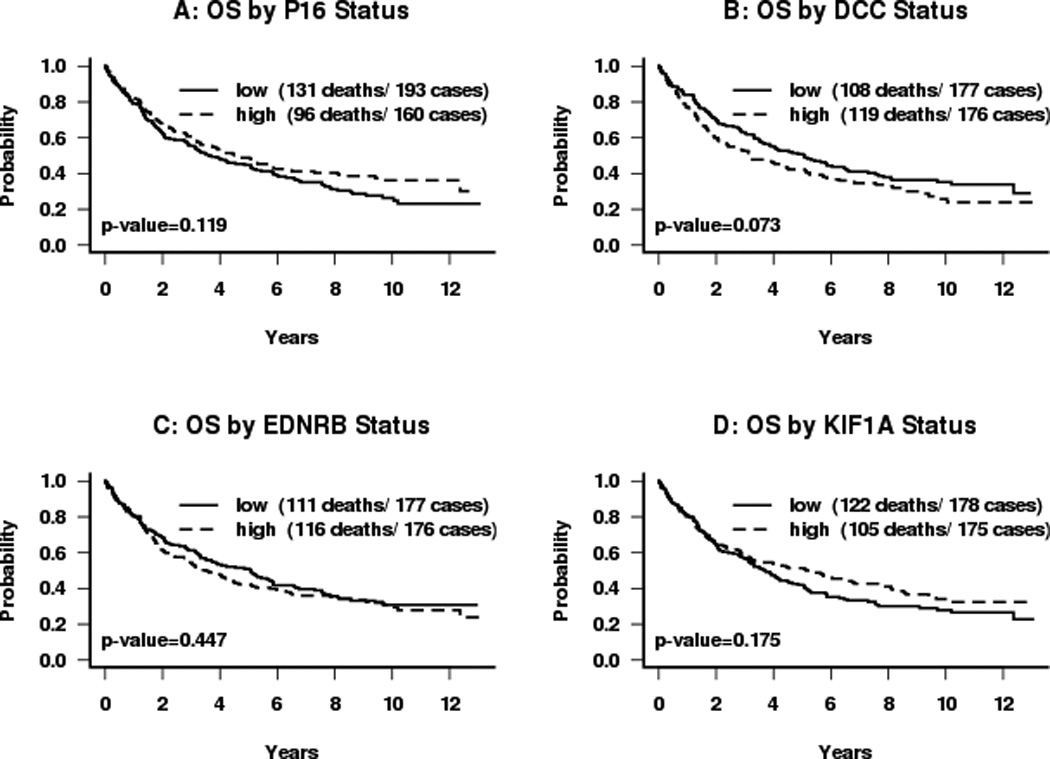
Kaplan-Meier survival curves for subjects categorized by dichotomous promoter hypermethylation status: (A) p16; (B) DCC; (C) EDNRB; (D) KIF1A. Median normalized methylation levels were used as cut-offs. P-values are obtained from the log-rank test.
Figure 4.
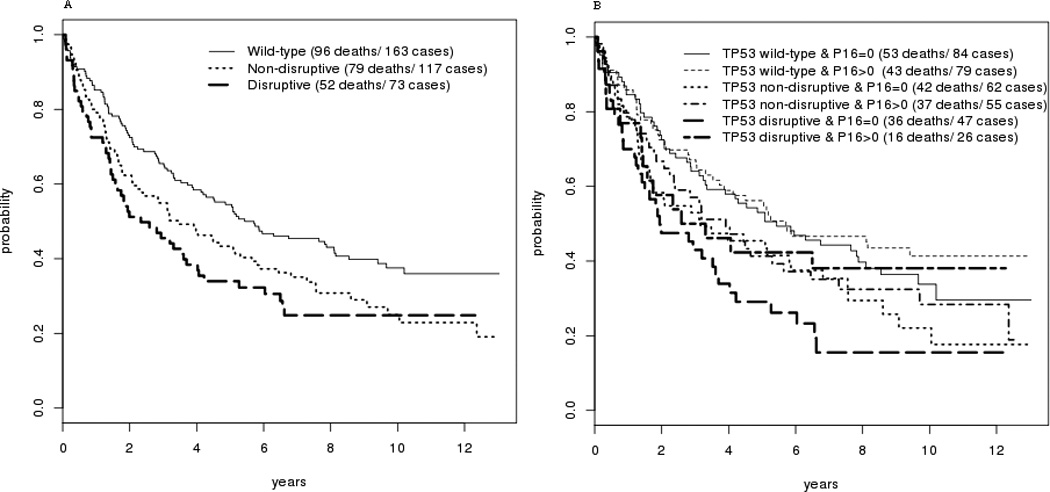
A: Kaplan-Meier Survival curve for subjects categorized by TP53 mutational status
B: Survival for subjects categorized by both TP53 mutational status and p16 methylation status.
DISCUSSION
Tumor-specific hypermethylation of the promoter region of genes is a well-recognized mechanism of abrogation of gene function that may contribute to the malignant transformation and phenotype of cancer. When hypermethylation of the promoter is present, the gene is not transcribed, reducing or eliminating the function of tumor suppressor genes such as p16. As a result, methylation of genes involved in key pathways that contribute to tumorigenesis may affect tumor behavior and prognosis. The potential for prognostic implication of methylation of individual genes has been explored for a variety of human cancers including head and neck squamous cell carcinoma (HNC). The power and reliability of studies exploring prognostic effect of tumor markers is dependent on sample size, length of follow-up and integrity of clinical outcome data. We have looked for association between clinical outcome and promoter hypermethylation of four tumor-specific target genes (p16, DCC, EDNRB and KIF1A) in a large, prospectively collected cohort of HNSCC from the multi-institutional cooperative group study ECOG 4393/RTOG 9614. This cohort is among the largest collection of HNC tumors ever evaluated for correlation between molecular alterations and clinical outcome. In the current study, the effect of methylation of each candidate gene was considered individually as well as in combination with the others and with mutation status of TP53 and the presence or absence of HPV DNA. Mutation status of TP53 in this cohort has previously been shown to have a significant correlation with overall survival. In particular, patients with tumors with disruptive mutation of TP53 have a greater than two-fold rate of death compared those who have HNC with either wild-type or nondisruptive TP53 mutation.(13)
It is increasingly apparent that the functional disruptions underlying tumorigenesis and the cancer phenotype are complex, affecting an interwoven system of key pathways that control functions such as apoptosis, cell growth regulation, signal transduction and differentiation. There are numerous gene products involved in each of these pathways, rendering multiple candidate treatment targets and detection/prognostic markers. Our results reflect the complexity and interdependence of the many genes involved in two ways. The methylation status of p16 was found to affect the outcome of patients particularly the subsets defined by the concomitant status of the TP53 gene. In particular, prognosis of the group of patients with tumors containing disruptive TP53 mutation, known to have a poor clinical outcome, was further refined by simultaneously considering the methylation status of p16.
Most published reports of the prognostic association of p16 methylation in cancer indicate a poorer outcome for tumors with this epigenetic event, in accordance with our findings for the overall cohort. A significant survival disadvantage was observed for p16 hypermethylated colorectal cancers (n=84) of which 28.6% were affected. Five year survival probability was 25% for methylated tumors and 72% for non-methylated tumors.(27) A larger study of 212 colorectal carcinoma cases showed an association between high p16 methylation and large tumor size, more frequent recurrence and shorter cancer-related survival (HR=3.38 p<0.001) (28) These results were not confirmed by a more recent cohort study and literature review indicating that promoter methylation of CDKN2A was not associated with colorectal-specific mortality. (29) In a study of 44 Stage 1A NSCLC cases, multivariate analysis demonstrated reduced relapse free survival associated with p16 methylation p=0.016.(30) However Wif-1 methylation was more strongly associated with overall survival in the NSCLC cohort, demonstrating the complexity of analysis of multiple parameters in a small study population. In another report, p16 methylation was again associated with poor prognosis of NSCLC in a study of 119 cases, showing a reduced 5 year survival rate for resectable cancers, particularly those with early stage. (31) Methylation of p16 has also been implicated as a poor prognostic marker for ovarian carcinoma as indicated in a study of 249 cases. Among the 100 cases with p16 methylation, there was a significantly higher risk of disease progression.(32)
Methylation of p16 has also been associated with poor outcome in HNC in several published reports. Among 52 oral squamous cell carcinoma cases, hypermethylated p16 was associated with higher rates of lymph node metastasis and distant metastasis and with shortened disease-free survival. However, these results were only seen in certain subsets of patients (older or younger) in this small study and were not confirmed with multivariate analysis.(33) In another report of 78 cases with oral epithelial dysplasia, the rate of progression to oral cancer in lesions with p16 methylation was significantly higher than when p16 was not methylated. This observed effect was stronger for older patients and moderate dysplasia cases, again demonstrating the limitation of the observation in that it applied only to very small subsets. (34)
In our cohort of HNC patients treated with surgery with or without post-operative radiation, p16 methylation status alone was found to be associated with a significant difference in survival when analyzing the entire group. However, when considering subsets of patients, the group with the worst prognosis based on p53 mutation status (disruptive p53 mutation), a high level of p16 methylation was associated with a better outcome. Individuals with disruptive p53 mutation who also had high p16 methylation had a rate of survival similar to those with non-disruptive mutation (Figure 4B). The cohort utilized for this study was of a size and follow-up duration to offer sufficient power and reliability. Compared to published results of smaller cohorts, our ability to perform multivariate analysis of subsets of the cohort is enhanced. The mechanism by which p16 methylation portends an improved prognosis for tumors with TP53 disruptive mutation, a group with a poor prognosis, has not been investigated. However, it is well documented that most HNC have some disruption of cell cycle control at the G1 checkpoint, including deletion or mutation of p16, presence of HPV16 E7, or rarely Rb mutation (35). The relationship with smoking and lower p16 methylation levels suggests that exposure to carcinogens likely results in a higher frequency of mutation and deletion of p16 and less likelihood of endogenous transformation through p16 methylation. The observed “protective” effect may reflect a better outcome from lower carcinogen exposure and a less damaged cancer genome. It may also be that p16 methylation represents partial abrogation of cell cycle control, sufficient for tumorigenesis but with less virulent tumor behavior as a result.
The prognostic implication of methylation of other individual genes has been reported in several studies of limited size. Methylation of DCC was correlated with aggressive phenotype for HNC in a study of 96 tumors with increased rate of bone invasion, invasive growth pattern and reduced survival (p=0.050). (36) Intriguingly, in this study, methylation of p14ARF was associated with a good prognosis (HR =0.30 for methylated tumors p=0.021), perhaps implicating a mechanism similar to the protective effect of p16 methylation in our cohort. Our group recently examined promoter hypermethylation of a larger panel of genes in DNA collected in salivary rinses from head and neck cancer patients as a biomarker for outcome. (20) Overall survival in a group of 61 HNC patients was significantly shorter if pre-treatment saliva was found to contain tumor-specific hypermethylated DNA of any of the target genes (DAPK; DCC; MINT-31; TIMP-3, p16, MGMT, CCNA1) p=0.015. Hypermethylated DNA in saliva was found to independently predict local recurrence in a multivariate analysis (p=0.010). However, in a subsequent study of 97 patients, only methylation of TIMP-3 in pretreatment salivary rinses remained predictive of local-recurrence (15). The inconsistency of these results may be due to the relatively small sample size as well as the indirect approach of sampling tumor-DNA that has been released into saliva of cancer patients.
In contrast to the result for p16 methylation, there was no association in the study cohort between survival and methylation of KIF1A, EDNRB or DCC alone. The presence of oncogenic HPV DNA in HNC has been reported to be a good prognostic indicator in numerous series (37–39). However, HPV is a factor only in the oropharynx since the presence of HPV DNA in tumors from other sites is vanishingly rare. HPV status did not appear to influence the clinical effect of promoter hypermethylation in this series.
Translational Relevance.
Clinical behavior of cancer appears to be attributable to the accumulation of molecular alterations underlying malignant transformation. Evaluation of only a few individual molecular events (TP53 mutation, presence of HPV16) has been shown to correlate with survival of head and neck cancer (HNC) in studies with adequate cohort size to power rigorous statistical evaluation. Clinical correlates for putative suppressor gene promoter hypermethylation have been evaluated in a large cooperative group HNC cohort. The results help refine established prognostic groups in that while p16 methylation was associated with a small negative prognostic impact on the overall cohort, a subset of cases with disruptive TP53 mutation is demonstrated to have improved prognosis when methylation of p16 is also present. These findings may help direct multimodality therapeutic options for HNC.
Acknowledgments
Grant support: Supported by grants from National Institute of Health and the National Institute of Dental and Craniofacial Research (R01 DE013152-11).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflict of interest relevant to this article was reported.
References
- 1.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–1157. doi: 10.1126/science.1206923. PMCID: 3162986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. PMCID: 3415217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- 4.Ha PK, Califano JA. Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. Lancet Oncol. 2006;7(1):77–82. doi: 10.1016/S1470-2045(05)70540-4. [DOI] [PubMed] [Google Scholar]
- 5.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60(6):376–392. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- 7.Aggerholm A, Guldberg P, Hokland M, Hokland P. Extensive intra- and interindividual heterogeneity of p15INK4B methylation in acute myeloid leukemia. Cancer Res. 1999;59(2):436–441. [PubMed] [Google Scholar]
- 8.Harbeck N, Nimmrich I, Hartmann A, Ross JS, Cufer T, Grutzmann R, et al. Multicenter study using paraffin-embedded tumor tissue testing PITX2 DNA methylation as a marker for outcome prediction in tamoxifen-treated, node-negative breast cancer patients. J Clin Oncol. 2008;26(31):5036–5042. doi: 10.1200/JCO.2007.14.1697. [DOI] [PubMed] [Google Scholar]
- 9.Weiss G, Cottrell S, Distler J, Schatz P, Kristiansen G, Ittmann M, et al. DNA methylation of the PITX2 gene promoter region is a strong independent prognostic marker of biochemical recurrence in patients with prostate cancer after radical prostatectomy. J Urol. 2009;181(4):1678–1685. doi: 10.1016/j.juro.2008.11.120. [DOI] [PubMed] [Google Scholar]
- 10.Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358(11):1118–1128. doi: 10.1056/NEJMoa0706550. [DOI] [PubMed] [Google Scholar]
- 11.Garcia MP, Garcia-Garcia A. Epigenome and DNA methylation in oral squamous cell carcinoma. Methods Mol Biol. 2012;863:207–219. doi: 10.1007/978-1-61779-612-8_12. [DOI] [PubMed] [Google Scholar]
- 12.Mikeska T, Bock C, Do H, Dobrovic A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Rev Mol Diagn. 2012;12(5):473–487. doi: 10.1586/erm.12.45. [DOI] [PubMed] [Google Scholar]
- 13.Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357(25):2552–2561. doi: 10.1056/NEJMoa073770. PMCID: 2263014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821. PMCID: 38513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun W, Zaboli D, Wang H, Liu Y, Arnaoutakis D, Khan T, et al. Detection of TIMP3 promoter hypermethylation in salivary rinse as an independent predictor of local recurrence-free survival in head and neck cancer. Clin Cancer Res. 2012;18(4):1082–1091. doi: 10.1158/1078-0432.CCR-11-2392. PMCID: 3288549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harden SV, Tokumaru Y, Westra WH, Goodman S, Ahrendt SA, Yang SC, et al. Gene promoter hypermethylation in tumors and lymph nodes of stage I lung cancer patients. Clin Cancer Res. 2003;9(4):1370–1375. [PubMed] [Google Scholar]
- 17.Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000;60(4):892–895. [PubMed] [Google Scholar]
- 18.Demokan S, Chang X, Chuang A, Mydlarz WK, Kaur J, Huang P, et al. KIF1A and EDNRB are differentially methylated in primary HNSCC and salivary rinses. Int J Cancer. 2010;127(10):2351–2359. doi: 10.1002/ijc.25248. PMCID: 2946472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carvalho AL, Chuang A, Jiang WW, Lee J, Begum S, Poeta L, et al. Deleted in colorectal cancer is a putative conditional tumor-suppressor gene inactivated by promoter hypermethylation in head and neck squamous cell carcinoma. Cancer Res. 2006;66(19):9401–9407. doi: 10.1158/0008-5472.CAN-06-1073. [DOI] [PubMed] [Google Scholar]
- 20.Carvalho AL, Henrique R, Jeronimo C, Nayak CS, Reddy AN, Hoque MO, et al. Detection of promoter hypermethylation in salivary rinses as a biomarker for head and neck squamous cell carcinoma surveillance. Clin Cancer Res. 2011;17(14):4782–4789. doi: 10.1158/1078-0432.CCR-11-0324. PMCID: 3215270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho AL, Jeronimo C, Kim MM, Henrique R, Zhang Z, Hoque MO, et al. Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14(1):97–107. doi: 10.1158/1078-0432.CCR-07-0722. [DOI] [PubMed] [Google Scholar]
- 22.Begum S, Brait M, Dasgupta S, Ostrow KL, Zahurak M, Carvalho AL, et al. An epigenetic marker panel for detection of lung cancer using cell-free serum DNA. Clin Cancer Res. 2011;17(13):4494–4503. doi: 10.1158/1078-0432.CCR-10-3436. PMCID: 3131425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2008;14(2):366–369. doi: 10.1158/1078-0432.CCR-07-1402. [DOI] [PubMed] [Google Scholar]
- 24.Zhao M, Rosenbaum E, Carvalho AL, Koch W, Jiang W, Sidransky D, et al. Feasibility of quantitative PCR-based saliva rinse screening of HPV for head and neck cancer. Int J Cancer. 2005;117(4):605–610. doi: 10.1002/ijc.21216. [DOI] [PubMed] [Google Scholar]
- 25.Kaplan ELMP. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 26.DR C. Regression models and lifetables. J R Stat Soc [B] 1972;34:187–220. [Google Scholar]
- 27.Liang JT, Chang KJ, Chen JC, Lee CC, Cheng YM, Hsu HC, et al. Hypermethylation of the p16 gene in sporadic T3N0M0 stage colorectal cancers: association with DNA replication error and shorter survival. Oncology. 1999;57(2):149–156. doi: 10.1159/000012023. [DOI] [PubMed] [Google Scholar]
- 28.Mitomi H, Fukui N, Tanaka N, Kanazawa H, Saito T, Matsuoka T, et al. Aberrant p16((INK4a)) methylation is a frequent event in colorectal cancers: prognostic value and relation to mRNA expression and immunoreactivity. J Cancer Res Clin Oncol. 2010;136(2):323–331. doi: 10.1007/s00432-009-0688-z. [DOI] [PubMed] [Google Scholar]
- 29.Shima K, Morikawa T, Baba Y, Nosho K, Suzuki M, Yamauchi M, et al. MGMT promoter methylation, loss of expression and prognosis in 855 colorectal cancers. Cancer Causes Control. 2011;22(2):301–309. doi: 10.1007/s10552-010-9698-z. PMCID: 3278857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshino M, Suzuki M, Tian L, Moriya Y, Hoshino H, Okamoto T, et al. Promoter hypermethylation of the p16 and Wif-1 genes as an independent prognostic marker in stage IA non-small cell lung cancers. Int J Oncol. 2009;35(5):1201–1209. doi: 10.3892/ijo_00000437. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Lee JJ, Wang L, Liu DD, Lu C, Fan YH, et al. Value of p16INK4a and RASSF1A promoter hypermethylation in prognosis of patients with resectable non-small cell lung cancer. Clin Cancer Res. 2004;10(18 Pt 1):6119–6125. doi: 10.1158/1078-0432.CCR-04-0652. [DOI] [PubMed] [Google Scholar]
- 32.Katsaros D, Cho W, Singal R, Fracchioli S, Rigault De La Longrais IA, Arisio R, et al. Methylation of tumor suppressor gene p16 and prognosis of epithelial ovarian cancer. Gynecol Oncol. 2004;94(3):685–692. doi: 10.1016/j.ygyno.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 33.Su PF, Huang WL, Wu HT, Wu CH, Liu TY, Kao SY. p16(INK4A) promoter hypermethylation is associated with invasiveness and prognosis of oral squamous cell carcinoma in an age-dependent manner. Oral Oncol. 2010;46(10):734–739. doi: 10.1016/j.oraloncology.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Cao J, Zhou J, Gao Y, Gu L, Meng H, Liu H, et al. Methylation of p16 CpG island associated with malignant progression of oral epithelial dysplasia: a prospective cohort study. Clin Cancer Res. 2009;15(16):5178–5183. doi: 10.1158/1078-0432.CCR-09-0580. [DOI] [PubMed] [Google Scholar]
- 35.Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res. 1996;56(16):3630–3633. [PubMed] [Google Scholar]
- 36.Ogi K, Toyota M, Ohe-Toyota M, Tanaka N, Noguchi M, Sonoda T, et al. Aberrant methylation of multiple genes and clinicopathological features in oral squamous cell carcinoma. Clin Cancer Res. 2002;8(10):3164–3171. [PubMed] [Google Scholar]
- 37.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92(9):709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 38.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. PMCID: 2943767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100(4):261–269. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]