

Abstract
Background:
c-Met mutations play a critical role in the development and progression of primary tumors and metastases. Activation of the HGF/SF-c-Met pathway determines a poor prognosis in non-small-cell and small-cell lung cancer (SCLC) patients. Missense mutations of c-Met have been identified in SCLC patients located in the juxtamembrane (JM) and in the Sema domain. To determine the role of the c-Met pathway in SCLC, we have investigated the presence of c-Met mutations in SCLC patients.
Patients and methods:
Forty-four tumor tissue samples from SCLC patients were obtained with bronchoscopy before beginning treatment. Analysis of c-Met mutations was performed in exon 2 and exon 14.
Results:
Of the 44 patients included in this study, 23 were classified as limited disease and were treated with sequential or concurrent chemotherapy and thoracic radiotherapy. Twenty-one patients with extensive disease received chemotherapy alone, the majority with cisplatin or carboplatin plus etoposide. The median survival was 14 months (95% CI: 9.4 to 18.5 months) and the 2- and 5-year survival rates were 24% and 15%, respectively. Previously identified missense mutations E168D, R988C and T1010I in c-Met were not found in our study. However, novel mutations were identified, including T995I in the juxtamembrane domain (T995I) and a mutation which does not change amino acid in codon 178 in the Sema domain.
Conclusion:
In SCLC patients, the presence of mutations in c-Met gene is a rare event. Other genetic alterations involved in the HGF/SF-c-Met pathway should be assessed to define the role of this signaling pathway in SCLC.
Despite efforts against smoking, lung cancer remains the leading cause of cancer deaths in Western countries. Small-cell lung cancer (SCLC) accounts for approximately 20% of all lung cancers. SCLC is characterized by its rapid doubling time and early development of widespread metastases (Elias, 1997). SCLC is typically staged according to a two-stage system, which was developed by the Veterans Administration Lung Cancer Study Group, as limited disease (LD) or extensive disease (ED). Patients with LD have involvement restricted to one hemithorax and its regional lymph nodes within a single radiation port; all other tumors are characterized as ED. At presentation, 60% to 70% of all SCLC patients will have ED (Murren JR, 2005).
The aggressive course of SCLC determines that the median survival of patients receiving only supportive care is 12 weeks for those with LD and 5 weeks for those with ED (Zelen, 1973). Combination chemotherapy has become the mainstream of therapy for SCLC. In patients with ED, chemotherapy produces response rates of 50% to 60% and median survival of 7 to 11 months. However, despite initial sensitivity to chemotherapy, less than 3% of patients are alive at 3 years (Albain, 1990). In patients with LD, the combination of chemotherapy plus radiotherapy achieves a response rate over 80% with a median survival around 20 months, whereas the 5-year survival rate is 15% to 25% in the recent phase III trials (Takada, 2002; Turrisi, 1999). Since the 1980s, etoposide in combination with cisplatin or carboplatin has been the standard treatment in patients with LD or ED, although other regimens like anthracycline-based combinations are equally effective (Roth, 1992). In a recent phase III trial, etoposide plus cisplatin (EP) demonstrated better results than cyclophosphamide, epirubicin and vincristine in patients with LD, whereas in ED the efficacy of both regimens was similar (Sundstrom, 2002). In fact, relatively little progress has been made in SCLC in the past two decades. The most important advances in patients with LD have been obtained by integrating chemotherapy with thoracic radiotherapy (TRT). Two meta-analyses demonstrated a 14% improvement in median survival by adding TRT to chemotherapy (Pignon, 1992; Warde, 1992). More recently, several randomized studies suggested a benefit for concurrent chemoradiotherapy compared to sequential treatment (Takada, 2002; Murray, 1993). In addition, the use of prophylactic cranial irradiation in LD patients with complete response after chemo-radiotherapy appears to provide a significant improvement in 3-year survival (Auperin, 1999). In contrast, the prognosis of patients with ED has been improved only minimally. Data from the Surveillance, Epidemiology and End Results (SEER) database showed a modest improvement in median survival from 7 months to 8.9 months in these patients from the period 1972–1994 (Chute, 1999). Thus, new active therapies to improve the prognosis for SCLC patients are required, and agents like taxanes, gemcitabine, topotecan, and irinotecan have demonstrated significant single agent activity. However, the impact of these agents in the prognosis of SCLC patients has not been established in randomized trials. For example, the addition of paclitaxel to EP for ED SCLC increased toxicity without improving survival (Niell, 2005). The phase III study carried out by the Japanese Cooperative Oncology Group was the only trial to demonstrate a significant improvement in survival over the EP regimen in ED SCLC patients. In this study, the cisplatin/irinotecan (IP) combination showed a significant increase in median survival (12.8 vs 9.4 months) and 2-year survival rate (19.5% vs 5.2%) compared to EP regimen (Noda, 2002). However, in a recent published randomized trial, the IP regimen has not demonstrated a benefit compared to standard EP. In this study, the median survival was 9.3 months in patients treated with IP and 10.2 months in patients treated with EP, whereas the 2-year survival was 8% in both arms (Hanna, 2006).
Thus, the suboptimal results obtained with chemotherapy in SCLC patients, emphasize the need to find new potential therapeutic targets based on genetic profiles.
Several abnormalities in both oncogenes and tumor suppressor genes are present in SCLC, as well as MYC DNA amplification, p53 mutation, Rb inactivation, and loss of alleles on chromosome 3p. Tyrosine kinase receptors (RTKs) are key molecules in normal cellular differentiation. RTKs have been implicated in the etiology of multiple tumors, and they may be important therapeutic targets. RTKs play an important role in lung cancer oncogenesis, especially in SCLC, including c-Met. Previous studies have shown that HGF/c-Met signaling is functional and important in SCLC.
The c-Met receptor is located on chromosome 7, band 7q21–7q31, and spans more than 120 kb in length, consisting of 21 exons (Seki, 1991). The c-Met oncogene encodes a transmembrane tyrosine kinase receptor (Met) for the hepatocyte growth factor (HGF), also known as scatter factor (SF). The extracellular portion includes a conserved “Sema” domain and the intracellular portion presents three functional domains: a juxtamembrane (JM) domain, the tyrosine kinase catalytic domain, and a C-terminal tail. (Fig. 1). The Sema domain is necessary for the c-Met receptor binding to hepatocyte growth factor (HGF), dimerization and activation, while the JM domain is crucial for catalytic functions.
Figure 1.
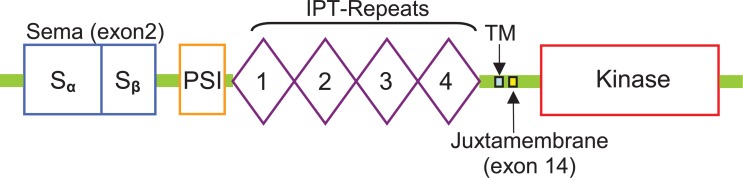
Functional domains of the Met receptor.
Abbreviations: Sema domain (semaphorin like), PSI domain (found in plexins, semaphorins and integrins), IPT repeat domains ( found in immunoglobulin-like regions, plexins and transcription factors),TM transmembrane and Tyrosine kinase domain (located intracellularly).
Several studies suggest that activation of the HGF/SF-c-Met signaling pathway mediates a diversity of biological functions, for instance cell proliferation, survival, scattering, cell adhesion and motility, induction of cell polarity, tissue regeneration, invasion, tumor metastasis and angiogenesis (Rubin, 1993).
The first direct evidence linking c-Met directly to human oncogenesis was reported in 1997, when Schmidt et al. (Schmidt, 1997) identified missense mutations located in the tyrosine kinase domain of c-Met gene in the germline of affected members of hereditary papillary renal carcinoma (HPRC) families and in a subset of sporadic papillary renal carcinomas.
Aberrant Met activation, by binding with its high-affinity ligand HGF/SF or by autophosphorylation as a result of c-Met mutations, provokes a cytoplasmic signals cascade, resulting in activation of multiple signal transducers (Grb2, Gab1, PI3k, Stats, ERK ½, FAK). This activated signaling pathway plays a critical role in the development and progression of primary tumors and metastases (Jeffers, 1996). High levels of c-Met expression have been correlated with the metastatic spread of tumors and poor prognosis in patients with breast carcinoma, gastric cancer, endometrial carcinoma, hepatocellular carcinoma, colorectal cancer, renal carcinoma and bladder carcinoma (Di Renzo, 1995; Natali, 1996; Ueki, 1997; Ghoussoub, 1998; Taniguchi, 1998; Wagatsuma, 1998; Camp, 1999; Hida, 1999; Nakajima, 1999). Likewise, activation of the HGF/SF-c-Met pathway has been associated with shorter survival in non-small-cell lung cancer (NSCLC) and SCLC patients (Harvey, 1996; Siegfried, 1998; Bharti, 2004; Masuya, 2004). Many missense mutations of c-Met, which mainly are present in the tyrosine kinase domain, are involved in invasion and metastasis of tumor cells and have been identified in various solid tumors such as hereditary papillary renal carcinomas (Schmidt, 1998), childhood hepatocellular carcinoma (Park, 1999) and in a small percentage of squamous cell carcinomas of the head and neck (Di Renzo, 2000). Interestingly, c-Met mutations were more frequently identified in human carcinoma metastases compared to primary tumors, suggesting a role for the activation of c-Met in tumor progression (Lorenzato, 2002).
A growing body of evidence suggests a potential role for the HGF/SF-c-Met signaling pathway in establishing new effective therapies. Rasola et al. showed that HGF sensitizes ovarian cancer cells treated with paclitaxel or cisplatin to apoptosis (Rasola, 2004). Furthermore, small molecule inhibitors such as SU11274, specifically targeting c-Met can be effective in patients carrying mutations in this gene (Ma, 2005). Finally, the geldan-amycin family of anisomycin antibiotics have been shown to downregulate c-Met expression, reducing phosphorylation of signaling proteins, cell motility and viability of SCLC cells (Maulik, 2002). These findings suggest that tumors with aberrant HGF/SF-c-Met signaling could be considered for gelda-mycin-based therapy.
In the present study, we have examined potential mutations of c-Met in the JM domain (R988C and T1010I) and the Sema domain (E168D) in SCLC tumor samples.
Materials and Methods
Tumor samples and mutation analysis
The study included a total of 44 paraffin-embedded tumor samples from SCLC patients, obtained by bronchoscopy. Informed consent was obtained from all patients prior to analysis. Genomic DNA was extracted using phenol-chloroform extraction method.
For mutational analysis, the coding regions of exon 2 (Sema domain) and exon 14 (JM domain) of c-Met protooncogene (GenBank accession number NM_000245; http://www.ncbi.nlm.nih.gov) was performed by nested PCR (Polymerase chain reaction) and direct sequencing. Exons were amplified individually. In the sequencing analysis we used the same primers as were used for the nested PCR. Table1 lists the primers used.
50μl PCR reaction mixture consisted of 1X PCR Buffer (16.6 mM [NH4]2 SO4, 67 mM Tris-HCl pH 8.8, 0.01% Tween-20),1mM MgCl2, 0,1mM each of four deoxynucleotide triphosphates (dNTPs) (Roche Diagnostics, Germany), 0,5μM of each primer, one unit of HotStarTaq DNA Polymerase (Qiagen, Hilden, Germany). The PCR reaction was carried out as follows: initial denaturation, 5 min at 95C then 20 cycles of 95C for 30sec, each Tm for 30sec, 72C for 30 sec.
3 microliters of PCR product from the first reaction mixture was used as template for the second PCR. The conditions of the nested PCR were the same than the first PCR with 35 cycles.
PCR products were electrophoresed on a 2% agarose gel with ethidiun bromide (2mg/ml) and visualized with the Gelprinter Plus Transilluminator (Biorad, Hercules, CA, U.S.).
Samples were sequenced using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, U.S.) according to the manufacturer’s manual, and analyzed on a 3130 Genetic Analyzer. All PCR products were sequenced in both directions forward and reverse. The sequences were aligned with the published genomic sequence using the DNA Sequencing Analysis Software version 5.1.1 (Applied Biosystem).
Results
Forty-four SCLC patients treated in our institution between October 1996 and September 2005 were enrolled onto the study. Patient characteristics are detailed in Table 2. Thirty-nine patients were male and five were women. The median age was 63 years (range, 42–79) and 65% of patients had a Karnofsky performance score of 70 or better. Twenty three (52%) patients were classified as LD and twenty-one (48%) patients as ED. The 23 (52%) patients with LD received chemotherapy and TRT; in 12 patients treatments were given sequentially and in 11 patients chemoradiotherapy was given concurrently. The 21 patients with ED were treated with chemotherapy alone, the majority with cisplatin or carboplatin plus etoposide. Fourteen (32%) patients who achieved a complete response after chemoradiotherapy received prophylactic cranial irradiation and 10 (23%) patients required palliative radiotherapy. Sixty (36%) patients achieved a complete response and 18 (41%) patients achieved a partial response for an overall response rate of 77%. The median progression-free survival time was 10 months (95% confidence interval [CI], 8.1–11.9 months). The median survival was 14 months (95% CI, 9.4–18.5 months). The 2- and 5-year overall survival rates were 24% and 14%, respectively (Fig. 2). At present, 35 of the 44 patients included in the study have died. The median survival for patients with LD was 18 months (95% CI, 10.9–25 months) versus 13 months (95% CI, 7.3–18.6 months) for those with ED.
Table 2.
Patient baseline characteristics.
| Characteristic |
Patients (N= 44)
|
|
|---|---|---|
| No | % | |
| Sex | ||
| Male | 39 | 89 |
| Female | 5 | 11 |
| Age, years | ||
| Median | 63 | |
| Range | 42–79 | |
| ≥ 65 | 21 | 47 |
| Performance status | ||
| 0 / 1 | 19 | 43 |
| 2 | 10 | 22 |
| 3–4 | 13 | 29 |
| Unknown | 2 | 6 |
| Stage | ||
| Limited-stage disease | 23 | 52 |
| Extensive-stage disease | 21 | 48 |
| Lactate deshydrogenase | ||
| Normal levels | 28 | 64 |
| High levels | 14 | 32 |
| Unknown | 2 | 5 |
| Treatment | ||
| Chemoradiotherapy | 23 | 52 |
| Sequential | 12 | 52 |
| Concurrent | 11 | 48 |
| Chemotherapy alone | 21 | 48 |
| Cisplatin / Etoposide | 11 | 52 |
| Carboplatin / Etoposide | 6 | 28 |
| Etoposide | 2 | 10 |
| Others | 2 | 10 |
Figure 2.

Kaplan-Meier plot of survival. The median survival was 14 months (95% CI, 9.4 to 18.5 months).
We performed a mutation analysis of the Sema domain (E168D) and JM domain (R988C and T1010I), as reported previously (Ma, 2003) in the SCLC tumor samples. By sequencing PCR products corresponding to exon 2 (Sema domain), we studied 41 patients, and in exon 14 (JM domain) we analyzed 38 patients. Both exons were examined in 35 patients. We screened a total of 44 paraffin-embedded tumor samples from SCLC patients (Table 2). We did not find any of the mutations previously reported (Fig. 3). However, in two cases, we identified an AGC→AGT substitution in codon 178 of the Sema domain that did not give rise to any amino acid change; this could be a polymorphism but we were unable to test this as it was not possible to obtain normal DNA. In addition, we observed one ACT→ATT substitution in codon 995 of the JM domain that converted Thr to Ile (T995I) (Fig. 4).
Figure 3.

Example of Wt sequences of c-Met in SCLC. The forward and reverse DNA sequences are shown. Red circles indicate A. E168D position (GAG>GAT) in the Sema domain and B. R988C (CGC>TGC) and C. T1010I (ACT>ATT) in the JM domain, which have been detected by different authors but not identified in our samples.
Figure 4.

Novel mutations of c-Met in SCLC. The forward and reverse DNA sequences ar shown. A. Sema domain AGC to AGT change does not modify amino acid in codon 178. B. JM domain T995I mutation (ACT>ATT) of c-Met in SCLC sample. Mutations are indicated with arrows.
Discussion
The c-Met receptor tyrosine kinase is involved in oncogenesis in many human cancers. Also, activation of the HGF/SF-c-Met pathway has been associated with shorter survival in NSCLC and SCLC patients (Harvey, 1996; Siegfried, 1998; Bharti, 2004; Masuya, 2004). Numerous missense mutations of the c-Met protooncogene, mostly in the tyrosine kinase domain, are associated with invasion and metastasis of tumor cells. These mutations have been identified in different solid tumors (Schmidt, 1998; Park, 1999; Di Renzo, 2000).
Several mutations have been reported in the JM domain, including T1010I and P1009S in human gastric cancer (Lee, 2000). The T1010I mutation was previously identified in a tumor biopsy of hereditary renal papillary cancer and a papillary renal cancer cell line, ACHN, and appeared to be a polymorphism (Schmidt, 1999). This mutation caused constitutive phosphorylation of MET when transfected into NIH3T3 cell line. (Schmidt, 1999) The T1010I mutation has also been detected in the large cell lung cancer cell line, Hop-92 and in breast cancer (Lee, 2000). Lee et al. (Lee, 2000) screened 30 breast cancer tumor tissues. They focused on exon 14 in the JM domain. The T1010I mutation was found in 1/30 (3%) tissues. This mutation was also in the DNA from a tumor cell-negative lymph node of the same patient, indicating it was a germline mutation. Lee et al. also checked 50 tumor cell lines from different origins (stomach, lung, kidney, prostate and skin) and detected a C to T sequence change (T1010I) in a large cell lung cancer cell line (Hop-62).
In a recent study, Rossi et al. (Rossi, 2005) evaluated 83 paraffin-embedded tissues from patients with pure pulmonary large-cell neuroendocrine carcinoma to examine the mutational status in exon 14 encoding for the relevant JM domain of the c-Met receptor tyrosine kinase. No mutations were detected in any of the tumor cells.
In contrast, Ma et al. (Ma, 2003), found the novel Sema and JM domain missense mutations of c-Met identified for the first time in SCLC, but the frequency of these mutations was low. Ma et al. examined 10 SCLC cell lines and 32 paired SCLC/normal tissues for the E168D missense mutation in the Sema domain (exon 2) and the T1010I and R988C missense mutations in the JM domain (exon 14). They found the E168D in only one tumor tissue sample (3%), whereas the R988C missense mutation was found in the H69 and H249 cell lines, both of which were originally derived from patients with ED SCLC (Phelps, 1996). The T1010I missense mutation was found in 1/32 (3%) tumor tissue sample. In 2005, this same group (Ma, 2005), evaluated 4 NSCLC cell lines and 127 lung adenocarcinoma tumor tissue for the presence of the c-Met mutation, located at the extracellular Sema and JM domains, in exon 2 and 14, respectively. Within the Sema domain they identified the E168D mutation in one tumor tissue (0.8%) and the R988C in the JM domain mutation in one NSCLC cell line (H1437). Both the R988C and T1010I mutations were detected in one tumor tissue (0.8%).
In the present study, we examined missense mutations T1010I and R988C in the JM domain (exon 14) and E168D in the Sema domain (exon 2). These mutations were not found in the DNA from any of the 44 SCLC paraffin-embedded tissues studied. These results confirm the low frequency of T1010I, R988C and E168D missense mutations as previously reported in other studies in SCLC (Ma, 2003), NSCLC (Ma, 2005), breast (Lee, 2000), and large-cell neuroendocrine carcinoma (Rossi, 2005). Interestingly, we have identified two novel mutations. In the Sema domain, we observed an AGC→AGT substitution in codon 178 that does not give rise to an amino acid change in two samples, and in the JM domain, we observed a novel missense mutation (T995I) in one sample. This study confirms that the presence of c-Met mutation is a rare event in SCLC patients, highlighting the need to study other genetic changes in the HGF/SF-c-Met pathway in order to dissect this signaling pathway in SCLC.
Table 1.
PCR primer sequences, annealing temperatures and PCR length products.
| EXON2 | Sequence 5′- 3′ | Tm | Size |
|
| |||
| Forward I | GGACCTGCCAGCGACATGT | 60 C | 355 bp |
| Reverse I | TTGTTGCTTTCAAAGGCATGG | ||
| Forward nested | CTTTCCCCACAATCATACTGCTGA | 60 C | 320 bp |
| Reverse nested | GCATGGACATACTTAATGGGGTAAGA | ||
|
| |||
| EXON14 | Sequence 5′- 3′ | Tm | Size |
|
| |||
| Forward I | TGGGCACTGGGTCAAAGTCTC | 60.5 C | 281 bp |
| Reverse I | AACAATGTCACAACCCACTGAGGTA | ||
| Forward nested | CCATGATAGCCGTCTTTAACAA | 59 C | 193 bp |
| Reverse nested | TATACCTTCTGGAAAAGTAGCTCG | ||
Acknowledgments
Research supported by Spanish Ministry of Health grants, through the Red Temática de Investigación Cooperativa de Centros de Cáncer (CO3-010), by La Fundación Badalona Contra el Cáncer, and by La Fundación Carvajal.
References
- Albain KS, Crowley JJ, et al. “Determinants of improved outcome in small-cell lung cancer: an analysis of the 2,580-patient Southwest Oncology Group data base.”. J. Clin. Oncol. 1990;8(9):1563–74. doi: 10.1200/JCO.1990.8.9.1563. [DOI] [PubMed] [Google Scholar]
- Auperin A, Arriagada R, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N. Engl. J. Med. 1999;341(7):476–84. doi: 10.1056/NEJM199908123410703. [DOI] [PubMed] [Google Scholar]
- Bharti A, Ma PC, et al. “Haptoglobin alpha-subunit and hepatocyte growth factor can potentially serve as serum tumor biomarkers in small cell lung cancer.”. Anticancer Res. 2004;24(2C):1031–8. [PubMed] [Google Scholar]
- Camp RL, Rimm EB, et al. “Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma”. Cancer. 1999;86(11):2259–65. doi: 10.1002/(sici)1097-0142(19991201)86:11<2259::aid-cncr13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Chute JP, Chen T, et al. “Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress.”. J. Clin. Oncol. 1999;17(6):1794–801. doi: 10.1200/JCO.1999.17.6.1794. [DOI] [PubMed] [Google Scholar]
- Di Renzo MF, Olivero M, et al. “Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer.”. Clin. Cancer Res. 1995;1(2):147–54. [PubMed] [Google Scholar]
- Di Renzo MF, Olivero M, et al. “Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas”. Oncogene. 2000;19(12):1547–55. doi: 10.1038/sj.onc.1203455. [DOI] [PubMed] [Google Scholar]
- Elias AD. “Small cell lung cancer: state-of-the-art therapy in 1996”. Chest. 1997;112(4 Suppl):251S–258S. doi: 10.1378/chest.112.4_supplement.251s. [DOI] [PubMed] [Google Scholar]
- Ghoussoub RA, Dillon DA, et al. “Expression of c-met is a strong independent prognostic factor in breast carcinoma”. Cancer. 1998;82(8):1513–20. doi: 10.1002/(sici)1097-0142(19980415)82:8<1513::aid-cncr13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Hanna N, Bunn PA, Jr, et al. “Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer.”. J. Clin. Oncol. 2006;24(13):2038–43. doi: 10.1200/JCO.2005.04.8595. [DOI] [PubMed] [Google Scholar]
- Harvey P, Warn A, et al. “Immunoreactivity for hepatocyte growth factor/scatter factor and its receptor, met, in human lung carcinomas and malignant mesotheliomas.”. J. Pathol. 1996;180(4):389–94. doi: 10.1002/(SICI)1096-9896(199612)180:4<389::AID-PATH685>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Hida Y MT, Fujita M, et al. “Clinical significance of hepatocyte growth factor and c-Met expression in extrahepatic biliary tract cancers.”. Oncol. Rep. 1999;6:1051–1056. doi: 10.3892/or.6.5.1051. [DOI] [PubMed] [Google Scholar]
- Jeffers M, Rong S, et al. “Hepatocyte growth factor/scatter factor-Met signaling in tumorigenicity and invasion/metastasis.”. J. Mol. Med. 1996;74(9):505–13. doi: 10.1007/BF00204976. [DOI] [PubMed] [Google Scholar]
- Lee JH, Han SU, et al. “A novel germ line juxtamembrane Met mutation in human gastric cancer”. Oncogene. 2000;19(43):4947–53. doi: 10.1038/sj.onc.1203874. [DOI] [PubMed] [Google Scholar]
- Lorenzato A, Olivero M, et al. “Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion.”. Cancer Res. 2002;62(23):7025–30. [PubMed] [Google Scholar]
- Ma PC, Jagadeeswaran R, et al. “Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer.”. Cancer Res. 2005;65(4):1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- Ma PC, Kijima T, et al. “c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions.”. Cancer Res. 2003;63(19):6272–81. [PubMed] [Google Scholar]
- Masuya D, Huang C, et al. “The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients.”. Br. J. Cancer. 2004;90(8):1555–62. doi: 10.1038/sj.bjc.6601718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulik G, Kijima T, et al. “Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer.”. Clin. Cancer Res. 2002;8(2):620–7. [PubMed] [Google Scholar]
- Murray N, Coy P, et al. “Importance of timing for thoracic irradiation in the combined modality treatment of limited-stage small-cell lung cancer. The National Cancer Institute of Canada Clinical Trials Group.”. J. Clin. Oncol. 1993;11(2):336–44. doi: 10.1200/JCO.1993.11.2.336. [DOI] [PubMed] [Google Scholar]
- Murren TA., JR . Cancer Principles and Practice of Oncology. Philadelphia, PA: 2005. [Google Scholar]
- Nakajima M, Sawada H, et al. “The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas”. Cancer. 1999;85(9):1894–902. doi: 10.1002/(sici)1097-0142(19990501)85:9<1894::aid-cncr3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Natali PG, Prat M, et al. “Overexpression of the met/HGF receptor in renal cell carcinomas.”. Int. J. Cancer. 1996;69(3):212–7. doi: 10.1002/(SICI)1097-0215(19960621)69:3<212::AID-IJC11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Niell HB, Herndon JE, 2nd, et al. “Randomized phase III intergroup trial of etoposide and cisplatin with or without paclitaxel and granulocyte colony-stimulating factor in patients with extensive-stage small-cell lung cancer: Cancer and Leukemia Group B Trial 9732.”. J. Clin. Oncol. 2005;23(16):3752–9. doi: 10.1200/JCO.2005.09.071. [DOI] [PubMed] [Google Scholar]
- Noda K, Nishiwaki Y, et al. “Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer.”. N. Engl. J. Med. 2002;346(2):85–91. doi: 10.1056/NEJMoa003034. [DOI] [PubMed] [Google Scholar]
- Park WS, Dong SM, et al. “Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas.”. Cancer Res. 1999;59(2):307–10. [PubMed] [Google Scholar]
- Phelps RM, Johnson BE, et al. “NCI-Navy Medical Oncology Branch cell line data base.”. J. Cell. Biochem. Suppl. 1996;24:32–91. doi: 10.1002/jcb.240630505. [DOI] [PubMed] [Google Scholar]
- Pignon JP, Arriagada R, et al. “A meta-analysis of thoracic radiotherapy for small-cell lung cancer.”. N. Engl. J. Med. 1992;327(23):1618–24. doi: 10.1056/NEJM199212033272302. [DOI] [PubMed] [Google Scholar]
- Rasola A, Anguissola S, et al. “Hepatocyte growth factor sensitizes human ovarian carcinoma cell lines to paclitaxel and cisplatin.”. Cancer Res. 2004;64(5):1744–50. doi: 10.1158/0008-5472.can-03-2383. [DOI] [PubMed] [Google Scholar]
- Rossi G, Cavazza A, et al. “Role of chemotherapy and the receptor tyrosine kinases KIT, PDGFRalpha, PDGFRbeta, and Met in large-cell neuroendocrine carcinoma of the lung.”. J. Clin. Oncol. 2005;23(34):8774–85. doi: 10.1200/JCO.2005.02.8233. [DOI] [PubMed] [Google Scholar]
- Roth BJ, Johnson DH, et al. “Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group.”. J. Clin. Oncol. 1992;10(2):282–91. doi: 10.1200/JCO.1992.10.2.282. [DOI] [PubMed] [Google Scholar]
- Rubin JS, Bottaro DP, et al. “Hepatocyte growth factor/scatter factor and its receptor, the c-met proto-oncogene product.”. Biochim. Biophys. Acta. 1993;1155(3):357–71. doi: 10.1016/0304-419x(93)90015-5. [DOI] [PubMed] [Google Scholar]
- Schmidt L, Duh FM, et al. “Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas.”. Nat. Genet. 1997;16(1):68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- Schmidt L, Junker K, et al. “Novel mutations of the MET proto-oncogene in papillary renal carcinomas”. Oncogene. 1999;18(14):2343–50. doi: 10.1038/sj.onc.1202547. [DOI] [PubMed] [Google Scholar]
- Schmidt L, Junker K, et al. “Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene.”. Cancer Res. 1998;58(8):1719–22. [PubMed] [Google Scholar]
- Seki T, Hagiya M, et al. “Organization of the human hepatocyte growth factor-encoding gene”. Gene. 1991;102(2):213–9. doi: 10.1016/0378-1119(91)90080-u. [DOI] [PubMed] [Google Scholar]
- Siegfried JM, Weissfeld LA, et al. “The clinical significance of hepatocyte growth factor for non-small cell lung cancer.”. Ann. Thorac. Surg. 1998;66(6):1915–8. doi: 10.1016/s0003-4975(98)01165-5. [DOI] [PubMed] [Google Scholar]
- Sundstrom S, Bremnes RM, et al. “Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up.”. J. Clin. Oncol. 2002;20(24):4665–72. doi: 10.1200/JCO.2002.12.111. [DOI] [PubMed] [Google Scholar]
- Takada M, Fukuoka M, et al. “Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104.”. J. Clin. Oncol. 2002;20(14):3054–60. doi: 10.1200/JCO.2002.12.071. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Yonemura Y, et al. “The relation between the growth patterns of gastric carcinoma and the expression of hepatocyte growth factor receptor (c-met), autocrine motility factor receptor, and urokinase-type plasminogen activator receptor”. Cancer. 1998;82(11):2112–22. [PubMed] [Google Scholar]
- Turrisi AT, 3rd, Kim K, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N. Engl. J. Med. 1999;340(4):265–71. doi: 10.1056/NEJM199901283400403. [DOI] [PubMed] [Google Scholar]
- Ueki T, Fujimoto J, et al. “Expression of hepatocyte growth factor and its receptor c-met proto-oncogene in hepatocellular carcinoma”. Hepatology. 1997;25(4):862–6. doi: 10.1002/hep.510250413. [DOI] [PubMed] [Google Scholar]
- Wagatsuma S, Konno R, et al. “Tumor angiogenesis, hepatocyte growth factor, and c-Met expression in endometrial carcinoma”. Cancer. 1998;82(3):520–30. doi: 10.1002/(sici)1097-0142(19980201)82:3<520::aid-cncr14>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Warde P, Payne D. “Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis.”. J. Clin. Oncol. 1992;10(6):890–5. doi: 10.1200/JCO.1992.10.6.890. [DOI] [PubMed] [Google Scholar]
- Zelen M. “Keynote address on biostatistics and data retrieval.”. Cancer Chemother. Rep. 1973;34(2):31–42. [PubMed] [Google Scholar]