

Abstract
Sclerosing rhabdomyosarcoma is a unique rhabdomyosarcoma variant, characterized by a prominent hyalinizing matrix. A notable pitfall is the potential for the unusual matrix and often pseudovascular growth pattern of this lesion to lead to confusion with other sarcoma types, including osteosarcoma, chondrosarcoma, and angiosarcoma. Here we report a case of sclerosing rhabdomyosarcoma arising in a 40-year old male. The tumor was centered in the pterygomaxillary fossa with extensive infiltration into adjacent structures. Fine needle aspiration yielded a preliminary diagnosis of high-grade pleomorphic undifferentiated sarcoma, for which he received neoadjuvant chemotherapy and surgical resection. Microscopic examination showed a malignant spindled to round cell neoplasm with prominent osteoid-like, hyaline stroma. Focal rhabdomyoblastic differentiation and diffuse immunoreactivity for desmin and myogenin aided in diagnosis. Nineteen months status post primary resection, the patient expired with multiple lung and bony metastases. Among 39 cases reported thus far (including the present case), there is a broad age range (0.3–79 years), with an average age at presentation of 27 years. The most commonly involved sites are the extremities (n = 19) and head and neck (n = 15). Most cases have been treated by resection, often combined with radiation and/or chemotherapy. Out of 31 cases with follow-up information provided, 6 patients developed local recurrence, 7 patients developed regional or distant metastasis, and 5 patients died of disease. Herein we discuss the ongoing controversy regarding how sclerosing rhabdomyosarcoma might best fit into existing rhabdomyosarcoma classification schemes, based upon current clinicopathologic and molecular genetic evidence.
Keywords: Sclerosing rhabdomyosarcoma, Rhabdomyosarcoma, Sarcoma, Head and neck
Introduction
Rhabdomyosarcoma comprises a group of malignant soft tissue neoplasms with skeletal muscle differentiation. The current World Health Organization classification system combines both histologic and molecular features to categorize rhabdomyosarcoma into three main types: embryonal, alveolar, and pleomorphic [1]. Most commonly seen in young children, embryonal rhabdomyosacoma (ERMS) includes botryoid and spindle cell variants, which collectively possess no distinct molecular signature. Alveolar rhabdomyosarcoma (ARMS) is diagnosed most often in older children and young adults, has a poorer prognosis than ERMS, and often has an associated fusion product involving the FOX01a gene. Pleomorphic rhabdomyosarcoma (PRMS) typically occurs in adults, also carries a poor prognosis, and demonstrates complex cytogenetic aberrations [2].
Sclerosing rhabdomyosarcoma (SRMS) is an unusual variant of rhabdomyosarcoma that was first described in 2000 by Mentzel and Katenkamp [3]. These authors reported a series of unusual rhabdomyosarcoma cases, exhibiting prominent hyaline sclerosis and a pseudovascular growth pattern. Additional cases have been reported since in both adult and pediatric patients, with a marked predilection for the extremities and head and neck region [4–17]. Here we report a new case of SRMS, centered within the pterygomaxillary fossa of an adult male. In addition, we review the literature regarding the clinicopathologic features and emerging molecular genetic characterization of this rare entity.
Case Presentation
A 40 year-old black male was referred by the emergency department to a dentist for extraction of a right mandibular molar. The dentist noted swelling of the adjacent mucosa, which was thought to represent an abscess. However, despite tooth extraction, the swelling worsened; over the course of a month, the patient developed headaches, nausea, vomiting, throbbing cheek pain, dysphagia, shortness of breath, and chills.
The patient was admitted to the hospital for further evaluation. His past medical history was significant for facial radiation more than 20 years prior for an unknown type of nasopharyngeal cancer. He also had a 20 pack-year history of cigarette smoking. Clinical examination showed a large, right facial swelling. Intraorally, a necrotic mass involving the right buccal mucosa, retromolar region, and palate was evident (Fig. 1a). A panoramic radiograph showed marked resorption of the right mandibular ramus, coronoid process, and condyle (Fig. 1b). Magnetic resonance imaging (MRI) revealed a 7.6 cm mass centered in the right pterygomaxillary fossa, with extension into the lateral skull base, infratemporal fossa, masticator space, lateral pterygoid plate, buccal soft tissue, and mandible (Fig. 1c). Full body computed tomography (CT) demonstrated no metastatic disease. Fine needle aspiration showed pleomorphic, spindled to epithelioid malignant cells. Immunohistochemical stains performed on a core needle biopsy exhibited positivity for vimentin, an increased Ki-67 proliferation index, and negativity for p63 and cytokeratin (AE1/AE3). A preliminary diagnosis of high-grade pleomorphic undifferentiated sarcoma was rendered.
Fig. 1.
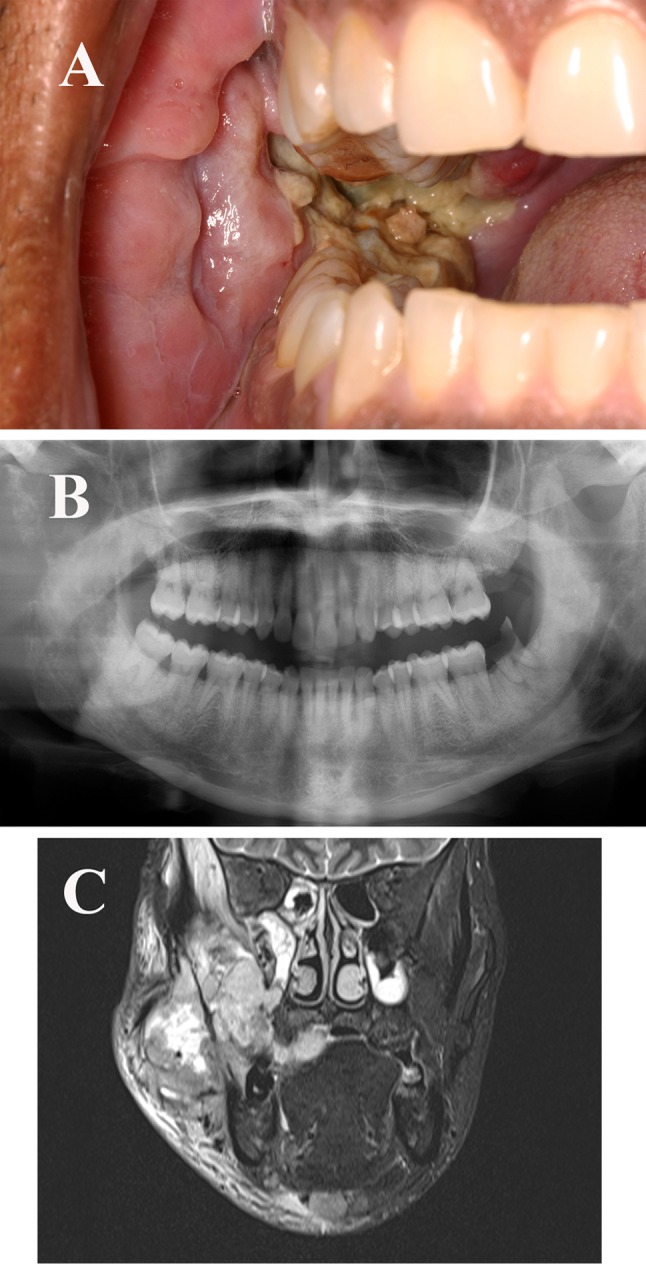
a Intraoral examination showed an extensive necrotic mass involving the right buccal mucosa, retromolar region, and palatal mucosa. b Panoramic radiograph showing marked resorption of the right mandibular ramus, coronoid process, and condyle. c Magnetic resonance imaging showed an infiltrative 7.6 cm mass centered in the right pterygomaxillary fossa
The patient received two cycles of gemcitabine and docetaxel followed by composite resection with reconstruction. Gross examination of the surgical specimen showed an infiltrative, firm, lobulated mass. The cut surface was tan-yellow and focally hemorrhagic. Microscopic examination showed a proliferation of spindle-shaped to ovoid cells within a prominent hyalinizing matrix, often mimicking osteoid or chondroid (Fig. 2a). In some areas, the dense stroma compressed the tumor cells into cords or microalveolar structures. In addition, tumor cells were found within or lining cleft-like spaces between hyalinized bands; however, a prominent pseudovascular growth pattern was not evident. In focal areas, there was a fascicular growth pattern without a hyaline matrix (Fig. 2b). The tumor cells often exhibited abundant eosinophilic cytoplasm, ovoid nuclei, and vesicular chromatin. Focal “strap cells” with rhabdomyoblastic differentiation and cross striations were identified (Fig. 2c). Also noted were occasional cells with small, round nuclei and scant cytoplasm. Scattered osteoclast-like giant cells were identified as well. The mitotic count was 4 per 10 high-power fields.
Fig. 2.

a Prominent hyalinizing matrix, mimicking osteoid (hematoxylin and eosin, original magnification ×400). b Proliferation of malignant spindle-shaped to ovoid cells arranged in intersecting fascicles. The tumor cells exhibited abundant eosinophilic cytoplasm (hematoxylin and eosin, original magnification ×200). c Focal “strap cells” with rhabdomyoblastic differentiation and cross striations were identified (hematoxylin and eosin, original magnification ×600). d Immunohistochemical stain showing diffuse positivity for desmin (original magnification ×400). e Immunohistochemical stain showing diffuse positivity for myogenin (original magnification ×400)
Immunohistochemical stains showed the neoplastic cells to be diffusely positive for desmin and myogenin, focally positive for CD99 and WT-1, and negative for epithelial membrane antigen and cytokeratin (AE1/AE3) (Fig. 2d, e). The Ki-67 proliferation index was 60 %. Fluorescence in situ hybridization (FISH) was negative for translocations involving the FOXO1a, EWSR1 and SS18 loci. Based on the above findings, a diagnosis of sclerosing rhabdomyosarcoma was made.
Per his request, the patient initially received only one cycle of doxorubicin and 1 week of radiotherapy. Two months later, multiple lung metastases were detected. His disease continued to progress despite additional therapy, including two cycles of doxorubicin/ifosfamide/mesna, eight cycles of decarbazine, and a clinical trial with trabectedin. Nineteen months status post primary resection, the patient expired as a result of complications from widely metastatic disease.
Review of the Literature
A review of the English language literature reveals 38 previously reported cases of sclerosing rhabdomyosarcoma [3–17]. A summary of clinicopathologic features for the 39 total cases (including the current case) is provided in Tables 1 and 2.
Table 1.
Clinicopathologic features of 39 reported cases (including the current case) of sclerosing rhabdomyosarcoma
| Authors | Year of publication | Age (yrs.) | Sex | Location | Desmin | Myogenin | MyoD1 | Treatment | Follow-up | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Mentzel and Katenkamp [3] | 2000 | 40 | M | Left lower leg | + | + | − | Excision, RT | NED at 11 mos | Trauma to left tibia 10 yrs. prior |
| 41 | F | Muscle in area of left upper jaw | (+) | + | (+) | Wide excision, RT | Local recurrence at 8 mos. (treated with chemotherapy and additional RT) | |||
| 56 | F | Proximal body of sacrum | + | − | + | Incomplete excision, RT, chemotherapy | Progression of pulmonary metastases; alive with disease at 7 mos | Pulmonary metastases evident at presentation | ||
| Folpe et al. [4] | 2002 | 40 | M | Forearm | (+) dotlike pattern | (+) | + | Re-excision, chemotherapy, RT | NED at 26 mos | |
| 50 | M | Hand | + | (+) | + | Amputation, chemotherapy | NED at 5 mos | |||
| 18 | M | Orbit and skull base | (+) dotlike pattern | (+) | + | Chemotherapy | Developed intra-abdominal and lymph node metastases at 48 mos.; died of disease at 60 mos | |||
| 21 | M | Nasopharynx | (+) | (+) | + | NA | Recent case with no follow-up information provided | |||
| Chiles et al. [5] | 2004 | 6 | M | Arm | + | (+) | + | NA | Local recurrence at 1.69 yrs.; alive 3.28 yrs | |
| 11 | M | Elbow | NA | NA | NA | NA | Died of disease after 3.09 yrs.; developed metastasis after 1.68 yrs | |||
| 7 | F | Pharynx | (+) | (+) | + | NA | Local recurrence at 0.33 yrs.; alive at 3.09 yrs | |||
| 10 | F | Orbit | + | (+) | + | NA | Alive at 1.52 yrs | |||
| 3 | M | Leg | + | (+) | + | NA | Alive at 0.82 yrs | |||
| 8 | F | Sinus | + | (+) | (+) | NA | Alive at 0.01 yrs | |||
| 16 | F | Thigh | (+) | (+) | + | NA | Alive at 2.11 yrs | |||
| 4 | F | Scapula | + | + | + | NA | NA | |||
| 12 | M | Masseter | + | (+) | + | NA | Alive at 0.07 yrs | |||
| 9 | M | Orbit | (+) | (+) | (+) | NA | Alive at 3.58 yrs | |||
| 4 | M | Neck | + | + | + | NA | Alive at 3.12 yrs | |||
| 0.3 | M | Retroperito-neum | (+) | (+) | (+) | NA | NA | |||
| 5 | M | Scrotum | (+) | (+) | (+) | NA | Alive at 1.97 yrs | |||
| Vadgama et al. [6] | 2004 | 3 | F | Left ischiorectal fossa with extension into left buttock | (+) | (+) | + | Excision, postoperative chemotherapy | NED at 4 mos | |
| Croes et al. [7] | 2005 | 79 | F | Soft tissue of right lower leg with infiltration into the tibia | + | (+) | (+) | Amputation of distal femur and lower leg | Multiple skeletal and lung metastases and a solitary breast metastasis at 12 mos | |
| Knipe et al. [8] | 2005 | 66 | F | Base of tongue | + | + | NA | Chemotherapy, RT | NED 5 mos. after treatment | |
| Kuhnen et al. [9] | 2006 | 62 | M | Deep soft tissue of left lower leg | (+) | (+) | + | Resection | Developed lung metastases and pleural spread at 6 mos. after resection; alive with disease 10 mos. after diagnosis | |
| Zambrano et al. [10] | 2006 | 8 | F | Abdomen | + | (+) | (+) | Chemotherapy followed by resection | NA | |
| 18 | M | Soft tissue of right thigh and groin | + | (+) | + | NA | NA | |||
| 17 | F | Soft tissue of left lower leg adjacent to the fibula | (+) | (+) | (+) | Chemotherapy followed by resection | NA | |||
| Sakayama et al. [11] | 2008 | 19 | M | Right leg | NA | + | NA | 4 courses of chemotherapy, resection, additional chemotherapy, and PBSCT | NED at 12 years | Incidental trauma to tumor area 1 month prior to presentation; initial impression based on MRI was intramuscular hematoma; multiple pulmonary metastases evident at time of diagnosis |
| Wang et al. [12] | 2008 | 54 | M | Left wrist | (+) for 4 cases; − for 1 case | (+) for 2 cases; − for 2 cases; NA for 1 case | + | Resection, RT | NA | |
| 52 | F | Right thigh | + | Resection, RT | Recurred at 6 mos.; recurrence treated by radical excision and chemotherapy; NED at 36 mos | |||||
| 20 | F | Right side of face | + | Tumor debulking, chemotherapy | NED at 26 mos | |||||
| 40 | M | Right cheek | + | Wide local excision, RT | NED at 16 mos | |||||
| 12 | F | Right nasal cavity | + | Simple excision | Recurred after 3 mos.; recurrence treated by wide local excision and chemotherapy; NED at 5 mos | |||||
| Lamovec et al. [13] | 2009 | 60 | M | Right parotid gland | (+) | + | + | Parotidectomy, RT | 3 local recurrences (at 30 mos., 42 mos., and 48 mos.) treated by excision, modified neck dissection. and adjuvant chemotherapy; alive with disease | |
| Soglio et al. [14] | 2009 | 7 | M | Left deltoid | + | (+) | NA | Resection, chemotherapy | Axillary lymph node metastasis at 2 yrs.; surgery for locoregional recurrence at 6 yrs. followed by nonmyeloablative allogeneic stem cell transplantation; subsequent surgery for pulmonary metastasis | |
| Cantley et al. [15] | 2010 | 55 | F | Right popliteal fossa | + | (+) | NA | NA | NA | Right inguinal lymph node metastasis present at time of diagnosis |
| Gavino et al. [16] | 2010 | 31 | F | Right lower leg | (+) | (+) | (+) | RT, radical resection, adjuvant chemotherapy | Left gluteal metastasis resected 1 month after surgery; died of disease 16 mos. after initial diagnosis with metastases to the lungs, peritoneum, pelvis, spine, abdominal wall, and right forearm | |
| Martorell et al. [17] | 2010 | 37 | M | Soft tissue posterior to the right elbow | SRMS areas − (ERMS +) | SRMS areas −(ERMS +) | SRMS areas + (ERMS +) | Excision with wide margins | Metastasis to right testis detected and treated by orchiectomy 1 year after excision,; died of disease 8 mos. after orchiectomy with multiple metastases to the mediastinum, lung, and right thigh | Primary in right elbow classified as SRMS with focal fusocellular ERMS; testicular metastasis purely comprised of fusocellular ERMS |
| Robinson et al. | 2012 | 40 | M | Pterygomax-illary fossa and adjacent structures | + | + | NA | Chemotherapy, resection | Metastases to bone and lung treated by RT and additional chemotherapy; died of disease at 19 mos | Patient had RT > 20 years prior for a nasopharyngeal tumor of unknown type |
ERMS embryonal rhabdomyosarcoma; F female, M male, mos. months, MRI magnetic resonance imaging; NA not available or not stated; NED no evidence of disease, yrs. years, RT radiation therapy, PBSCT peripheral blood stem cell transplantation; SRMS sclerosing rhabdomyosarcoma; + positive staining (includes cases reported as >50 % of cells staining, “diffuse,” “strong and diffuse,” or “positive”); (+) focal or weakly positive staining (includes cases reported as ≤ 50 % of cells staining, “patchy,” or “faint”); − negative staining
Table 2.
Summary of clinicopathologic data for 39 reported cases (including the current case) of sclerosing rhabdomyosarcoma [3–17]
| Feature | Value |
|---|---|
| Age at presentation | |
| Mean | 27 years |
| Range | 0.3–79 years |
| Peak decades | 1st decade (n = 13), 2nd decade (n = 9) |
| Sex | |
| Males | 22 |
| Females | 17 |
| Most common primary locations | |
| Head and neck | n = 15 |
| Lower extremity | n = 11 |
| Upper extremity | n = 8 |
| Maximum diameter | “bean sized” to 11.8 cm |
| Signs and symptoms | |
| pain or discomfort | n = 8 |
| dysphagia | n = 2 |
| dysarthria | n = 1 |
| overlying skin ulceration | n = 1 |
| nerve paralysis | n = 1 |
| induration | n = 1 |
| nasal obstruction | n = 1 |
| difficulty opening mouth | n = 1 |
| headache, nausea, vomiting, and chills | n = 1 |
| Patient outcomes | |
| Follow-up information provided | n = 31 |
| Local recurrence | n = 6 |
| Development of regional or distant metastasis | n = 7 |
| Died of disease | n = 5 |
Characteristic histopathologic findings included spindle-shaped, round, or polygonal tumor cells embedded in a prominent hyalinizing matrix. The lesions typically were infiltrative, although one case exhibited well-demarcated borders [14]. A pseudovascular growth pattern was noted in 11 cases [3, 6, 9, 10, 12, 17]. Fascicular, nested, cordlike, trabecular, solid, and rarely alveolar architectural patterns also were observed. The hyalinized collagenous stroma often mimicked osteoid or chondroid. In fact, in one of the cases reported by Zambrano et al. [10], tumor cells were observed in empty spaces within a densely collagenized background, thereby simulating cartilaginous lacunae. The round or polygonal tumor cells typically were small, with scant cytoplasm, hyperchromatic nuclei, and inconspicuous nucleoli. In approximately one-third of cases, there were spindle cells with rhabdomyoblastic features, including abundant granular eosinophilic cytoplasm and eccentric to centrally placed nuclei. Focal cross striations and nuclear pseudoinclusions were noted as well. Rhabdomyoblastic strap cells were described in a few cases [4, 10, 12], and spider rhabdomyoblasts were identified in a single case [3]. In one of the cases reported by Zambrano et al. [10], there were occasional large, anaplastic cells with prominent microvesicular steatosis, mimicking lipoblasts. Multinucleated giant cells were seen in the current case and one case described by Zambrano et al. [10]. Necrosis was present in 6 cases and was more often focal than diffuse [4, 7, 9, 11, 17]. Mitotic activity ranged from 0.5 to 25 mitoses per 10 high-power fields.
Immunohistochemical stains showed the neoplastic cells to exhibit variable degrees of positivity for muscle markers, including desmin, MyoD1, myogenin, myoglobin, sarcomeric actin, fast myosin, smooth muscle actin, and muscle specific actin (Table 1). In addition, the tumor cells often demonstrated positivity for vimentin, WT-1, and CD99; in rare cases, expression of CD56, calponin, p53, MDM2, and Bcl-2 was reported as well [13, 14, 16]. The neoplastic cells were negative for cytokeratins, EMA, vascular markers, neuroendocrine markers, and melanoma markers. The Ki-67 proliferation index ranged from 10 to 60 %, with the highest index reported in the current case.
Discussion
There is some debate in the literature whether SRMS represents a distinct rhabdomyosarcoma type versus a variant of ERMS or ARMS. Indeed, the clinicopathologic features of SRMS overlap with those of both ERMS and ARMS. Similar to ERMS, SRMS exhibits a peak in the first decade. However, SRMS occurs over a broad age range, and the mean age at diagnosis is somewhat older for SRMS (27 years) and ARMS (16 years) than ERMS (7 years) [18]. In addition, both SRMS and ARMS exhibit a predilection for the extremities, although the head and neck region is the second most commonly involved site in SRMS and is a favored site in ERMS as well. Morphologically, the observation of overt myogenesis with the presence of rhabdomyoblasts in one-third of SRMS cases would favor an association with ERMS. However, alveolar or microalveolar architecture likened to ARMS occasionally has been reported in SRMS as well [4, 6, 12]. Although prominent extracellular matrix production generally is rare in RMS, some cases of ARMS have thin fibrous bands, which somewhat resemble the hyalinizing matrix in SRMS [2, 4]. Furthermore, in two cases of SRMS, including the present case, multinucleated giant cells have been identified [10]; such cells occasionally may be seen in ARMS but are rare in ERMS.
Because there are so many shared clinicopathologic features among SRMS, ARMS, and ERMS, there is growing interest in how molecular genetic characterization of SRMS may aid in its classification. Molecular and cytogenetic studies performed on a limited number of SRMS cases exhibit complex and varied profiles; this lack of a distinct molecular genetic signature favors but does not allow for definitive classification as a variant of ERMS. Six SRMS cases have been karyotyped and found to exhibit aneuploidy with multiple nonspecific losses and gains of chromosomes [5, 7, 10]. In addition, by reverse transcriptase polymerase chain reaction, Chiles et al. [5] found that only one of six SRMS cases possessed the PAX3-FOXO1a or PAX7-FOXO1a fusion transcript, which is most closely associated with ARMS. Similarly, in the present case, we were unable to detect a FOXO1a translocation by FISH. A complicating factor is a report by Kuhnen et al. [9], who performed comparative genomic hybridization analysis on a single case of SRMS; these investigators demonstrated losses of 10q22 and chromosome Y and gain of chromosome 18, which is a pattern similar to that observed in PRMS. Of additional interest, single nucleotide polymorphism microarray analysis performed on a single case of SRMS by Bouron-dal Solgio et al. [14] showed amplification of MDM2 and HMGA2, which also has been described in well-differentiated and dedifferentiated liposarcoma. In summary, current evidence is too limited to support the use of testing for FOXO1a translocations, specific chromosomal losses or gains, or amplification of specific genes in making a diagnosis of SRMS.
It is important for pathologists to be familiar with SRMS, because it potentially may be mistaken for other entities. Indeed, in 5 previously reported cases, the osteoid-like or chondroid-like matrix led to a misdiagnosis of osteosarcoma or chondrosarcoma [4, 10, 11]. In several cases, a pseudovascular growth pattern has been described, which Mentzel and Katenkamp commented may lead to confusion with angiosarcoma [3]. In addition, Folpe et al. [4] commented that areas of SRMS with cords and individual tumor cells embedded within a hyaline matrix may mimic sclerosing epithelioid fibrosarcoma. Recognition of at least focal rhabdomyoblastic differentiation and inclusion of muscle markers in the immunohistochemical panel should help to differentiate SRMS from these other entities.
With regard to immunohistochemical markers supportive of skeletal muscle differentiation, MyoD1 generally has been viewed as a more sensitive marker for SRMS than desmin and myogenin, based upon observations by Folpe et al. and subsequent authors [4, 6, 9, 12, 17]. Nevertheless, we caution that strong, diffuse reactivity for MyoD1 is not a uniform finding (focal or weak staining: n = 9, negative: n = 1) (Table 1). Furthermore, strong or diffuse expression of desmin has been reported in a substantial minority of cases (n = 17) (Table 1). Therefore, when considering a diagnosis of SRMS, we recommend the inclusion of several muscle markers in the immunohistochemical panel. MyoD1 and desmin appear to be the most sensitive among these markers; however, in the current case, MyoD1 was not available in our laboratory, and so we selected desmin and myogenin.
An intriguing finding in the present case was the patient’s history of radiation therapy during adolescence for treatment of a nasopharyngeal malignancy. This finding may be incidental, and we were unable to determine what type of nasopharyngeal malignancy the patient had. Nevertheless, considering the possible scope of the radiation field and the passage of an adequate latency period, one might conjecture that in this case SRMS developed as a radiation-induced sarcoma. The development of RMS as a sequela of radiation therapy is exceedingly rare, with pleomorphic undifferentiated sarcoma (malignant fibrous histiocytoma) and osteosarcoma representing the most common types of radiation-induced sarcoma [19, 20]. The etiology of RMS is largely unknown, although in some cases genetic factors have been implicated by an association with various heritable conditions, such as Li Fraumeni syndrome, Beckwith-Wiedemann syndrome, and neurofibromatosis type 1[18]. Interestingly, a single case of radiation-induced RMS in a patient with neurofibromatosis type 2 has been described as well [21]. Therefore, the possibility that environmental factors, such as ionizing radiation, may contribute to tumor formation in conjunction with genetic factors may be considered.
In summary, we have presented an additional case of SRMS, arising in the head and neck of an adult. SRMS exhibits a predilection for the head and neck and extremities. A prominent hyalinized matrix is characteristic of these lesions and potentially may lead to confusion with other neoplasms, such as osteosarcoma or chondrosarcoma. Based upon current evidence, it is unclear whether SRMS is best regarded as a distinct type of RMS or a variant of ERMS or ARMS. Clinicopathologic and molecular genetic characterization of additional cases may help resolve how to classify this lesion in the future.
Conflict of interest
The authors declare they have no conflict of interest.
References
- 1.Parham DM, Barr FG, Montgomery E. Skeletal muscle tumors. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumours: pathology and genetics of tumours of soft tissue and bone. 3. Lyon: IARC Press; 2006. pp. 141–154. [Google Scholar]
- 2.Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006;130:1454–1465. doi: 10.5858/2006-130-1454-RIAACA. [DOI] [PubMed] [Google Scholar]
- 3.Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000;436:305–311. doi: 10.1007/s004280050451. [DOI] [PubMed] [Google Scholar]
- 4.Folpe AL, McKenny JK, Bridge JA, et al. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–1183. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Chiles MC, Parham DM, Qualman SJ, et al. Sclerosing rhabdomyosarcomas in children and adolescents: a clincopathologic review of 13 cases from the intergroup rhabdomyosarcoma study group and children’s oncology group. Pediatr Dev Pathol. 2004;7:583–594. doi: 10.1007/s10024-004-5058-x. [DOI] [PubMed] [Google Scholar]
- 6.Vadgama B, Sebire NJ, Malone M, et al. Sclerosing rhabdomyosarcoma in childhood: case report and review of the literature. Pediatr Dev Pathol. 2004;7:391–396. doi: 10.1007/s10024-003-9453-5. [DOI] [PubMed] [Google Scholar]
- 7.Croes R, Debiec-Rychter M, Cokelaere K, et al. Adult sclerosing rhabdomyosarcoma: cytogenetic link with embryonal rhabdomyosarcoma. Virchows Arch. 2005;446:64–67. doi: 10.1007/s00428-004-1131-0. [DOI] [PubMed] [Google Scholar]
- 8.Knipe TA, Chandra RK, Bugg F. Sclerosing rhabdomyosarcoma: a rare variant with predilection for the head and neck. Laryngoscope. 2005;115:48–50. doi: 10.1097/01.mlg.0000150676.75978.3c. [DOI] [PubMed] [Google Scholar]
- 9.Kuhnen C, Herter P, Leuschner I, et al. Sclerosing pseudovascular rhabdomyosarcoma-immunohistochemical, ultrastructural, and genetic findings indicating a distinct subtype of rhabdomyosarcoma. Virchows Arch. 2006;449:572–578. doi: 10.1007/s00428-006-0282-6. [DOI] [PubMed] [Google Scholar]
- 10.Zambrano E, Pérez-Atayde AR, Ahrens W, et al. Pediatric sclerosing rhabdomyosarcoma. Int J Surg Pathol. 2006;14:193–199. doi: 10.1177/1066896906290558. [DOI] [PubMed] [Google Scholar]
- 11.Sakayama K, Tauchi H, Sugawara Y, et al. A complete remission of sclerosing rhabodmyosarcoma with multiple lung and bone metastases treated with multi-agent chemotherapy and peripheral blood stem cell transplantation (PTSCT): a case report. Anticancer Res. 2008;28:2361–2368. [PubMed] [Google Scholar]
- 12.Wang J, Tu X, Sheng W. Sclerosing rhabdomyosarcoma: a clinicopathologic and immunohistochemical study of five cases. Am J Clin Pathol. 2008;29:410–415. doi: 10.1309/EP3FRRY6GMP555QC. [DOI] [PubMed] [Google Scholar]
- 13.Lamovec J, Volavsek M. Sclerosing rhabdomyosarcoma of the parotid gland in an adult. Ann Diagn Pathol. 2009;13:334–338. doi: 10.1016/j.anndiagpath.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Bouron-Dal Soglio D, Rougemont AL, Absi R, et al. SNP genotyping of a sclerosing rhabdomyosarcoma: reveals highly aneuploid profile and a specific MDM2/HMGA2 amplification. Hum Pathol. 2009;40:1347–1352. doi: 10.1016/j.humpath.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 15.Cantley RL, Cimbaluk D, Reddy V, et al. Fine needle aspiration diagnosis of a metastatic adult sclerosing rhabdomyosarcoma in a lymph node. Diagn Cytopathol. 2010;38:761–764. doi: 10.1002/dc.21325. [DOI] [PubMed] [Google Scholar]
- 16.Gavino ACP, Spears MD, Peng Y. Sclerosing spindle cell rhabdomyosarcoma in an adult: report of a new case and review of the literature. Int J Surg Pathol. 2010;18:394–397. doi: 10.1177/1066896908327166. [DOI] [PubMed] [Google Scholar]
- 17.Martorell M, Ortiz CM, Garcia JA. Testicular fusocellular rhabdomyosarcoma as a metastasis of elbow sclerosing rhabdomyosarcoma: a clinicopathologic, immunohistochemical and molecular study of one case. Diagn Pathol. 2010;5:52. doi: 10.1186/1746-1596-5-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiss SW, Goldblum JR. Chapter 21 Rhabdomyosarcoma. In: Weiss SW, Goldblum JR, editors. Enzinger and Weiss’s soft tissue tumors. 5. Philadelphia: Mosby; 2008. pp. 595–631. [Google Scholar]
- 19.Bjerkehagen B, Smeland S, Walberg L, et al. Radiation-induced sarcoma: 25-year experience from The Norwegian Radium Hospital. Acta Oncol. 2008;47:1475–1482. doi: 10.1080/02841860802047387. [DOI] [PubMed] [Google Scholar]
- 20.Patel SR. Radiation-induced sarcoma. Curr Treat Options Oncol. 2000;1:258–261. doi: 10.1007/s11864-000-0037-6. [DOI] [PubMed] [Google Scholar]
- 21.Carlson ML, Babovic-Vuksanovic D, Messiaen L, et al. Radiation-induced rhabdomyosarcoma of the brainstem in a patient with neurofibromatosis type 2. J Neurosurg. 2010;112:81–87. doi: 10.3171/2009.6.JNS09105. [DOI] [PubMed] [Google Scholar]