

Abstract
We present a carcinosarcoma ex non-recurrent pleomorphic adenoma composed of a large cell neuroendocrine carcinomatous component and a spindle cell sarcoma with myofibroblastic differentiation. The tumor contained a hyalinized transition zone where the classical PA appeared to acquire two different histopathological patterns of malignant transformation of the epithelial component. The carcinomatous component was strongly and diffusely positive for low-molecular weight cytokeratins (AE1-3), synaptophysin, thyroid transcription factor-1 and focally positive for chromogranin A. All these markers were negative in the sarcomatous component. The sarcomatous component displayed immunoreactivity for smooth muscle actin with a predominantly linear, subplasmalemmal pattern. No expression of CD31, S100 protein, h-caldesmon, desmin, CD34, p63, myogenin, Myo D1 and c-kit was detected. Strong immunohistochemical expression of p53 was documented in both the carcinomatous and sarcomatous components as well as in the atypical epithelial component in the transition zone associated with the hyalinized pleomorphic adenoma.
Keywords: Carcinoma ex pleomorphic adenoma, Carcinosarcoma, Large cell neuroendocrine carcinoma, Salivary glands, Parotid
Introduction
Carcinosarcoma (CS) arising in salivary glands is a very rare tumor accounting for less than 0.5 % of all malignant salivary gland tumors [1]. CS may or may not be associated with a concurrent or recurrent pleomorphic adenoma (PA). Although rare, CS frequently displays a multitude of histological patterns corresponding to varying lines of histological differentiation in both the carcinomatous and sarcomatous components. Reportedly, the most common epithelial malignancy is adenocarcinoma NOS and the most common sarcomatous component is chondrosarcoma [2]. Salivary gland CS with large cell neuroendocrine carcinoma has, to the best of our knowledge, only been reported in one previous case [3]. This case was not associated with a previous or concurrent PA and was associated with a sarcomatous component which featured rhabdomyosarcomatous differentiation. Herein we present a unique case of a parotid gland CS ex (non-recurrent) PA which was composed of a large cell neuroendocrine carcinoma and spindle cell sarcoma showing myofibroblastic differentiation.
Case Report
A 50 year old man with diabetes mellitus, hypertension and a previous mild stroke 5 years ago presented with a rapidly (months) expanding mass in the left parotid region. There had been a stable lump in that place for a period of 5 years.
On clinical examination, there was an 8 cm hard left parotid mass with left incomplete facial nerve palsy affecting the buccal and mandibular branches of the VII nerve. No palpable lymph nodes were detected. A computerized tomography (CT) scan showed a large necrotic tumor mass with ipsilateral nodal enlargements in level II.
A fine needle aspiration cytology was performed which showed necrotic debris admixed with cohesive and discohesive atypical cells with hyperchromatic, irregular nuclei and scant pale to finely vacuolated cytoplasm. The features were consistent with a high-grade carcinoma. A chest X-ray showed no tumorous lesions.
Subsequently, a left sided radical parotidectomy, facial nerve resection and ipsilateral modified radical neck dissection (levels 2–4) resected en bloc with the parotid gland and level 5 separately were performed. (The neck dissection yielded 55 lymph nodes, of which 3 (level 1 and 2) harbored metastatic tumor; carcinomatous component). Separate biopsies (positive for malignancy) from the trunk of the facial nerve were taken. Postoperatively, the patient was treated with adjuvant radio- and chemotherapy. The case was recently diagnosed and we have no meaningful follow up.
Materials and Methods
The tissue was fixed in formalin and embedded in paraffin. Four-micron thick sections were cut and stained with Hematoxylin and Eosin (H&E). The tumor was sampled with 15 tissue blocks. An immunohistochemical study was performed with the following commercial antibodies using the manufacturers’ protocols: Smooth muscle actin (SMA), desmin, CD34, S-100 protein, pan-cytokeratin (AE1-3), chromogranin A, synaptophysin, thyroid transcription factor 1 (TTF-1), p53, HER2, cytokeratin 20, Ki-67, CD31, Myo D1, myogenin, p16, p63, cyclin D1, h-caldesmon and CD117/c-kit.
Results
Gross pathological examination: The specimen contained parotid tissue with a tumor and measured 13 × 8.5 × 4.5 cm. The cut sections revealed a relatively well circumscribed tumor measuring 6.5 × 6 × 4 cm with a variegated firm whitish appearance and softer tan, fleshy areas with necrosis and haemorrhage (Fig. 1). The neck dissection (level 2–4) contained multiple enlarged lymph nodes (the largest measuring 4 cm in maximum dimension; level 2, grossly involved by metastatic tumor).
Fig. 1.
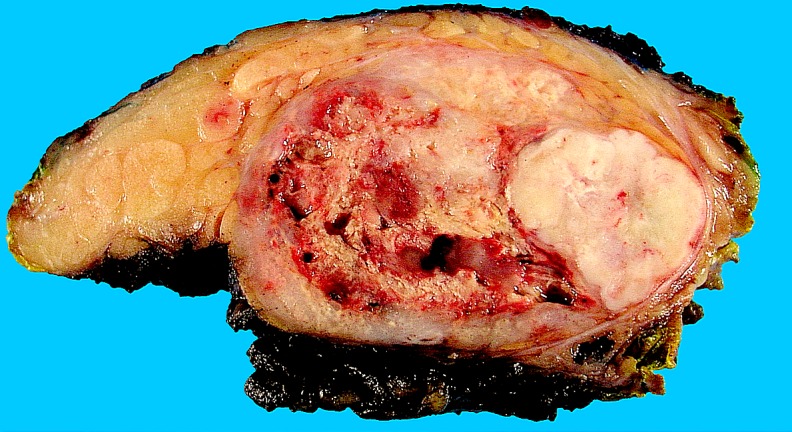
Photomicrograph of the gross surgical specimen showing a relatively well circumscribed tumor with a white–gray to tan cut surface. Large parts of the tumor showed haemorrhagic necrosis
Routine histological examination: The tumor was widely invasive extending into the periglandular adipose tissue. The majority of the neoplasm (90 %) revealed a distinctive biphasic appearance composed of an epithelial component with variably sized nests and an intervening spindle cell component (Fig. 2a). The epithelial nests were highly cellular and composed of pleomorphic large cells with high nuclear to cytoplasmic ratio and overlapping, irregular round to ovoid hyperchromatic nuclei with coarse chromatin (Fig. 2b). Abundant mitotic figures and apoptotic bodies were present in the carcinomatous nests which frequently displayed central necrosis. The sarcomatous component displayed moderately pleomorphic spindle cells with readily identified mitotic figures including atypical forms set in a variably collagenized stroma (Fig. 2c). No true (Flexner-Wintersteiner) or pseudo- (Homer-Wright) rosettes were identified and no rhabdomyoblastic differentiation, chondroid matrix or osteoid was identified.
Fig. 2.

Haematoxylin and eosin stained sections revealed a distinctly biphasic appearance with a nested, carcinomatous component intimately intermingled with a spindle cell, sarcomatous component (a). The carcinomatous component showed high-grade malignant histological features including prominent nuclear atypia, high nuclear to cytoplamic ratio and abundant mitotic activity (b). The sarcomatous component displayed significant nuclear atypia and mitotic activity, including atypical forms (arrow; c). Areas with the classical features of a pleomorphic adenoma were present (d). The pleomorphic adenoma was associated with areas of hyalinization. The hyalinized areas of the pleomorphic adenoma (right) merged with a component of the tumor that had a “less orderly” appearance (left; e). This “less orderly” area contained a clearly atypical epithelial component. The epithelium displayed in addition to nuclear atypia a clear to vacuolated cytoplasm and a mostly readily identifiable peripheral myoepithelial layer (f). A mammary type of intraductal dysplasia/ductal carcinoma in situ-like proliferation which was invested by myoepithelial cells was also identified (g)
In 2 out of 15 blocks a component of PA was identified (Fig. 2d). In addition to classical features of PA, these areas also displayed a less orderly appearance with stromal hyalinization where the epithelial cells had increased amounts of clear to vacuolated cytoplasm and varying degrees of nuclear atypia, scattered mitotic figures and a variably preserved bilayered configuration (Fig. 2e, f). Focally this atypical epithelial component (in the hyalinized areas) not only merged with the classical PA component, but also with areas of necrosis which were contiguous with the high-grade malignant component of the tumor. In addition, another pattern of epithelial atypia was present in the PA component. This was manifested as a mammary type of intraductal dysplasia/ductal carcinoma in situ-like proliferation invested by myoepithelial cells (Fig. 2g). Multifocal perineural growth, including involvement of large nerve trunks, was identified and vascular invasion was present. Focally the surgical margin was involved by the malignant tumor component.
Immunohistochemistry: The malignant epithelial component was strongly and diffusely positive for low-molecular weight cytokeratins (AE1-3), synaptophysin and focally positive for chromogranin A. There was strong and diffuse nuclear expression of TTF-1. All these markers were negative in the sarcomatous component (Fig. 3a–c). No immunoreactivity for CK 20, HER2 was detected. The sarcomatous component displayed immunoreactivity for SMA with a predominantly linear, subplasmalemmal pattern (Fig. 3d). No expression of CD31, S100 protein, h-caldesmon, desmin, CD34, p63, myogenin, Myo D1 and c-kit was detected.
Fig. 3.

Photomicrographs from the immunohistochemical study showing that the carcinomatous component was positive for cytokeratins (a), synaptophysin (b) and TTF-1 (c), while the sarcomatous component was negative. The sarcomatous component displayed immunoreactivity for SMA (d). The presence of smooth muscle actin positive myoepithelial cells displayed a gradual reduction to complete absence in the atypical epithelial component in the hyalinized area (e)
Smooth muscle actin immunoreactive cells were identified in the myoepithelial component of the non-hyalinized PA component. The presence of SMA positive myoepithelial cells displayed a gradual reduction to complete absence in the atypical epithelial component in the hyalinized area (Fig. 3e). This gradual loss of myoepithelial cells was also demonstrated by the staining pattern for S100 protein which was virtually identical to that of SMA (data not shown).
Strong nuclear immunoreactivity for p53 was identified in the majority of the neoplastic cells in both the carcinomatous and sarcomatous components (data not shown). In addition, focal significant nuclear p53 immunoreactivity was detected in the atypical epithelial areas (data not shown). The proliferative activity (Ki-67) was consistently exceedingly high (>90 %) in the epithelial component and more variable (but still high) in the sarcomatous component, on average 50 %, but with hot spots where up to 80 % of the sarcomatous cells featured nuclear expression of Ki-67 (data not shown). No immunoreactivity for cyclin D1 was identified. p16 was moderate to strongly positive (both cytoplasmic and nuclear) throughout both the carcinomatous and sarcomatous components. In addition moderate to strong nuclear and cytoplasmic positivity for p16 was present in the hyalinized areas with epithelial atypia (data not shown).
Discussion
Carcinosarcoma (CS) of salivary glands is a very rare neoplasm accounting for a significant minority (0.04–0.16 %) of all salivary gland tumors and <0.5 % of malignant salivary gland neoplasms as summarized by Gnepp [1] in the most comprehensive review (comprising 43 cases) on this topic to date. CS arising in a salivary gland was first reported by Kirklin et al. [4] and since then, about 75 cases have been reported in the English literature, mostly in the form of case reports and a few small series but also as a small number of cases presented as parts of a series of “carcinoma ex PA” (Ca ex PA). The majority (about two-thirds) of salivary gland CSs have been located in the parotid, about one-fifth in the submandibular gland and 15 % in the palate [2]. CS became incorporated in the WHO classification of salivary gland tumors in 1991, not as a specific entity, but as a variant of carcinoma arising in PA. Generally, CS is a neoplasm that affects older adults (mean age 58 years) albeit with a wide age range (14–87 years) and with no gender difference. CS of salivary glands is a very aggressive neoplasm with high frequency of local recurrence and both lymphatic and hematogenous spread. Reportedly, around 60 % of patients die from the disease with a median survival of 29 months [1]. The rarity of CS in salivary glands is highlighted by the fact that in sizeable series on Ca ex PA, only a few examples of CS have been documented. For example, in a recent series on 41 cases of Ca ex PA, only 2 cases (5 %) were labeled as CS [5], in a series comprising 73 cases, 1 case (1.4 %) was diagnosed as sarcomatoid carcinoma [6] and in the series by Katabi et al. [7] of 43 cases of Ca ex Pa, 1 case (2.3 %) was a CS. Surprisingly, in a recent review on Ca ex PA, the occurrence of CS in this setting was not mentioned at all [8]. The series on malignant transformation of PA with the highest frequency of CS and sarcomatoid carcinoma was from Japan [9]. In this study of 19 cases, 2 tumors (11 %) were diagnosed as CS and sarcomatoid carcinoma, respectively.
In general (and understandably so), series on Ca ex PA with CS cases are not as detailed in their characterization of these cases as those which deal with “pure” CS. Although CS may show a bewildering array of histological appearances reflecting different degrees of various lines of histological differentiation, the most common epithelial malignant components are adenocarcinoma NOS and squamous cell carcinoma. The sarcomatous components have been labeled, in decreasing order of frequency, as chondrosarcoma, fibrosarcoma, leiomyosarcoma, osteosarcoma, rhabdomyosarcoma and liposarcoma [10]. The presence of the latter (liposarcoma) has only been described in 2 cases [1, 11]. Interestingly, and in contrast to CSs occurring in the uterus (malignant mixed Műllerian tumors), rhabdomyoblastic differentiation is unusual and has only been reported in a handful of cases [3, 10–13]. The spindle cell sarcomatous component of our tumor showed high-grade histological features. The stroma of the sarcoma was variably collagenized and the tumor cells expressed SMA in a linear, sublemmal fashion and were negative for desmin and h-caldesmon, CD31, S100 protein, CD34, p63, myogenin, Myo D1 and c-kit; features which conform to a myofibroblastic phenotype. The neoplastic spectrum of myofibroblasts have relatively recently been reviewed in an excellent paper by Eyden and Fisher [14].
The fact that CS may show multiple lines of differentiation (reflecting a pronounced degree of presumed inherent genetic instability of this entity) is highlighted by the case published by Tanahashi et al. [11], where the malignant epithelial component contained poorly differentiated adenocarcinoma, squamous cell carcinoma, salivary duct carcinoma and undifferentiated carcinoma while the sarcomatous component displayed areas of “spindle cell sarcoma”, “chondrosarcoma, liposarcoma and rhabdomyosarcoma”. Other unusual forms of carcinoma that may rarely be associated with CS of salivary glands are undifferentiated carcinoma and large cell neuroendocrine carcinoma. In contrast to the case presented by Tanahashi et al., our case displayed a rather “monotonous” biphasic appearance composed of a spindle cell sarcoma showing myofibroblastic differentiation and a high-grade, large cell neuroendocrine component. This type of epithelial differentiation in a salivary gland CS has, to the best of our knowledge, only been reported in one previous case [3]. In contrast to our case, this case did show multiple lines of differentiation. The carcinomatous component showed in addition to neuroendocrine features, areas of squamous cell carcinoma, adenocarcinoma NOS while the sarcomatous component harbored areas of “spindle cell sarcoma, myxosarcoma and rhabdomyosarcoma”.
Large cell neuroendocrine carcinoma (LCNEC) is an established diagnostic category first described by Travis et al. [15] in the lung and has, since then, been described in various organs such as cervix uteri, ovary, gastrointestinal tract, urinary bladder and others. Regarding the Head and Neck region, LCNEC is not an accepted separate neoplastic entity but classified as a variant of atypical carcinoid in the salivary glands by the latest (2005) WHO edition on Head and Neck tumors. However, a few cases of bona fide LCNEC of salivary gland have been reported [16, 17]. The situation regarding salivary glands and LCNEC in relation to the WHO classification is analogous to that of the larynx, i.e. that the tumor seems to exist at these sites in reality but not in the WHO “blue book” [18–20].
Regarding the histogenesis/origin of CS in salivary glands, two main categories have been suggested, namely (1) those that occur in the setting of malignant transformation of PA (“CS ex PA”) and (2) tumors that are not associated with any precursor lesion (“de novo”). The current wisdom of text books is that the majority of CSs in salivary glands arise “de novo” [21, 22]. It is still an open question whether CS derived from PA and de novo cases are genetically separate or not. In our case we could observe a transformation from bona fide PA to a component of the neoplasm (PA) with (1) increasing nuclear atypia, (2) cytoplasmic clearing, (3) progressively complete loss of the myoepithelial layer both on light microscopy and immunohistochemically (S100 protein and SMA), (4) Increased proliferative activity (both increased number of mitotic figures and immunohistochemical Ki-67 expression) and (5) nuclear expression of p53. This is undoubtedly strong evidence for not only “atypia”, but frank malignant transformation (where these changes were most pronounced). This is in line with previous reports that emphasize stromal hyalinization as an ominous sign, frequently accompanying (early) malignant transformation of PA [6, 23–26]. However, notwithstanding the fact that we could not observe any histomorphological transformation from the hyalinized areas to either the high-grade neuroendocrine carcinomatous or sarcomatous components, the atypical clear cell/hyalinized areas merged with areas of tumor necrosis. Moreover, we also observed an intraductal, “mammary type”, dysplastic/carcinomatous component. This intraductal carcinomatous stage in the evolution of Ca ex PA was found as the sole malignant feature in 15/41 (37 %) patients and was present in 31/37 (76 %) cases of Ca ex PA in the series by Weiler et al. [5]. These and other authors [27, 28] have recently proposed that this “mammary-type” of intraductal carcinoma represents the earliest stage of malignant transformation in Ca ex PA. Based on the evidence presented by these authors, it appears undoubtedly that this is a common pathway for the development of malignancy in PA. However, even though a stage of “mammary-type” intraductal carcinoma may be a significant event in the malignant transformation, there is no reason a priori that this should be the only histomorphologically identifiable stage in the malignant progression of a PA. Based on the findings presented in our case, it appears that the acquisition of malignant features of the luminal epithelium occurred both (1) simultaneously with the loss of the myoepithelial layer and (2) with preserved myoepithelium around the ducts. The idea of several possible routes for malignant transformation in PA is supported by the fact that in their series, Weiler et al. [5] noted that no intraductal carcinoma was detected in all five cases of myoepithelial carcinomatous transformation of PA. Similar findings have been reported by Altemani et al. [29]. Whether this difference in malignant transformation is something that differs between “carcinoma ex” versus “carcinosarcoma ex” on a general scale needs to be studied in a larger series of cases.
The absence of HER2 expression in the tumor presented herein is not surprising since significant membranous protein expression (often with an underlying gene amplification) is associated with salivary duct carcinoma. Also in breast carcinomas with neuroendocrine differentiation HER2 is typically not overexpressed.
Regarding the histogenesis of carcinosarcomas of salivary glands, at least two broad categories come to mind, namely (1) those derived from PA and (2) de novo cases. There is ample molecular evidence for a clonal relation in CSs arising in the uterine corpus and this pattern has increasingly been demonstrated in CS of other organs [30, 31]. The literature on this topic regarding CS of salivary glands is scarce, but there is mounting evidence that a clonal relation between the carcinomatous and sarcomatous components also hold true in the salivary glands [32–35]. Speculatively, but in line with current thinking regarding tumorigenesis, the difference between the two categories of CS would then be that the malignant transformation in (1) occurs in a neoplastic, albeit benign, cell in contrast to an uncommitted stem cell in (2), which then through “genetic instability” gives rise to two or more lines of differentiation. Indirect evidence that this may be the case stems from the fact that a substantial number of salivary gland CSs appear to show multiple lines of differentiation in both the carcinomatous and sarcomatous components, a phenomenon that is related to the fact that uterine CSs regularly display, in addition to varying grades, several histologic types/lines of differentiation in both parts of the tumors. Whether different genetic mechanisms are separately activated in de novo and CSs associated with PA is not known. An extreme example of genetic instability, giving rise to various lines of differentiation in Ca ex PA, is the recently published case by Karpowicz et al. [36] where the malignant component, in addition to harboring salivary duct carcinoma, adenocarcinoma and squamous cell carcinoma, also featured a component of melanoma (positive for S100 protein, HMB-45, melan A, MART-1 and tyrosinase in the immunohistochemical study.
The TTF-1 positivity in the neuroendocrine carcinomatous component in our case is not surprising. Several studies have demonstrated the expression of TTF-1 in malignant neuroendocrine extrapulmonary tumors of various types. A tumor to tumor metastasis (from a pulmonary LCNEC or medullary thyroid carcinoma) was considered, but the absence of a lung tumor on chest X-ray, absence of a thyroid mass and the intimate intermingling of the two components throughout the tumor mass argued strongly against this. As mentioned above, few cases of primary LCNEC of salivary glands have been reported. The relatively more common, although still rare, primary small cell (neuroendocrine) carcinoma of salivary glands in the setting of a collision tumor had to be considered. Again, the very intimate intermingling of the components was not consistent with this. Moreover the nuclear morphology and the size of the tumor cells argued against this. The fact that the primary Merkel cell type of small cell carcinoma may show slightly larger neoplastic cells (than the “pulmonary/oat cell type”) and is the most common type in salivary glands (60 and 73 % in two studies) [37, 38] prompts consideration, but this subtype of neuroendocrine carcinoma consistently expresses cytokeratin 20 which was completely negative in the neuroendocrine carcinomatous component in our tumor.
The role of TP53 in malignant transformation of PA is complex and has been nicely summarized in a recent paper in this journal [8]. A few points of interest are: (1) Frequent loss of 17p (where TP53 is located) has been reported [34] and (2) both overexpression of p53 and TP53 mutations have been reported [27, 39]. However, the relation between immunohistochemically detected p53 and the presence of mutation is not simple. In one study [27], the protein was detected in 79 % of cases of Ca ex PA while TP53 mutation was only detected in 37 % and, in addition, some of the cases with protein overexpression did not reveal any mutation in TP53. Moreover, TP53 mutation was not detected in any of the 11 Ca ex PA studied by Gedlicka [40]. In view of this data we cannot firmly establish a role for a mutated TP53 in the malignant progression of the PA in our case. However, based on the strong, coarse staining pattern in the vast majority of malignant neoplastic cell nuclei, (including in focal areas of the atypical epithelial cells which displayed progressive loss of the myoepithelial layer in direct continuity with the conventional PA), it is a tempting interpretation that TP53 was mutated in our case.
Based on the highly malignant behavior of salivary gland CSs, the prognosis for our patient (despite adjuvant treatment) is guarded. The tumor presented herein was large, locally advanced, infiltrated widely, including into the extra parotid adipose tissue, had focal positive margins, exhibited both vascular and perineural invasion (both large vessels and large nerve trunks with a separate positive biopsy from the facial nerve) and had metastasized to cervical lymphnodes (with the largest metastasis measuring 4 cm), all of which are features associated with poor prognosis. In line with the typical behaviour of carcinosarcomas, it was the carcinomatous component which displayed a propensity for lymphatic spread.
In conclusion, we herein present, to the best of our knowledge, the first case of a salivary gland carcinosarcoma ex non-recurrent pleomorphic adenoma composed of a large cell neuroendocrine carcinomatous component strongly expressing TTF-1 and a spindle cell sarcoma with myofibroblastic differentiation. We also identified a hyalinized transition zone where the classical PA appeared to acquire two different histopathological patterns of malignant transformation in the epithelial component.
References
- 1.Gnepp DR. Malignant mixed tumors of the salivary glands: a review. Pathol Annu. 1993;28(Pt 1):279–328. [PubMed] [Google Scholar]
- 2.Gnepp DR. WHO classification of tumors. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. Pathology and genetics. Head and neck tumors. Lyon: IARC Press; 2005.
- 3.Ueo T, Kaku N, Kashima K, et al. Carcinosarcoma of the parotid gland: an unusual case with large-cell neuroendocrine carcinoma and rhabdomyosarcoma. Apmis. 2005;113:456–464. doi: 10.1111/j.1600-0463.2005.apm_231.x. [DOI] [PubMed] [Google Scholar]
- 4.Kirklin JW, Mc DJ, Harrington SW, et al. Parotid tumors: histopathology, clinical behavior, and end results. Surg Gynecol Obstet. 1951;92:721–733. [PubMed] [Google Scholar]
- 5.Weiler C, Zengel P, van der Wal JE, et al. Carcinoma ex pleomorphic adenoma with special reference to the prognostic significance of histological progression: a clinicopathological investigation of 41 cases. Histopathology 2011;59:741–50. [DOI] [PubMed]
- 6.Lewis JE, Olsen KD, Sebo TJ. Carcinoma ex pleomorphic adenoma: pathologic analysis of 73 cases. Hum Pathol. 2001;32:596–604. doi: 10.1053/hupa.2001.25000. [DOI] [PubMed] [Google Scholar]
- 7.Katabi N, Gomez D, Klimstra DS, et al. Prognostic factors of recurrence in salivary carcinoma ex pleomorphic adenoma, with emphasis on the carcinoma histologic subtype: a clinicopathologic study of 43 cases. Hum Pathol. 2010;41:927–34. [DOI] [PubMed]
- 8.Antony J, Gopalan V, Smith RA, et al. Carcinoma ex Pleomorphic Adenoma: a comprehensive review of clinical, pathological and molecular data. Head Neck Pathol. 2012;6(1):1–9. doi: 10.1007/s12105-011-0281-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harada H. Histomorphological investigation regarding to malignant transformation of pleomorphic adenoma (so-called malignant mixed tumor) of the salivary gland origin: special reference to carcinosarcoma. Kurume Med J. 2000;47:307–323. doi: 10.2739/kurumemedj.47.307. [DOI] [PubMed] [Google Scholar]
- 10.Kwon MY, Gu M. True malignant mixed tumor (carcinosarcoma) of parotid gland with unusual mesenchymal component: a case report and review of the literature. Arch Pathol Lab Med. 2001;125:812–815. doi: 10.5858/2001-125-0812-TMMTCO. [DOI] [PubMed] [Google Scholar]
- 11.Tanahashi J, Daa T, Kashima K, et al. Carcinosarcoma ex recurrent pleomorphic adenoma of the submandibular gland. Apmis. 2007;115:789–794. doi: 10.1111/j.1600-0463.2007.apm_647.x. [DOI] [PubMed] [Google Scholar]
- 12.Gandour-Edwards RF, Donald PJ, Vogt PJ, et al. Carcinosarcoma (malignant mixed tumor) of the parotid: report of a case with a pure rhabdomyosarcoma component. Head Neck. 1994;16:379–382. doi: 10.1002/hed.2880160414. [DOI] [PubMed] [Google Scholar]
- 13.Gogas J, Markopoulos C, Karydakis V, et al. Carcinosarcoma of the submandibular salivary gland. Eur J Surg Oncol. 1999;25:333–335. [PubMed] [Google Scholar]
- 14.Eyden B, Banerjee SS, Shenjere P, et al. The myofibroblast and its tumours. J Clin Pathol. 2009;62:236–249. doi: 10.1136/jcp.2008.061630. [DOI] [PubMed] [Google Scholar]
- 15.Travis WD, Linnoila RI, Tsokos MG, et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol. 1991;15:529–553. doi: 10.1097/00000478-199106000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Larsson LG, Donner LR. Large cell neuroendocrine carcinoma of the parotid gland: fine needle aspiration, and light microscopic and ultrastructural study. Acta Cytol. 1999;43:534–536. [PubMed] [Google Scholar]
- 17.Nagao T, Sugano I, Ishida Y, et al. Primary large-cell neuroendocrine carcinoma of the parotid gland: immunohistochemical and molecular analysis of two cases. Mod Pathol. 2000;13:554–561. doi: 10.1038/modpathol.3880096. [DOI] [PubMed] [Google Scholar]
- 18.Greene L, Brundage W, Cooper K. Large cell neuroendocrine carcinoma of the larynx: a case report and a review of the classification of this neoplasm. J Clin Pathol. 2005;58:658–661. doi: 10.1136/jcp.2004.019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis JS, Jr., Spence DC, Chiosea S, et al. Large cell neuroendocrine carcinoma of the larynx: definition of an entity. Head Neck Pathol. 2010;4:198–07. [DOI] [PMC free article] [PubMed]
- 20.Lewis JS, Jr., Ferlito A, Gnepp DR, et al. Terminology and classification of neuroendocrine neoplasms of the larynx. Laryngoscope 2011;121:1187–93. [DOI] [PubMed]
- 21.Wenig BM. Atlas of head and neck pathology. Amsterdam: Saunders Elsevier; 2008.
- 22.Gnepp DR. Diagnostic surgical pathology of the head and neck. Amsterdam: Saunders Elsevier; 2009.
- 23.Foote FW, Jr, Frazell EL. Tumors of the major salivary glands. Cancer. 1953;6:1065–1133. doi: 10.1002/1097-0142(195311)6:6<1065::AID-CNCR2820060602>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 24.Beahrs OH, Woolner LB, Kirklin JW, et al. Carcinomatous transformation of mixed tumors of the parotid gland. AMA Arch Surg. 1957; 75:605–13; (discussion 13–4). [DOI] [PubMed]
- 25.Brandwein M, Huvos AG, Dardick I, et al. Noninvasive and minimally invasive carcinoma ex mixed tumor: a clinicopathologic and ploidy study of 12 patients with major salivary tumors of low (or no?) malignant potential. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:655–664. doi: 10.1016/S1079-2104(96)80071-0. [DOI] [PubMed] [Google Scholar]
- 26.Auclair PL, Ellis GL. Atypical features in salivary gland mixed tumors: their relationship to malignant transformation. Mod Pathol. 1996;9:652–657. [PubMed] [Google Scholar]
- 27.Ihrler S, Weiler C, Hirschmann A, et al. Intraductal carcinoma is the precursor of carcinoma ex pleomorphic adenoma and is often associated with dysfunctional p53. Histopathology. 2007;51:362–371. doi: 10.1111/j.1365-2559.2007.02736.x. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto K, Yamamoto H, Shiratsuchi H, et al. S100P expression in ductal type of carcinoma ex pleomorphic adenoma. Am J Surg Pathol. 2011;35:346–55. [DOI] [PubMed]
- 29.Altemani A, Martins MT, Freitas L, et al. Carcinoma ex pleomorphic adenoma (CXPA): immunoprofile of the cells involved in carcinomatous progression. Histopathology. 2005;46:635–641. doi: 10.1111/j.1365-2559.2005.02157.x. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto T, Fujii H, Arakawa A, et al. Loss of heterozygosity analysis shows monoclonal evolution with frequent genetic progression and divergence in esophageal carcinosarcoma. Hum Pathol. 2004;35:322–327. doi: 10.1016/j.humpath.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 31.Rossi G, Cavazza A, Sturm N, et al. Pulmonary carcinomas with pleomorphic, sarcomatoid, or sarcomatous elements: a clinicopathologic and immunohistochemical study of 75 cases. Am J Surg Pathol. 2003;27:311–324. doi: 10.1097/00000478-200303000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Gotte K, Riedel F, Coy JF, et al. Salivary gland carcinosarcoma: immunohistochemical, molecular genetic and electron microscopic findings. Oral Oncol. 2000;36:360–364. doi: 10.1016/S1368-8375(00)00016-6. [DOI] [PubMed] [Google Scholar]
- 33.Thompson L, Chang B, Barsky SH. Monoclonal origins of malignant mixed tumors (carcinosarcomas). Evidence for a divergent histogenesis. Am J Surg Pathol. 1996;20:277–285. doi: 10.1097/00000478-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 34.Fowler MH, Fowler J, Ducatman B, et al. Malignant mixed tumors of the salivary gland: a study of loss of heterozygosity in tumor suppressor genes. Mod Pathol. 2006;19:350–355. doi: 10.1038/modpathol.3800533. [DOI] [PubMed] [Google Scholar]
- 35.Vekony H, Leemans CR, Ylstra B, et al. Salivary gland carcinosarcoma: oligonucleotide array CGH reveals similar genomic profiles in epithelial and mesenchymal components. Oral Oncol. 2009;45:259–265. doi: 10.1016/j.oraloncology.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Karpowicz MK, Shalmon B, Molberg KH, et al. Melanoma in a carcinoma ex pleomorphic adenoma of the parotid gland: a case report and putative histogenesis. Hum Pathol. 2011;42:1355–8. [DOI] [PubMed]
- 37.Chan JK, Suster S, Wenig BM, et al. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol. 1997;21:226–234. doi: 10.1097/00000478-199702000-00014. [DOI] [PubMed] [Google Scholar]
- 38.Nagao T, Gaffey TA, Olsen KD, et al. Small cell carcinoma of the major salivary glands: clinicopathologic study with emphasis on cytokeratin 20 immunoreactivity and clinical outcome. Am J Surg Pathol. 2004;28:762–770. doi: 10.1097/01.pas.0000126776.65815.48. [DOI] [PubMed] [Google Scholar]
- 39.Righi PD, Li YQ, Deutsch M, et al. The role of the p53 gene in the malignant transformation of pleomorphic adenomas of the parotid gland. Anticancer Res. 1994;14:2253–2257. [PubMed] [Google Scholar]
- 40.Gedlicka C, Item CB, Wogerbauer M, et al. Transformation of pleomorphic adenoma to carcinoma ex pleomorphic adenoma of the parotid gland is independent of p53 mutations. J Surg Oncol. 2011;101:127–30. [DOI] [PubMed]