

Abstract
Herein we test the following hypotheses: (1) High-risk Human Papillomavirus (HR-HPV) may be involved in the etiology of mucoepidermoid carcinoma (MEC), and (2) The detection rate of HR-HPV in MEC has been increasing over time. Ninety-eight archival MEC specimens from three institutions spanning three decades were studied for HPV16/18 E6/E7 transcripts. RNA was extracted from formalin-fixed paraffin embedded specimens and HPV16/18 E6/E7 expression assessed by nested reverse transcription polymerase chain reaction (RT-PCR). A subset of MEC were also studied for MECT1-MAML2 fusion transcripts by nested RT-PCR and amplicon sequencing. The HPV expression data was validated by immunofluorescence (IF) with monoclonal HPV16/18 E6 antibody, PCR with the GP5+/6+ consensus primers, and sequencing of RT-PCR amplicons. HPV genome was localized by in-situ hybridization with the Ventana Inform HPVIII Family 16 probe. P16INK4a overexpression and aberrant p53 expression were assessed by immunohistochemistry. HPV16 E6/E7 transcripts were demonstrated in (29/98) 30 % of MEC by RT-PCR. HPV18 E6/E7 transcripts were demonstrated in 13/98 (13 %) of MEC by RT-PCR. Seven of 98 tumors (7 %) demonstrated both HPV16/18. No significant association was found between HPV status and gender, age, and tumor site. All 13 HPV18+ MEC were diagnosed between 2001 and 2010, whereas 45 MEC diagnosed from 1977 to 2000 were negative for HPV18 (p = 0.002). By contrast, there was no significant difference with respect to HPV16 detection and date of diagnosis. All MEC that were positive for E6 protein were also HPV16/18 positive by RT-PCR. Sequencing a subset of RT-PCR amplicons confirmed HPV type- and region-specific sequences. PCR using GP5+/6+ consensus primers demonstrated HPV status concordance in 9 of 10 cases. DNA degradation was present in the last case; the RT-PCR amplicons were sequenced from this case which confirmed the presence of HPV type- and region-specific sequences. Strong (+4/+4) and diffuse (>50 %) nuclear and cytoplasmic p16 expression was seen in 64 % of MEC in the glandular regions, and 18 % of MEC in the solid, squamoid regions. No correlation was seen between p16 expression and HPV status. Twenty-nine MEC (22 HPV+ and 7 HPV-negative) were selected for further evaluation for p53 expression. Strong aberrant nuclear p53 expression was present in only 2/22 HPV + MEC (9 %, both Grade 3); no HPV-negative MEC demonstrated aberrant p53 expression. MECT1-MAML2 fusion transcripts were demonstrated in 23/37 (62 %) MEC. No significant association was found between the presence of the MECT1-MAML2 fusion transcripts and tumor grade, HPV status, gender, era of diagnosis (2000 and earlier vs. 2001–2010) or tumor site. We demonstrate for the first time that transcriptionally active HPV16/18 is common to MEC. These findings were validated by demonstrating concordant results by separate PCR with consensus primers, and/or confirming the presence of HPV type- and region-specific sequences in the RT-PCR amplicons. We also visualized E6 viral oncoprotein and HPV genome within tumor cells. HR-HPV is thus potentially implicated in the pathogenesis of MEC. The frequency of HPV18 detection is significantly increased in MEC diagnosed after 2001, whereas we found no differences in the HPV16 detection rates per era of diagnosis.
Keywords: Mucoepidermoid carcinoma, Human Papillomavirus, HPV16/18, E6
Introduction
The significantly increased incidence of oropharyngeal cancers over the last three decades, especially in young women, is known to be caused by high-risk Human Papillomavirus (HR-HPV)-mediated carcinogenesis. The SEER 9 data also demonstrates a trend of increased incidence of mucoepidermoid carcinoma (MEC) in women, ages 15–34 years [1]. This raises the interesting question as to whether HR-HPV can also be involved in MEC carcinogenesis. The issue of HPV-mediated promotion of salivary tumors has been addressed only in a limited manner. Vageli demonstrated HPV16/18 genome in seven of nine parotid tumors, including an oncocytoma, acinic cell carcinoma, Warthin’s tumor, and a pleomorphic adenoma. Detection of viral DNA supports the idea that HPV is “associated” with a specific cancer. However, it does not distinguish whether HPV is transcriptionally active and a potential carcinogenic promoter (e.g. “driver” infection) versus transcriptionally inactive (referred to as “passenger” or “bystander” infection). Relatively high viral copy number was demonstrated in some tumors by quantitative real-time PCR, which suggests an etiologic association [2]. Our approach in this study was to first rigorously assess for HPV16/18 transcripts, as (1) these are the most commonly detected HPV types in oropharyngeal carcinoma, and (2) detection of transcripts supports the idea of a “driver infection” promoting carcinogenesis, and (3) this approach is potentially more sensitive than initially screening samples using PCR and the consensus GP5+/GP6+ general primers. The GP5+/GP6+ primers detect sequences in the L1 region which may be lost upon HPV integration; if HPV is both integrated and episomal forms, one would expect positive results. However if HPV is entirely integrated, PCR using the GP5+/GP6+ primers may result in a false negative reaction [3].
Here we test the following hypotheses: (1) HR-HPV may be implicated in the etiology of MEC, and (2) The detection rate of HR-HPV in MEC has been increasing over time.
Materials and Methods
This study was approved by the Institutional Review Boards at Montefiore Medical Center (MMC), University of Alabama at Birmingham (UAB), and University of the Pacific (UOP).
Sample Procurement
All tumor blocks were recut and the hematoxylin and eosin (H + E) slides were examined. The diagnoses of MEC were confirmed in all tumors studied for HPV; we have previously detailed inclusion criteria and discussed the most common sources for misdiagnoses of MEC [4]. All resection specimens were graded according to the modified grading criteria [4, 5]. The UOP cohort consisted of biopsy specimens, therefore tumor grading for this group was assigned as originally reported. The procurement of tumor specimens was morphologically guided using the H + E slides from corresponding formalin-fixed paraffin embedded (FFPE) blocks as a map. Specimens were procured from the blocks using either sterile punch biopsy needles or sterile razors. Sterile technique was used to prevent contamination between specimens and great care was taken to avoid contaminating samples with adjacent squamous mucosa when present.
Assessment of HPV16/18 Transcriptional Activity: RNA Extraction
All surfaces and equipment were cleaned with RNaseZap (Ambion, Life Technologies, Austin, TX, USA). Procured samples were deparaffinized in 1 ml xylene for 5 min in room temperature. Samples were centrifuged, maximum speed, for 2 min, twice, forming pellets; xylene was removed and samples were rehydrated with graded ethanol (100 %, then 70 %) and centrifuged at 12,000 rpm. The pellets were rinsed in diethyl pyrocarbonate (DEPC) (Sigma, St. Louis, MO, USA) treated water (0.1 %, v/v) and digested in 250 μL of lysis buffer containing 0.1 M EDTA, 0.2 M Tris–HCl at pH 8.5, and Proteinase K (Sigma, St. Louis, MO, USA) stock 20 μg/ml, final concentration of 400 μg/ml and sodium dodecyl sulfate at 1 % final concentration. The lysis buffer was incubated at 37 °C for 30 min prior to addition. Samples were incubated at 55 °C overnight to maximize digestion. RNA extraction was performed by incubating the samples in TRIzol reagent at room temperature for 5 min, adding 200 μl of chloroform to the mixture, shaking vigorously for 15 s, then incubating at room temperature for 3 min, and centrifuging at 12,000 rpm at 4 °C for 15 min. The upper aqueous RNA phase was removed and transferred to a new tube. Ten microliters of 1 mg/mL glycogen (Roche Applied Science) and 1 ml of isopropanol were added and stored overnight at −20 °C. This overnight step promotes efficient precipitation of the fragmented RNA. The RNA was further purified by centrifuging and washing the pellets with 1 mL 70 % ethanol (4 °C), at 12,000 rpm for 30 min at 4 °C. The supernatant was removed and the 70 % ethanol washing step was repeated. The supernatant was again removed, and the RNA pellet was dried for 15 min at room temperature and resuspended in 30 μL RNase free water. Any remaining residual genomic DNA was digested by DNase using the Turbo DNA-Free kit (Ambion) for 15 min at 37 °C, for a final volume of 50 μL. The extracted total RNA was stored at −80 °C. Total RNA concentrations were measured by NanoDrop Epoch Spectrophotometer System (BioTech US). Specimen preparation was performed in a room separate from the nucleotide extraction area, with regular decontamination (DNA AWAY and RNaseAWAY Surface Decontaminants, Molecular BioProducts) of all surfaces and pipettes.
Reverse Transcription, First Amplification, and Nested Real-time Amplification
Table 1 details the primers used; each HPV assessment included eight different primer pairs. For initial assay development, primer specificity was confirmed with RNA isolated from samples of HeLa (HPV18) and SiHa (HPV16) that were embedded in paraffin blocks, and five formalin-fixed paraffin-embedded (FFPE) samples of oropharyngeal carcinomas which were determined to be HPV16+ by HPV Genotyping PCR performed in an independent commercial laboratory, and two FFPE samples of pancreatic carcinoma (HPV negative).
Table 1.
Primer designs for detection of HPV16/18 E6 and E7 cDNA for mucoepidermoid carcinoma
| Primer | Reaction | HPV genome | Amplicon (bp) | Primer (bp) | Primer sequence |
|---|---|---|---|---|---|
| HPV18E6 FP | First PCR | 331 | 250 | 24 | 5′-GAG AAT TAA GAC ATT ATT CAG ACT-3′ |
| HPV18E6 RP | First PCR | 581 | 24 | 5′-TTA TAC TTG TGT TTC TCT GCG TCG-3′ | |
| HPV18E6 FP | Nested real time PCR | 428 | 142 | 24 | 5′-CCA GAA ACC GTT GAA TCC AGC AGA-3′ |
| HPV18E6 RP | Nested real time PCR | 570 | 24 | 5′-TTT CTC TGC GTC GTT GGA GTC GTT-3′ | |
| HPV18E7 FP | First PCR | 644 | 240 | 24 | 5′-AAT GAA ATT CCG GTT GAC CTT CTA-3′ |
| HPV18E7 RP | First PCR | 884 | 24 | 5′-GAC ACA CAA AGG ACA GGG TGT TCA-3′ | |
| HPV18E7 FP | Nested real time PCR | 725 | 135 | 24 | 5′-CAT CAA CAT TTA CCA GCC CGA CGA-3′ |
| HPV18E7 RP | Nested real time PCR | 860 | 24 | 5′-GAA ACA GCT GCT GGA ATG CTC GAA-3′ | |
| HPV16E6 FP | First PCR | 299 | 257 | 24 | 5′-TGT TTA AAG TTT TAT TCT AAA ATT-3′ |
| HPV16E6 RP | First PCR | 556 | 24 | 5′-CAG CTG GGT TTC TCT ACG TGT TCT-3′ | |
| HPV16E6 FP | Nested real time PCR | 361 | 164 | 24 | 5′-AAC ATT AGA ACA GCA ATA CAA CAA-3′ |
| HPV16E6 RP | Nested real time PCR | 525 | 24 | 5′-CTG CAA CAA GAC ATA CAT CGA CCG-3′ | |
| HPV16E7 FP | First PCR | 591 | 249 | 24 | 5′-ATA TAT GTT AGA TTT GCA ACC AGA-3′ |
| HPV16E7 RP | First PCR | 840 | 20 | 5′-GAT GGG GCA CAC AAT TCC TA-3′ | |
| HPV16E7 FP | Nested real time PCR | 681 | 127 | 24 | 5′-TCC AGC TGG ACA AGC AGA ACC GGA-3′ |
| HPV16E7 RP | Nested real time PCR | 831 | 24 | 5′-GCA CAC AAT TCC TAG TGT GCC CAT-3′ | |
| GAPDH FP | PCR | 1003 | 179 | 24 | 5′-ACT GAG CAC CAG GTG GTC TCC TCT-3′ |
| GAPDH RP | PCR | 1182 | 20 | 5′-TTA CTC CTT GGA GGC CAT GT-3′ |
The Bio-Rad iScript cDNA synthesis kit was used for reverse transcription of total RNA into cDNA. Samples were first screened for GAPDH expression. All samples which were GAPDH negative after first PCR were submitted to a second round of PCR. Only GAPDH positive samples were studied for HPV. Cross contamination was prevented by using separate sterile tips for each sample; for nested PCR, RT-PCR amplicon tubes were spun before the tubes were opened, and separate Eppendorf tube openers were used for transferring RT-PCR products to the nested PCR mix.
The first amplifications for HPV16E6, HPV16E7, HPV18E6 and HPV18E7 were performed using 30 cycles of polymerase chain reaction (PCR); the 50 μl samples contained at least 50 ng of cDNA, 0.3 μM final concentration of each of the first primer set (around 250 bp) and 1.25 U of Terra PCR Direct Polymerase Mix (Clontech, USA). The following thermocycler conditions were used: denaturation at 94 °C for 2 min, 40 cycles of denaturation at 94 °C for 30 s, annealing at 57 °C for 60 s, extension at 72 °C for 2 min, and final extension at 72 °C for 10 min. The initial denaturation step in “hot start” RT-PCR occasionally results in sample drop-out due to evaporation; therefore all reactions were run in triplicate. Samples were deemed positive if two of three reaction wells were positive. Every reaction included the following controls: HeLa (HPV18), SiHa (HPV16), FFPE HPV16+ oropharyngeal carcinoma, FFPE pancreatic carcinoma (HPV negative), and a blank sample without template.
The second amplifications used real-time PCR (RT-PCR), the Maxima SYBR Green/Fluorescein qPCR master Mix (Fermentas) and the Opticon2 detection system (Bio-Rad). This reaction contained 1/50 volumes of the first direct PCR reaction, 0.3 μM of the second set of nested primers and the Maxima SYBR Green/Fluorescein qPCR Master Mix. These assays were performed in triplicate under the following conditions: 10 min pre-incubation at 95 °C, 40 cycles of denaturation at 95 °C for 15 s, annealing at 58.6 °C for 30 s, and extension at 72 °C for 30 s and final extension at 72 °C for 5 min. The same positive and negative controls were used as described above. Amplicons were verified by delta cycle threshold (delta CT).
We avoided false positive results by (1) adhering to sterile technique, (2) requiring the detection of both type-concordant transcripts (E6 and E7) in order to designate specimens as positive, and (3) performing melting curve analysis on the amplicons from the final reaction. All negative control reactions were appropriate, including HeLa (HPV18) as a negative control for HPV16 RT-PCR and SiHa (HPV16) as a negative control for HPV18 RT-PCR.
Immunofluorescence for HPV16/18 E6
Paraffin-embedded tumor tissues were serially sectioned at 5 μm, deparaffinized and rehydrated through graded ethanol. Antigen retrieval was achieved by citrate-based antigen unmasking solution (Vector Labs. Inc. CA, USA) incubation in a 90 °C water bath for 10 min. Slides were blocked with 5 % normal goat serum and 5 % BSA in PBS for 1 h at room temperature, and then incubated overnight at 4 °C with mouse monoclonal HPV16/18 E6 (C1P5) primary antibody (Santa Cruz Inc., CA, USA) in blocking buffer, at a dilution of 1:50. After extensive washing, sections were incubated with goat anti-mouse secondary antibody conjugated to Alexa 488 (Invitrogen, CA, USA) for 45 min, at 1:500 dilution. Diamidino-2-phenylindole hydrochloride (DAPI) staining was performed to localize nuclei. Positive controls were FFPE section of HeLa and SiHa cells. Negative control tissues were treated in the same way, but incubated only with secondary Alexa 488-labeled antibody. Slides were washed and mounted in Vectashield medium for fluorescence (Vector Labs. Inc., CA, USA). Slides were examined using an immunofluorescence microscope, blinded to HPV status. Tumors were classified as either positive or negative based on a detection cut-off of strong nuclear and/or cytoplasmic staining in tumor cells.
Validation of HPV+ Samples by PCR
For purposes of validation, ten MEC were selected (3 HPV16+/18+, 3 HPV18+, 1 HPV16+, 3 HPV-negative) for auto-nested PCR using the general consensus primers GP5+/6+ to amplify a conserved region in the HPV L1 gene [6–8]. Samples were procured as described above, deparaffinized in xylene, rehydrated with graded ethanol, and air-dried. Pellets were resuspended in 300 μl of digestion buffer (10 mM Tris–HCl pH 8.3, 1 mM EDTA, 0.5 % Tween 20, 20 mg/ml proteinase K) and incubated at 56 °C while rocking for 16 h, until the tissue was completely digested. Proteinase K was inactivated by incubation for 20 min at 95 °C. DNA was extracted using phenol/chloroform/isoamyl alcohol (25:24:1; v/v) which was added to each tube at a volume equal to that of the digestion mixture (300 μl). The tubes were vortexed for 10–15 s and centrifuged at 14,000 rpm for 5 min and the supernatant was transferred to new tubes and mixed with 1/10 volume of 3 M sodium acetate and 2.5 volume of 99 % ice-cold ethanol, incubated at −20 °C for 60 min, then centrifuged at 14,000 rpm for 5 min. Pellets were washed with 1 ml of 70 % ethanol, centrifuged for 5 min at 14,000 rpm, after which the ethanol was removed and the specimen air-dried. Dry pellets were resuspended in the DNase/RNase free water. The final PCR mixture contained 0.02 U/μl of iProof HF DNA Polymerase, 200 μmol dNTP, 0.5 μM of each primer, 0.4 mM Mg2+, 1× high-fidelity PC buffer and 200 ng of DNA template. The following thermocycler conditions were used for 40 cycles: initial denaturation at 94 °C for 4 min, subsequent denaturation at 94 °C for 60 s, annealing at 42 °C for 60 s, and extension at 72 °C for 30 s with final extension at 72 °C for 4 min. 10 μl of the reaction mixture were electrophoresed through 2 % agarose gel containing 0.5 μg/ml ethidium bromide and visualized under an ultraviolet transilluminator.
DNA samples negative after 40 cycles were re-amplified by auto-nested PCR using the same GP5+/6+ primer pair and 2.5 μl template. The same thermocycler conditions as above will be used for 30 cycles. 10 μl of the reaction mixture were again electrophoresed through 2 % agarose gel containing 0.5 μg/ml ethidium bromide and visualized under an ultraviolet transilluminator. All PCR reactions included DNA from appropriate positive controls (HeLa, SiHa, and FFPE HPV16+ oropharyngeal SCC), negative controls (FFPE pancreatic carcinoma) and a template-free negative control.
Validation of HPV+ Samples by Sequencing
For purposes of validation, eight random HPV16+ MEC and two HPV16/18 + MEC were selected for sequencing of PCR amplicons. The PCR products from Siha and Hela cells were used as HPV type-specific, region-specific controls. PCR products were excised from 2 % Tris–acetate-EDTA (TAE) agarose gels and purified with a gel extraction kit (QIAGEN) according to the manufacturer’s instructions. Purified PCR products were sequenced using 5 pmol of either forward or reverse type specific E6 and E7 primers on an ABI 3730 sequencer analyzer (Applied Biosystems, Foster City, CA, USA). DNA sequences were analyzed by using the BLASTN algorithm (National Center for Biotechnology Information).
In-situ Hybridization (ISH) for HR-HPV
For purposes genome localization, twelve MEC (2 HPV16+, 4 HPV18+, 3 HPV16/18+, and 3 HPV negative by RT-PCR) were selected for in-situ hybridization (ISH) with the Inform HPVIII Family 16 Probe (B) (Ventana Medical Systems Inc, USA) following the HPVIII iview Blue + V3 protocol. This probe detects the following high-risk genotypes: HPV-16, -18, -31, -33, -35, -39, -45, -51, -52, -56, -58, and -66. ISH was performed in the UAB clinical immunohistochemistry facility on a Benchmark XT Autostainer (Ventana Medical Systems Inc, USA) following the manufacturer’s suggested protocol. Nitroblue tetrazolium chloride/5-Bromo-4-chloro-3-indolyl phosphate was the chromogenic reagent; slides are conterstained with Red Counterstain II. The positive control consisted of HPV16+ oropharyngeal carcinoma; negative controls consisted of brain tissue. The slides were read blinded to HPV status. MEC with an intranuclear dot staining pattern comprised of either single or multiple signals, and/or a distinctly strong granular cytoplasmic staining pattern, were deemed as positive [9, 10].
Immunohistochemistry for p16
Epitope retrieval was performed with 0.02 M concentration of citrate buffer (pH 6.0) heated at 97 °C for 20 min. Immunostaining was performed with a semi-automated immunostainer (Thermo Scientific, Labvision 720- Fremont, CA, USA) and the UltraVision LP polymer system. The E6H4 clone of p16 antibody was used (MTM laboratories, Westborough, MA, USA) at 1:2 dilution. Diaminobenzidine tetrachloride (DAB) chromagen was used to visualize the antibody-antigen complex; slides were counterstained with hematoxylin. Positive controls consisted of a HPV16+ tonsillar carcinoma with strong diffuse nuclear and cytoplasmic p16 expression. The negative control slides consisted of tissue sections processed without primary antibody. P16 expression was categorized for both staining intensity (scale 0–4) and percent staining distribution, for both the nuclei and cytoplasm of both squamoid and mucinous tumor elements. The slides were examined blinded to HPV status.
Immunohistochemistry for p53
Heat induced epitope retrieval was performed using 0.02 M citrate buffer (pH 6.0) at 120 °C for 2 min. Immunostaining was accomplished with a semi-automated immunostainer (Ventana Inc. Tucson, AZ, USA) and a biotin-free, multimer-based technology detection system. Prediluted anti-p53 antibodies (clone: Bp53-11, Ventana) was used, which detects both mutant and wild type p53 nuclear phosphoprotein. Diaminobenzidine tetrachloride was used to visualize the antibody-antigen complex; slides were counterstained with hematoxylin. Colonic adenocarcinoma was used as the positive control; the negative control slides consisted of tissue sections of each case processed without the addition of primary antibody. Strong nuclear staining was considered as positive/aberrant p53 expression.
RT-PCR Assay for the CRTC1/MAML2 Transcript
The first amplification was performed using 0.3 μg of MEC cDNA in the final mixture of iProof HF buffer, 200 μM dNTP mix, 0.02 U/μl of iProof High-Fidelity DNA polymerase, 0.5 mM of Mg2+ and 0.5 μM of each of the following primers: CRTC1: 5′-AAGATCGCGCTGCACAATCA-3′ and MAML2: 5′-GGTCGCTTGCTGTTGGCAGG-3′ [11]. The PCR cycling consisted of initial denaturation 98 °C for 30 s, 35 cycles of 98 °C denaturation for 10 s, annealing 60 °C for 30 s, extension 72 °C for 30 s, with one final extension 72 °C for 10 min. All samples were subjected to nested RT-PCR using 0.2 μl of amplified product from the initial PCR in Maxima® SYBR Green/ROX qPCR Master Mix (Fermentas, Inc.) The amplification conditions were: 98 °C for 5 min, 35 cycles of 98 °C denaturation for 30 s, annealing 55 °C for 30 s, extension 72 °C for 30 s, and final extension 72 °C for 5 min. The primers used for the second nested RT-PCR for CRTC1/MAML2 were CRTC1: 5′-GGAGGAGACGGCGGCCTTCG-3′ and MAML2: 5′-TTGCTGTTGGCAGGAGATAG-3′ [9]. The band size of the final amplicon was 117 bp, which was detected by 2 % agarose gel electrophoresis and visualized with ethidium bromide staining under UV light. The presence of intact RNA was verified for each sample by the simultaneous amplification for GAPDH expression. The positive control consisted of RNA extracted from a transcript positive MEC specimen which was confirmed by sequencing (see below). The negative control was comprised of RNA extracted from an oral cavity squamous cell carcinoma.
Sequencing of CRTC1/MAML2 Transcripts
For purposes of validation, the RT-PCR amplicons were purified from gels using QIAquick Gel Extraction Kit (Qiagen) and sequenced using ABI Big DyeTM v3.1 dye terminator cycle sequencing (UAB, Heflin Center for Genomic Sciences).
Statistical Analysis
Associations between HPV status (stratified as HPV16+, HPV18+, and either HPV16 and/or HPV18), p16 expression, p53 expression, MECT1-MAML2 transcription, gender, age, tumor grade, site, year of diagnosis were assessed by Student T test. Tumor site was stratified as either major versus minor salivary sites, oropharynx versus nonoropharynx, oropharynx including RMT versus others, and dependent intraoral sites versus nondependent sites. All tests were 2-tailed, with a probability value of less than 0.05 considered statistically significant.
Results
Assessment of HPV16/18 Transcriptional Activity
Ninety-eight MEC (58 from UP, 21 from UAB, and 19 from MMC) had amplifiable RNA suitable for HPV analysis confirmed by GAPDH expression. The HPV data is presented in Table 2. HPV16 E6/E7 expression was demonstrated 29/98 (30 %) of MEC (Fig. 1). HPV18 E6/E7 expression was demonstrated in 13/98 (13 %) of MEC. Seven of 98 tumors (7 %) tested positive for both HPV16/18. Only 2 and 5 %, of tumors had discordant E6/E7 results for HPV16 and HPV18, respectively, and were considered negative. No significant association was found between HPV status (HPV16+, HPV18+, either HPV16 and/or HPV18) and gender, age, and tumor site stratified four different ways: minor versus major salivary sites, oropharynx (tonsil, base of tongue) versus non-oropharynx, oropharynx plus retromolar trigone versus all others, and dependent sites versus nondependent sites (parotid and palate).
Table 2.
Summary of HPV data for 98 patients with mucoepidermoid carcinoma
| HPV16+ only (n = 22) | HPV18+ only (n = 6) | HPV16+/18+ (n = 7) | HPV-negative (n = 63) | |
|---|---|---|---|---|
| Female: male | 1.75:1 | 2:1 | 1:1.3 | 3.2:1 |
| Mean age | 48 | 48 | 48 | 51 |
| Major salivary sites | 27 % (6/22) | 33 % (2/6) | 43 % (3/7) | 17 % (11/63) |
| Oropharyngeal sites | 5 % (1/22) | 17 % (1/6) | 0 | 3 % (2/63) |
| Oral cavity sites | 68 % (15/22) | 50 % (3/6) | 57 % (4/7) | 79 % (50/63) |
| 1977–2000 | 50 % (11/22) | 0 | 0 | 54 % (34/63) |
| 2001–2010 | 50 % (11/22) | 100 % (6/6) | 100 % (7/7) | 46 % (29/63) |
| MEC1-MAML2 translocation | 77 % (10/13) | 66 % (2/3) | 25 % (1/4) | 59 % (10/17) |
Fig. 1.

Quantitative real time nested PCR for HPV16E6 cDNA in MEC and confirmation of amplicon size. Top This graph demonstrates the detection threshold of fluorescent amplicons (SYBR Green, Opticon2, BioRad) by PCR cycle. Four samples of MEC are positive. Samples 3 and 11 have high efficiency thresholds indicating relative high number of E6 transcripts. SiHa cells represent the positive control, HeLa the negative control. Bottom Gel electrophoresis confirmation of amplicon size. Lanes 1–6 represent MEC. The samples in lanes 1 and 3 are positive for both HPV16 E6 and E7, while the samples in lanes 2, 4, 5, and 6 are negative. The positive controls are represented in lane 8 (HPV16+ oropharyngeal squamous carcinoma) and SiHa cells. The negative controls are lane 10 (no template) and HeLa cells
Figure 2 shows a bar graph for HPV status over specimen year. All 13 HPV18 + MEC were diagnosed between 2001 and 2010, whereas 45 MEC diagnosed from 1977 to 2000 were negative for HPV18 (p = 0.002). By contrast, no significant difference was seen with respect to HPV16 detection and date of diagnosis: 11 HPV16 + MEC were diagnosed between 1989 and 2000, and 18 HPV16 + MEC were diagnosed between 2001 and 2010. HPV16+ was detected significantly more often in the San Francisco cohort (p = 0.0076) as compared to the patients from Alabama and New York.
Fig. 2.

Transcriptionally active HPV16/18 over era of diagnosis. This bar graph illustrates the HPV status by number of MEC studied per year of diagnosis. All 98 cases were studied for both HPV16 and HPV18. The purple and blue areas on the bars represent the number of cases that are negative for HPV18, and HPV16, respectively. The gold and red areas on the bars represent the number of cases that are positive for HPV16, and HPV18, respectively. Double positive cases are not indicated here. The first HPV16+ cases were detected in specimens from 1989. No significant difference was seen with respect to HPV16 detection and date of diagnosis: 11 HPV16+ MEC were diagnosed between 1989 and 2000, and 18 HPV16+ MEC were diagnosed between 2001 and 2010. All 13 HPV18+ MEC were diagnosed between 2001 and 2010, whereas 45 MEC diagnosed from 1977 to 2000 were negative for HPV18 (p = 0.002)
We then asked if there were any histological features associated with HPV-positive MEC. The tumors were re-reviewed blinded to HPV status, and classified with respect to basaloid tumor cell component. Three MEC were classified as predominantly basaloid; all three were HPV positive. Ten MEC were classified as mixed basaloid and nonbasaloid type; four of these tumors were HPV positive. While this finding is interesting, it was not predictive of HPV status as the remaining 22 HPV positive MEC were classified as “nonbasaloid”.
Assessment of E6 Viral Oncoprotein
Eighty-four MEC were studied by IF with the monoclonal C1P5 antibody which detects E6 protein of both HPV16/18. Eighteen tumors displayed nuclear and or cytoplasmic tumor staining; the protein was detected in both mucinous and squamoid elements (Figs. 3, 4). All cases positive by IF were HPV16/18 positive by RT-PCR. Fourteen additional tumors were negative by IF and positive by RT-PCR (sensitivity 55 %, specificity 100 %).
Fig. 3.

Immunofluorescence (IF) for HPV16/18 E6 protein in controls. Formalin fixed paraffin embedded HeLa (a, b, HPV18+) and SiHa cells (c, d, HPV16+) as positive (a, c) and negative (b, d) controls. The negative control tissues were incubated with secondary Alexa 488-labeled antibody only
Fig. 4.

Immunofluorescence (IF) for HPV16/18 E6 protein in MEC. a, b demonstrate low- and high-power, respectively, of an HPV-positive, cystic, low-grade MEC with proliferation of basaloid type cells. c, e demonstrate hematoxylin and eosin stained areas of a MEC which is HPV positive. d, f represent the corresponding regions demonstrating positive IF staining using an antibody to HPV16/18 E6 protein. Tumor nuclear and cytoplasmic staining is seen (bright green) which correlates with both the glandular and squamoid elements
Validation of HPV+ Samples by PCR
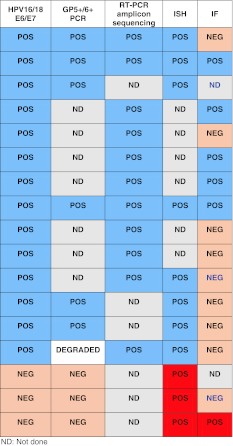
Six MEC that were positive by RT-PCR (three HPV16+/18+, two HPV18+, one HPV16+) were also positive by PCR with GP5+/6+ consensus primers; three MEC that were HPV-negative by RT-PCR were also negative by PCR. One MEC (HPV18+ by RT-PCR) was negative by PCR due to DNA degradation. This was resolved by amplicon sequencing from the corresponding RT-PCR, which confirmed HPV18–E6 and E7-region specific sequences (Table 3).
Table 3.
Validation of RT-PCR data by PCR and sequencing
Validation of HPV+ Samples by Sequencing
DNA sequencing of RT-PCR amplicons from the eight HPV16 + MEC and two HPV16/18+ MEC samples confirmed that the amplified product corresponded to the expected HPV-type-specific and region-specific sequences (Table 3).
In-situ Hybridization for HR-HPV
Eight of the nine HPV + MEC studied by ISH were deemed to have positive hybridization signals. (Table 3) Nuclear signals were typically single whereas cytoplasmic signals were typically prominent and multiple. Four MEC had both cytoplasmic and intranuclear signals, three MEC had only cytoplasmic signals, and one MEC had only nuclear signals. One HPV + MEC was ISH negative (Fig. 5). Three HPV-negative MEC also had positive signals; these cases are classified as false positives.
Fig. 5.

In-situ Hybridization with High-Risk HPV probe. a Positive control of HPV16+ oropharyngeal carcinoma demonstrating predominantly single dot signals indicative of integrated HPV. b MEC with multiple signals probably representing mixed episomal and integrated HPV. c MEC with single nuclear signals consistent with integrated HPV. d Strong granular cytoplasmic signals consistent with episomal HPV
p16INK4a Overexpression
Eighty-four MEC were studied by IHC for p16 expression; 15/84 cases (18 %) demonstrated strong (+4) nuclear and cytoplasmic p16 expression in ≥50 % of solid MEC components; 53/84 (64 %) of MEC demonstrated strong (+4) nuclear and cytoplasmic p16 expression in ≥50 % of the mucinous glandular components. No association was found between p16 expression and HPV status.
Aberrant P53 Expression
As HPV-mediated carcinogenesis in the oropharynx is usually associated with wild-type, non-mutated p53 tumor suppressor gene, we selected 29 MEC (22 HPV+ and 7 HPV-negative) for further evaluation for p53 expression. Aberrant p53 expression (defined as strong nuclear expression) was present in only 2/22 HPV + MEC (9 %, both Grade 3); no HPV-negative MEC demonstrated aberrant p53 expression. There was no association between the presence of HPV and aberrant p53 expression. There was a trend regarding association between aberrant p53 expression and Grade 3 MEC (p = 0.069).
MECT1-MAML2 Translocation
A subset of 37 samples were studied for MECT1-MAML2 fusion transcripts, which were detected in 23/37 (62 %) MEC. Amplicon sequencing in all 23 RT-PCR positive cases confirmed the presence of the t(11,19) MECT1-MAML2 fusion gene (Fig. 6). No significant association was found between MECT1-MAML2 fusion transcripts and HPV status, gender, tumor grade, tumor site, or era of diagnosis (2000 and earlier vs. 2001–2010).
Fig. 6.

MECT1-MAML2 fusion transcripts in MEC. a Gel electrophoresis from second nested RT-PCR reaction. a–f represent MEC. MEC in lanes A, C, and E demonstrate the MECT1-MAML2 fusion transcripts, at 117 bp, while the samples in lanes B, D, and F are negative. Lane G represents the positive control: MEC with MECT1-MAML2 fusion transcripts from a previous reaction, which was confirmed by sequencing. The negative controls are lane H (no template) and lane I (squamous cell carcinoma). Bottom Panels The MEC in panelB is negative for fusion transcripts; the reaction product did not migrate at 117 bp and the nucleotide sequences are different from those seen in panels C and D. The MEC in panels C and D are positive for the MECT1-MAML2 fusion transcript by RT-PCR. DNA nucleotide amplicon sequencing confirmed the presence of the t(11;19) CRTC1/MAML2 fusion gene :CGG*CTCCAGGGTTCCTTGAAAA (* is the breakpoint)
Discussion
The data demonstrate that transcriptionally active HR-HPV infection is common to MEC. We detected HPV16 E6/E7 transcripts in 30 % of MEC, HPV18 E6/E7 transcripts in 13 % of MEC, and 7 % of MEC contained transcripts for both HPV16/18. We were able to demonstrate a significantly increased HPV18 detection rate over the last decade, as compared to MEC diagnosed from 1977 to 2000. One might question if RNA degradation in the older specimens could be responsible for this finding. This is unlikely as the detection rates of HPV16 E6/E7 transcripts did not vary significantly over time, supporting that the increased detection of HPV18 in the last decade is a true phenomenon. One limitation of this study is that the E6 primers used did not detect the E6* splice variant which may have underestimated the rate of productive HPV16 infection.
The lack of association between HPV and MEC tumor site was somewhat surprising. Only four MEC arose from sites usually associated with HPV-mediated carcinogenesis (tonsil and tongue base); two of them were HPV positive. Twelve MEC from this cohort arose in the retromolar trigone (RMT, 12 %), which is greater than the reported distribution of 791 MEC from the Armed Forces Institute of Pathology (40/791 or 5 % RMT MEC) [12]. Six RMT MEC were HPV+. However no association between HPV and tumor site was demonstrated, despite different stratifications (major versus minor salivary sites, oropharynx versus nonoropharynx, oropharynx including RMT versus others, and dependent intraoral sites versus nondependent sites). The fact that HPV was detected in parotid and submandibular gland tumors is interesting and suggests a mechanism of systemic HPV exposure.
We demonstrate that HPV localization is not limited to MEC squamoid components; HPV is also localized in the mucinous/glandular tumor elements (Figs. 4 and 5). This is not surprising as HR-HPV also promotes uterine cervical adenocarcinomas [13] including rare uterine cervical MEC [14]. As previously mentioned, Vageli published a study in 2007 demonstrating HPV16/18 genome in seven of nine parotid tumors, including oncocytoma, acinic cell carcinoma, Warthin’s tumor, and pleomorphic adenoma [2]. A small number of recent publications have subsequently investigated HPV status in salivary tumors. Boland and colleagues found two of 27 (7 %) adenoid cystic carcinomas HR-HPV+ using the Ventana high-risk HPV family 16 in-situ hybridization probe [15]. Brunner and colleagues found two of six (33 %) MEC HR-HPV+ using the Dako Genepoint in-situ hybridization probe; 32 other salivary carcinomas were investigated and found to be HPV-negative [16]. Hafed and colleagues used the Digene HPV16/18 probe to investigate 34 salivary tumors, however their illustrations are concerning for nonspecific staining [17]. In this study, we used the Ventana probe to investigate the physical status of HPV; we demonstrate that HPV is more commonly episomal in than integrated in HPV + MEC (Fig. 5). Previously, we have reported that this probe has a sensitivity of 59 % (95 % CI: 39–78) and specificity of 58 % (95 % CI: 45–71) in the context of 110 head and neck squamous carcinomas (all sites) with an overall HR-HPV detection rate of 28 % by PCR [18]. Therefore, ISH is not the optimal for either primary investigation or validation of HPV status.
It is well accepted that transcriptionally active HR-HPV infection (“driver infection”) is etiologically responsible for a subset of oropharyngeal squamous carcinomas. What is the significance of this finding in MEC? The data suggest that HR-HPV may be involved in promoting MEC, however the data do not prove causation. The usual accepted criteria to support HPV-mediated carcinogenesis are: (1) The demonstration of transcriptionally active, and therefore biologically relevant, high-risk HPV infection, (2) The overexpression of p16 as a functional surrogate biomarker for functional abrogation of Rb tumor suppressor protein by E7, (3) Evidence of viral integration, and lastly (4) Wild type 53 protein. However, these criteria have important caveats. The p16 gene may be methylated and thus silenced in HPV-mediated cancers. HPV integration is not necessary for promoting carcinogenesis. Lastly, while the HR-HPV+/wild-type p53 profile is the expected genetic phenotype of never-smokers with HPV-mediated oropharyngeal cancers, this polarized relationship is not observed in patients with oral cavity cancer [19]. Furthermore, nondistruptive p53 mutations can be demonstrated in HPV-mediated head and neck cancers and are thought to have an additive impact on overall p53 functional loss [20]. With these caveats in mind, we queried as to whether p16INK4a overexpression correlated with HR-HPV transcriptional activity, as with the tonsillar cancer paradigm which typically demonstrate HR-HPV transcripts and p16INK4a overexpression. We found no significant association between HPV transcription and p16 overexpression. There are a number of possible explanations. Guo and colleagues studied MEC and demonstrated that p16INK4a promoter hypermethylation, homozygous deletions and point mutations in exons 1 and 2 are common [21].
We also queried as to whether there is an association between wild-type (unmutated) p53 and HPV status in a subgroup of MEC. Strong aberrant nuclear p53 expression was present in only 2/22 HPV + MEC (9 %, both Grade 3); no HPV-negative MEC demonstrated aberrant p53 expression. There was a trend regarding association between aberrant p53 expression and Grade 3 MEC (p = 0.069). There are few large studies regarding p53 status in MEC. Kiyoshima studied 27 MEC for mutations in exons 5–8 by PCR-SSCP (Single Strand Conformation Polymorphism) and aberrant nuclear p53 protein expression by IHC [22]. Mutations in exons 5–8 were demonstrated in four of 27 (14.8 %) of MEC. With respect to aberrant p53 expression, Kiyoshima specified percent distribution of positive cells, but failed to specify expression intensities; this is important as only strong nuclear staining may represent aberrant expression. Having said that, significant p53 expression (>50 %) was seen in five of 27 (18.5 %) and correlated significantly with high-grade. Wolfish and colleagues found strong nuclear aberrant p53 expression in 5–10 % of tumor cells, in six of 14 (43 %) sinonasal MEC; there was no correlation with grade [23]. Other studies on p53 status involve smaller numbers of specific salivary tumor types and are more anecdotal in nature [24, 25]. In the context of head and neck squamous cell carcinomas (HNSCC), as p53 mutations are common and a significant association between active HPV infection and wild-type (WT) p53 is observed, then HR-HPV is likely to promote carcinogenesis in HR-HPV +/WTp53 cancers via the E6 oncoprotein targeting of WTp53 for degradation through E6AP; this abrogates the functional pro-apoptotic pathway [26–28]. On the other hand, the published data support that an intact pro-apoptotic pathway is common to MEC. Thus functional p53 loss is not requisite for the promotion of MEC. In other words, the association of transcriptionally active HR-HPV with WTp53 in MEC does not have carry the same significance as it does with oropharyngeal carcinoma, as WTp53 is a common finding in MEC. But it is possible that functional abrogation of the p53 pathway may still have an additive impact on HR-HPV+/WTp53 MEC.
MEC is frequently associated with the reciprocal chromosome translocation t(11;19) (q14–21;p12–13) [11, 29–38]. The translocation breakpoint involves the MECT1 gene (also known as CRTC1, TORC1, or WAMOTP1) at chromosome 19p13, and the MAML2 gene at chromosome 11q21. MECT1 (mucoepidermoid translocated-1) expression is normally limited to fetal brain and liver, adult heart, skeletal muscle and liver, and normal salivary tissue [30]. The MECT1 protein activates CREB (cAMP response element binding protein) mediated transcription, which is involved in cell proliferation and differentiation in response to cytokines and growth factors [36]. The MAML (mastermind-like) protein is widely expressed in all tissues and is involved in the Notch signaling pathway. This pathway plays a complex, context-dependent role in cellular proliferation and differentiation. The MECT1-MAML2 translocation results in a novel fusion protein which enhances loss of contact inhibition in immortalized epithelial cells [29, 37]. Early experiments demonstrated that the MECT1-MAML2 fusion protein activated downstream Notch target proteins such as HES1, in a Notch ligand-independent manner [30]. However, expression array analyses have demonstrated that upregulated MECT1-MAML2 activates cAMP/CREB regulated genes, such as PEPCK1/PCK1, AREG, and NR4A3/NOR, but not Notch target genes such as the Hes family members [36, 37].
We demonstrated MECT1-MAML2 fusion transcripts in a subgroup of 23/37 (62 %) MEC. We found no association between MECT1-MAML2, HPV status or MEC grade. Larger studies on clinical samples have demonstrated the MECT1-MAML2 translocation in 34–81 % of MEC [11, 32–35]. By contrast, the MECT3-MAML2 translocation is present in a much smaller proportion (6 %) [37]. The MECT1-MAML2 translocation appears to be highly specific for MEC. While this translocation has been occasionally detected in Warthin’s tumors, [32, 33] other studies which included HNSCC, other salivary malignancies, and a large number of Warthin’s tumors [33, 37], have all been negative for the MECT1-MAML2 translocation.
Although we did not find an association between the translocation and HPV, it is still reasonable to question if an interaction between HPV-mediated carcinogenesis and the MECT1-MAML2 translocation is possible. As both CREB and MAML2 are widely expressed in all tissues, it has been suggested that the MECT1-MAML2 translocation is an early event in tumor initiation. We speculate a potential interaction between the MECT1-MAML2 translocation and HR-HPV-mediated transformation through the transcriptional co-activator p300, a CREB pathway regulatory molecule. The MECT1-MAML2 fusion protein has been demonstrated to upregulate p300 transcription [37]. Overexpression of p300 activates the HPV long control region (LCR) which in turn regulates E6/E7 transcription [39, 40]. Therefore, we speculate that the MECT1-MAML2 translocation may promote sustained E6/E7 overexpression through the effect of p300 on LCR.
Perhaps one of the most important developments in head and neck oncology of the past decade was the demonstration that patients with HPV-mediated oropharyngeal cancers enjoy significantly improved outcomes as compared to patients with HPV-negative counterpart cancers. This has become the basis for clinical trials investigating the impact on “treatment de-intensification” for patients with HPV-mediated oropharyngeal cancers. The significance of HPV in non-oropharyngeal HNSCC, such as the oral and laryngeal carcinomas, remains uncertain and is currently under investigation [41]. Could there be any prognostic significance to the presence of HR-HPV transcripts in salivary neoplasia? We doubt that HR-HPV is a significant prognosticator for MEC patients, given the greater inherent biological heterogeneity of MEC as compared to HNSCC.
In conclusion, we demonstrate the presence of transcriptionally active, biologically relevant, HR-HPV in approximately one-third of MEC. Given that MEC1-MAML2 fusion transcripts are thought to be an early event, it is possible that HR-HPV oncoproteins promote MEC as a later event in multistep carcinogenesis. This could be through the additive impact of the HR-HPV E6 and E7 oncoproteins on overall loss of tumor suppression function. This study broadens the scope of associations between HR-HPV and head and neck neoplasia, and can serve as the rationale for future studies furthering our understanding of the etiology of salivary tumors.
References
- 1.Bleyer A. Cancer of the oral cavity and pharynx in young females: increasing incidence, role of human papilloma virus, and lack of survival improvement. Semin Oncol. 2009;36:451–459. doi: 10.1053/j.seminoncol.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Vageli D, Sourvinos G, Ioannou M, Koukoulis GK, Spandidos DA. High-risk human papillomavirus (HPV) in parotid lesions. Int J Biol Markers. 2007;22:239–244. doi: 10.1177/172460080702200401. [DOI] [PubMed] [Google Scholar]
- 3.Duray A, Descamps G, Arafa M, et al. High incidence of high-risk HPV in benign and malignant lesions of the larynx. Int J Oncol. 2011;39:51–59. doi: 10.3892/ijo.2011.1031. [DOI] [PubMed] [Google Scholar]
- 4.Bai S, Clubwala R, Adler E, Sarta C, Schiff B, Smith RV, Gnepp D, Brandwein-Gensler M. Salivary mucoepidermoid carcinoma: a multi-institutional review of 76 patients. Head Neck Pathol 2012; doi:10.1007/s12105-012-0405. [DOI] [PMC free article] [PubMed]
- 5.Brandwein MS, Ivanov K, Wallace DI, Hille JJ, Wang B, Fahmy A, Bodian C, Urken ML, Gnepp DR, Huvos A, Lumerman H, Mills SE. Mucoepidermoid carcinoma: a clinicopathologic study of 80 patients with special reference to histological grading. Am J Surg Pathol. 2001;25:835–845. doi: 10.1097/00000478-200107000-00001. [DOI] [PubMed] [Google Scholar]
- 6.de Roda Husman AM, Walboomers JM, van den Brule AJ, Meijer CJ, Snijders PJ. The use of general primers GP5 and GP6 elongated at their 3′ ends with adjacent highly conserved sequences improves human papillomavirus detection by PCR. J Gen Virol. 1995, 76:1057–1062. [DOI] [PubMed]
- 7.Jacobs MV, de Roda Husman AM, van den Brule AJ, Snijders PJ, Meijer CJ, Walboomers JM. Group-specific differentiation between high- and low-risk human papillomavirus genotypes by general primer-mediated PCR and two cocktails of oligonucleotide probes. J Clin Microbiol. 1995;33:901–905. doi: 10.1128/jcm.33.4.901-905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans MF, Adamson CS, Simmons-Arnold L, Cooper K. Touchdown General Primer (GP5+/GP6+) PCR and optimized sample DNA concentration support the sensitive detection of human papillomavirus. BMC Clin Pathol. 2005 doi: 10.1186/1472-6890-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pannone G, Rodolico V, Santoro A, Lo Muzio L, Franco R, Botti G, Aquino G, Pedicillo MC, Cagiano S, Campisi G, Rubini C, Papagerakis S, De Rosa G, Tornesello ML, Buonaguro FM, Staibano S, Bufo P. Evaluation of a combined triple method to detect causative HPV in oral and oropharyngeal squamous cell carcinomas: p16 Immunohistochemistry, Consensus PCR HPV-DNA, and In Situ Hybridization. Infect Agent Cancer. 2012 doi:10.1186/1750-9378-7-4. [DOI] [PMC free article] [PubMed]
- 10.Hopman AH, Kamps MA, Smedts F, Speel EJ, Herrington CS, Ramaekers FC. HPV in situ hybridization: impact of different protocols on the detection of integrated HPV. Int J Cancer. 2005;115:419–428. doi: 10.1002/ijc.20862. [DOI] [PubMed] [Google Scholar]
- 11.Miyabe S, Okabe M, Nagatsuka H, Hasegawa Y, Inagaki A, Ijichi K, Nagai N, Eimoto T, Yokoi M, Shimozato K, Inagaki H. Prognostic significance of p27Kip1, Ki-67, and CRTC1-MAML2 fusion transcript in mucoepidermoid carcinoma: a molecular and clinicopathologic study of 101 cases. J Oral Maxillofac Surg. 2009;67:1432–1441. doi: 10.1016/j.joms.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Auclair PL, Ellis GL, Gnepp DR, Wenig BM, Janney CG: Salivary gland neoplasms: General considerations. In: Surgical pathology of the salivary glands. Ellis GL, Auclair PL, Gnepp DR editors, 1991, WB Saunders, Philadelphia.
- 13.Seoud M, Tjalma WA, Ronsse V. Cervical adenocarcinoma: moving towards better prevention. Vaccine. 2011;29:9148–9158. doi: 10.1016/j.vaccine.2011.09.115. [DOI] [PubMed] [Google Scholar]
- 14.Wilczynski SP, Bergen S, Walker J, Liao SY, Pearlman LF. Human papillomaviruses and cervical cancer: analysis of histopathologic features associated with different viral types. Hum Pathol. 1988;19:697–704. doi: 10.1016/S0046-8177(88)80176-X. [DOI] [PubMed] [Google Scholar]
- 15.Boland JM, McPhail ED, García JJ, Lewis JE, Schembri-Wismayer DJ. Detection of human papilloma virus and p16 expression in high-grade adenoid cystic carcinoma of the head and neck. Mod Pathol. 2012;25:529–536. doi: 10.1038/modpathol.2011.186. [DOI] [PubMed] [Google Scholar]
- 16.Brunner M, Koperek O, Wrba F, Erovic BM, Heiduschka G, Schoppper C, Thurnher D. HPV infection and p16 expression in carcinomas of the minor salivary glands. Eur Arch Otorhinolaryngol. 2012;269:2265–2269. doi: 10.1007/s00405-011-1894-2. [DOI] [PubMed] [Google Scholar]
- 17.Hafed L, Farag H, Shaker O, El-Rouby D. Is human papilloma virus associated with salivary gland neoplasms? An in situ-hybridization study. Arch Oral Biol. 2012;57:1194–1199. doi: 10.1016/j.archoralbio.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Schlecht NF, Brandwein-Gensler M, Nuovo GJ, Li M, Smith RV, Burk RD, Prystowsky MB. A comparison of clinically utilized human papillomavirus detection methods in head and neck cancer. Modern Pathol. 2011;24:1295–1305. doi: 10.1038/modpathol.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith EM, Rubenstein LM, Haugen TH, Pawlita M, Turek LP. Complex etiology underlies risk and survival in head and neck cancer human papillomavirus, tobacco, and alcohol: a case for multifactor disease. J Oncol. 2012;2012:571862. doi: 10.1155/2012/571862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2008;14:366–369. doi: 10.1158/1078-0432.CCR-07-1402. [DOI] [PubMed] [Google Scholar]
- 21.Guo XL, Sun SZ, Wang WX, Wei FC, Yu HB, Ma BL. Alterations of p16INK4a tumour suppressor gene in mucoepidermoid carcinoma of the salivary glands. Int J Oral Maxillofac Surg. 2007;36:350–353. doi: 10.1016/j.ijom.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Kiyoshima T, Shima K, Kobayashi I, Matsuo K, Okamura K, Komatsu S, Rasul AM, Sakai H. Expression of p53 tumor suppressor gene in adenoid cystic and mucoepidermoid carcinomas of the salivary glands. Oral Oncol. 2001;37:315–322. doi: 10.1016/S1368-8375(00)00083-X. [DOI] [PubMed] [Google Scholar]
- 23.Wolfish EB, Nelson BL, Thompson LD. Sinonasal tract mucoepidermoid carcinoma: a clinicopathologic and immunophenotypic study of 19 cases combined with a comprehensive review of the literature. Head Neck Pathol. 2012;6:191–207. doi: 10.1007/s12105-011-0320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomes CC, Diniz MG, Orsine LA, Duarte AP, Fonseca-Silva T, Conn BI, De Marco L, Pereira CM, Gomez RS. Assessment of TP53 mutations in benign and malignant salivary gland neoplasms. PLoS ONE. 2012;7:e41261. doi: 10.1371/journal.pone.0041261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Rawi NH, Omer H, Al Kawas S. Immunohistochemical analysis of P(53) and bcl-2 in benign and malignant salivary glands tumors. J Oral Pathol Med. 2010;39:48–55. doi: 10.1111/j.1600-0714.2009.00816.x. [DOI] [PubMed] [Google Scholar]
- 26.Jung AC, Briolat J, Millon R, de Reyniès A, Rickman D, Thomas E, Abecassis J, Clavel C, Wasylyk B. Biological and clinical relevance of transcriptionally active human papillomavirus (HPV) infection in oropharynx squamous cell carcinoma. Int J Cancer. 2010;126:1882–1894. doi: 10.1002/ijc.24911. [DOI] [PubMed] [Google Scholar]
- 27.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by HPV16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 28.Scheffner M, Whitaker NJ. HPV-induced carcinogenesis and the ubiquitin-proteasome system. Sem Canc Biol. 2003;13:59–67. doi: 10.1016/S1044-579X(02)00100-1. [DOI] [PubMed] [Google Scholar]
- 29.Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, O’Neil K, Stover K, El-Naggar A, Griffin JD, Kirsch IR, Kaye FJ. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003;33:208–213. doi: 10.1038/ng1083. [DOI] [PubMed] [Google Scholar]
- 30.Enlund F, Behboudi A, Andrén Y, Oberg C, Lendahl U, Mark J, Stenman G. Altered Notch signaling resulting from expression of a WAMTP1-MAML2 gene fusion in mucoepidermoid carcinomas and benign Warthin’s tumors. Exp Cell Res. 2004;292:21–88. doi: 10.1016/j.yexcr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Martins C, Cavaco B, Tonon G, Kaye FJ, Soares J, Fonseca I. A study of MECT1-MAML2 in mucoepidermoid carcinoma and Warthin’s tumor of salivary glands. J Mol Diagn. 2004;6:205–210. doi: 10.1016/S1525-1578(10)60511-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behboudi A, Enlund F, Winnes M, Andrén Y, Nordkvist A, Leivo I, Flaberg E, Szekely L, Mäkitie A, Grenman R, Mark J, Stenman G. Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer. 2006;45:470–481. doi: 10.1002/gcc.20306. [DOI] [PubMed] [Google Scholar]
- 33.Tirado Y, Williams MD, Hanna EY, Kaye FJ, Batsakis JG, El-Naggar AK. CRTC1/MAML2 fusion transcript in high grade mucoepidermoid carcinomas of salivary and thyroid glands and Warthin’s tumors: implications for histogenesis and biologic behavior. Genes Chromosomes Cancer. 2007;46:708–715. doi: 10.1002/gcc.20458. [DOI] [PubMed] [Google Scholar]
- 34.Seethala RR, Dacic S, Cieply K, Kelly LM, Nikiforova MN. A reappraisal of the MECT1/MAML2 translocation in salivary mucoepidermoid carcinomas. Am J Surg Pathol. 2010;34:1106–1121. doi: 10.1097/PAS.0b013e3181de3021. [DOI] [PubMed] [Google Scholar]
- 35.Fehr A, Meyer A, Heidorn K, Röser K, Löning T, Bullerdiek J. A link between the expression of the stem cell marker HMGA2, grading, and the fusion CRTC1-MAML2 in mucoepidermoid carcinoma. Genes Chromosomes Cancer. 2009;48:777–785. doi: 10.1002/gcc.20682. [DOI] [PubMed] [Google Scholar]
- 36.Coxon A, Rozenblum E, Park YS, Joshi N, Tsurutani J, Dennis PA, Kirsch IR, Kaye FJ. Mect1-Maml2 fusion oncogene linked to the aberrant activation of cyclic AMP/CREB regulated genes. Cancer Res. 2005;65:7137–7144. doi: 10.1158/0008-5472.CAN-05-1125. [DOI] [PubMed] [Google Scholar]
- 37.Wu L, Liu J, Gao P, Nakamura M, Cao Y, Shen H, Griffin JD. Transforming activity of MECT1-MAML2 fusion oncoprotein is mediated by constitutive CREB activation. EMBO J. 2005;24:2391–2402. doi: 10.1038/sj.emboj.7600719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakayama T, Miyabe S, Okabe M, Sakuma H, Ijichi K, Hasegawa Y, Nagatsuka H, Shimozato K, Inagaki H. Clinicopathological significance of the CRTC3-MAML2 fusion transcript in mucoepidermoid carcinoma. Mod Pathol. 2009;22:1575–1581. doi: 10.1038/modpathol.2009.126. [DOI] [PubMed] [Google Scholar]
- 39.Fontaine V, van der Meijden E, ter Schegget J. Inhibition of human papillomavirus-16 long control region activity by interferon-gamma overcome by p300 overexpression. Mol Carcinog. 2001;31:27–36. doi: 10.1002/mc.1036. [DOI] [PubMed] [Google Scholar]
- 40.Tidy J, Vousden KH, Mason P, Farrell PJ. A novel deletion within the upstream regulatory region of episomal human papillomavirus type 16. J Gen Virol. 1989;70:999–1004. doi: 10.1099/0022-1317-70-4-999. [DOI] [PubMed] [Google Scholar]
- 41.Isayeva T, Li Y, Maswahu D, Brandwein-Gensler M. Human papillomavirus in non-oropharyngeal head and neck cancers: a systematic literature review. Head Neck Pathol. 2012;6(Suppl 1):104–120. doi: 10.1007/s12105-012-0368-1. [DOI] [PMC free article] [PubMed] [Google Scholar]