

Background: Histidine kinase proteins can regulate bacterial behavior, including pathogenesis.
Results: Structural and functional features of two histidine kinases of Mycobacterium tuberculosis were determined.
Conclusion: Proper positioning of a short ATP lid for interaction with His phospho-accepting domain triggers ATP binding of DosS.
Significance: This work describes a novel structural regulatory mechanism of histidine kinases with a short ATP lid.
Keywords: Crystal Structure, Enzyme Mechanisms, Histidine Kinases, Mycobacterium tuberculosis, Phosphorylation, ATP Binding, Autophosphorylation
Abstract
The sensor histidine kinases of Mycobacterium tuberculosis, DosS and DosT, are responsible for sensing hypoxic conditions and consist of sensor and kinase cores responsible for accepting signals and phosphorylation activity, respectively. The kinase core contains a dimerization and histidine phosphate-accepting (DHp) domain and an ATP binding domain (ABD). The 13 histidine kinase genes of M. tuberculosis can be grouped based on the presence or absence of the ATP lid motif and F box (elements known to play roles in ATP binding) in their ABDs; DosS and DosT have ABDs lacking both these elements, and the crystal structures of their ABDs indicated that they were unsuitable for ATP binding, as a short loop covers the putative ATP binding site. Although the ABD alone cannot bind ATP, the kinase core is functional in autophosphorylation. Appropriate spatial arrangement of the ABD and DHp domain within the kinase core is required for both autophosphorylation and ATP binding. An ionic interaction between Arg440 in the DHp domain and Glu537 in the short loop of the ABD is available and may open the ATP binding site, by repositioning the short loop away from the site. Mutations at Arg440 and Glu537 reduce autophosphorylation activity. Unlike other histidine kinases containing an ATP lid, which protects bound ATP, DosS is unable to accept ATP until the ABD is properly positioned relative to the histidine; this may prevent unexpected ATP reactions. ATP binding can, therefore, function as a control mechanism for histidine kinase activity.
Introduction
Mycobacterium tuberculosis is one of the most dreaded pathogens in existence and has the ability to persist within its host (1). Its successful survival is due to alterations in gene expression in response to environmental changes (2). To sense these changes and transduce signals to control gene expression, M. tuberculosis utilizes two-component systems. Two-component systems are widely distributed signaling systems found in bacteria, eukaryotic microorganisms, and plants and consist of sensor histidine kinases (HKs)2 and response regulators (RRs) (3, 4). A typical bacterial two-component signal transduction system involves sensing of internal or external signals by a HK, leading to its autophosphorylation at a histidine residue, followed by phosphoryl transfer to an aspartate residue in an N-terminal phosphoacceptor (receiver; REC) domain of the cognate RR, which affects the properties of the C-terminal DNA binding domain. Activation of the RR by phosphorylation thereby regulates downstream gene expression and is deactivated by dephosphorylation. The HK is enzymatically active in autophosphorylation, phosphate transfer, and dephosphorylation.
M. tuberculosis has been demonstrated to have 12 two-component systems, involving 13 contributing HK (5). Among these HKs, two, DosS and DosT, function with a single RR, DosR, which functions to up-regulate downstream genes essential for the survival of M. tuberculosis in hypoxic conditions (6, 7). Activation of DosR induces a dormant state in the bacterium, allowing it to survive successfully within macrophages. DosS and DosT play roles in the recognition of hypoxic conditions as redox and oxygen sensors, respectively, and are structurally very similar to one another (8, 9). The architecture of these HKs can be divided into an N-terminal sensor core and a C-terminal kinase core (KC) (see Fig. 1A) (10). There are two GAF domains in the sensor cores; the first of these, GAF-A, has a heme group that functions to sense oxygen or redox status (11, 12), whereas the function of the second, GAF-B, is not yet known (13). KCs are responsible for transferring signals to the RR through their autophosphorylation and phosphate transfer to DosR. Full-length DosS and DosT are enzymatically inactive in the absence of a signal; however, the KC alone is sufficient for autophosphorylation and phosphate group transfer and is thought to be constitutively active when the sensor core is removed (8, 13).
FIGURE 1.
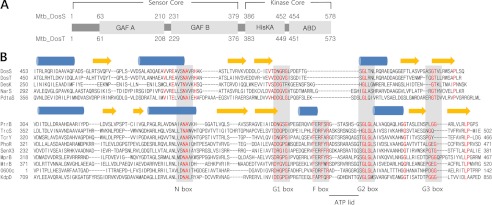
Domain arrangement and sequence alignment of DosS ABD and DosT ABD. DosS and DosT consist of two GAF domains, HisKA and ABD. Amino acid sequences of 13 ABDs from M. tuberculosis (Mtb) HKs and DesK ABD from B. subtilis were compared. These ABDs are divided into two groups based on the presence or absence of an F box. Members of both groups have a conserved N box and three G boxes. PrrB, TrcS, TcrY, PhoR, SenX3, MprB, MtrB, KdpD, and Rv0600c gene products have an ATP lid motif and conserved phenylalanine and arginine residues in their F boxes. The ATP lid motif is underlined and lies between the F and G2 boxes. DosS, DosT, NarS, PdtaS, and DesK have short sequences between the G1 and G2 boxes. Secondary structural elements (blue helices and yellow strands) above the sequences are based on DosS and PrrB structures for each group. Conserved regions are indicated by gray boxes, and conserved residues among the group members are highlighted by red. The multiple alignment was performed using ClustalW.
KC consists of an ATP binding domain (ABD) and a dimerization and histidine phospho-accepting (DHp) domain, which contains the phosphate-accepting histidine (10). Because the DHp domains of DosS and DosT belong to the HisKA_3 family, they are known as HisKA domains. Based on the structures of other HKs, such as DesK (Bacillus subtilis) and HK853 (Thermotoga maritima) (14, 15), it can be predicted that the HisKA domains consist of two long helices and that the KC forms a dimer through four helix bundle-like interactions of the two HisKAs, with two ABDs tethered to the HisKA dimer. During the autophosphorylation reaction of the KC, the ABD transfers a phosphate group from ATP to the histidine in the HisKA. The ABD not only binds ATP but also interacts with the HisKA dimer. A number of ABD structures have been determined revealing that, in general, ABDs of HKs have conserved N box and F box elements and three G boxes for ATP binding (16, 17). ABDs also usually have a long characteristic loop, known as an ATP lid, between the F and G2 boxes, which covers the nucleotide; however, this can exhibit a variety of conformations in the absence of nucleotide (16, 17). Conserved charged residues in the ATP lid interact with the nucleotide β-phosphate. The ATP lid is thought to play a role for kinase activity and to function to protect ATP from futile reactions (4, 18).
In this study, the amino acid sequences of DosS and DosT ABDs were analyzed, revealing that they do not contain an F box or a sufficiently large peptide region to form a complete ATP lid motif, as detected in other HKs. To understand how DosS and DosT are able to anchor ATP and guide it to the HisKA in the absence of an ATP lid, we determined the crystal structures of their ABDs and found that they are in a closed state for ATP binding. Structural analyses and autophosphorylation assays of wild-type and mutant DosS KC proteins suggest that an interaction between the HisKA region and ABD is required, not only for autophosphorylation, but also to facilitate opening of the ATP binding site. Ionic interactions between Arg440 and Glu537 residues may be involved in the activation step for ATP binding to DosS.
EXPERIMENTAL PROCEDURES
Gene Cloning and Protein Purification
The DosS (Rv3132c) and DosT (Rv2027c) genes were cloned previously from the chromosomal DNA of the M. tuberculosis H37Rv strain (11, 19). The regions encoding the DosS ABD (residues 454–578) and DosT ABD (residues 451–573) were amplified by PCR using primer sets carrying restriction sites. The PCR products for DosS ABD and DosT ABD digested with the restriction enzymes were cloned into the expression vectors, pHIS-parallel vector (20) and pET30a, respectively. Two mutations (L534M and V562M) were introduced to this DosS ABD clone for selenomethionine (SeMet) labeling, which was used for structural determination. The regions encoding the KC (residues 386–578) and HisKA (residues 380–446) of DosS were amplified by PCR and cloned into the expression vectors, pHIS-parallel vector and pET30a, respectively. Mutants of the KC, H396Q, C524S, C524S/R440C, C524S/E537C, and C524S/R440C/E537C were generated by QuikChange mutagenesis (Stratagene). The integrity of the insertion and mutations were verified by direct DNA sequencing.
The expression of the SeMet-labeled DosS ABD mutant was induced by 0.2 mm isopropyl-β-d-thiogalactopyranoside in the Escherichia coli strain B834 (Novagen) grown at log phase in M9 medium containing 25 μg/ml SeMet at 37 °C. The cells were then grown for an additional 24 h at 25 °C. For the wild-type protein, the E. coli strain BL21(DE3), carrying the recombinant plasmid, was grown in LB medium. The cells were grown for an additional 21 h at 18 °C after induction. The expressed protein was purified by affinity chromatography using a Ni-NTA column (Qiagen). The recombinant protein was digested using recombinant tobacco etch virus protease at 20 °C in the presence of 0.5 mm EDTA and 1 mm dithiothreitol. After complete digestion, the His6 tag was removed using a Ni-NTA column. Gel filtration was performed with a Superdex G75 column (GE Healthcare), equilibrated with 20 mm Tris-HCl, pH 7.5, containing 300 mm NaCl, and the fractions containing ABD were collected and concentrated using Centriprep YM10 (Millipore) for crystallization. The purified protein contains an additional four amino acids (GAMV) at the N terminus arising from the cloning procedure.
The DosT ABD was expressed in E. coli strain BL21(DE3). Cells grown to mid-log phase in LB broth containing kanamycin were induced by isopropyl-β-d-thiogalactopyranoside (0.2 mm) and cultivated overnight at 18 °C. The harvested cells were frozen at −20 °C and then thawed at 4 °C. The expressed protein was purified by affinity chromatography using a Ni-NTA column (Qiagen) and gel filtration chromatography with a Superdex G75 column (GE Healthcare), equilibrated with 20 mm Tris-HCl (pH 7.5), 300 mm NaCl, and the fractions containing ABD were collected and concentrated using Centriprep YM10 (Millipore) for crystallization. The purified protein contains an additional eight (LEHHHHHH) amino acids at the C terminus arising from the cloning procedure.
For autophosphorylation and ATP binding assays, the DosS HisKA domain was obtained in a similar manner to the DosT ABD and DosS KC, and its mutants were expressed and purified in the same way as the wild-type DosS ABD. Purified DosS HisKA and KC have the additional amino acids (LEHHHHHH or GAMDP) at the C and N termini, respectively.
Crystallization and Data Collection
Crystallization of the purified proteins was initially performed with commercially available sparse matrix screens (Hampton Research and Emerald Biostructures) using the sitting-drop vapor diffusion method at 21 °C. After optimization, the best crystals of the SeMet-substituted DosS ABD were obtained under conditions of 0.1 m sodium citrate (pH 6.0) containing 15% (w/v) PEG 1500 and 0.2 m zinc acetate, with a protein concentration of 20 mg/ml. The crystal was transferred to a cryo-protectant solution containing an additional 16% glycerol in the crystallization solution, prior to x-ray data diffraction collection. After a fluorescence scan, a single anomalous x-ray dispersion data for the SeMet crystal was collected at a wavelength corresponding to the selenium absorption peak (0.97908 Å) using a Quantum 210 CCD detector at the 6B beamline of the Pohang Accelerator Laboratory.
DosT ABD crystals suitable for data collection were obtained in drops where 1.25 μl of 18 mg/ml protein solution in storage buffer (20 mm Tris-HCl, pH 7.5, 300 mm NaCl) was mixed with 1.25 μl of reservoir solution (1.5 m NaCl, 10% ethanol). For data collection, 24% glycerol was added to the reservoir solution, and crystals were immediately placed in a −173 °C nitrogen-gas stream. X-ray diffraction data of native crystals were collected at a resolution of 1.9 Å at the 6B beamline of the Pohang Accelerator Laboratory. The detector distance was set to 150 mm, and diffraction data were collected each 1.0° oscillation per frame.
The collected data were indexed, integrated, and scaled using the HKL-2000 software package (21). The SeMet crystal of the DosS ABD belongs to the space group P41212, with unit cell parameters of a = 53.2, b = 53.2, and c = 184.6 Å. The crystal of the DosT ABD belongs to the space group P212121, with unit cell parameters of a = 50.9, b = 65.9, and c = 72.7 Å.
Structural Determination
The structure of the SeMet-substituted DosS ABD was determined by the single anomalous dispersion method, at a resolution of 1.8 Å. Six selenium atoms were identified in the asymmetric unit using SOLVE (22), and density modification and subsequent automated model building was performed with RESOLVE (23). The RESOLVE-built partial model was used as a guide to build the remainder of the protein manually into density-modified electron density maps with the program COOT (24). Refinement with isotropic displacement parameters was performed with Refmac5 (25) in the CCP4 suite (26). The Rwork and Rfree values of the refined structure were 0.186 and 0.236, respectively.
The structure of the native DosT ABD was determined using the crystal structure of the DosS ABD as the search model and the molecular replacement method with AMoRe software (27). The asymmetric unit contains two molecules and the Matthews coefficient, Vm, is calculated to be 2.13 Å3/Da, corresponding to a solvent content of 42.2%. Model building and structure refinement were carried out using COOT and Refmac5, respectively. The crystal structure was determined at a resolution of 1.9 Å, with Rwork = 0.194 and Rfree = 0.232. The final models have been deposited in the Protein Data Bank (PDB) (28) under PDB codes 3ZXO and 3ZXQ for the SeMet-substituted DosS ABD and DosT ABD, respectively.
Autophosphorylation Assays
Protein autophosphorylation reactions were carried out using 15 μm protein for 15 min at 30 °C in autophosphorylation buffer (50 mm Tris-HCl, pH 8.0, containing 50 mm KCl and 25 mm MgCl2). The reactions were initiated by the addition of [γ-32P]ATP (10 mCi/ml, 3000 Ci/mmol) and unlabeled ATP to a final concentration of 10 μm, terminated by the addition of 2× gel-loading buffer containing 4% SDS and 0.2 m dithiothreitol, and subjected to SDS-PAGE on 15% polyacrylamide gels. For the SDS-PAGE under non-reducing conditions, a protein sample was mixed with the gel-loading buffer without dithiothreitol and incubated for 2 min. To detect bound ATP in proteins, the autophosphorylation reaction mixtures were also subject to native PAGE on 12% polyacrylamide gels. For native PAGE after incubation with [γ-32P]ATP, gel-loading buffer without SDS was used for PAGE.
ADP Assay
To identify ATPase activity, purified wild-type and H395Q mutant KC and ABD were used in an ADP assay reaction using a colorimetric assay kit (Biovison, Mountain View, CA). The reaction mixture for ATP hydrolysis contains 50 mm Tris-HCl, pH 8.0, 50 mm KCl, 25 mm MgCl2, 30 nmol ATP, and various amounts of proteins (7.2 nmol, 14.4 nmol, and 21.6 nmol). Mixtures were incubated at 30 °C for 15 min, and the ADP colorimetric reaction completed by the addition of the reaction components (ADP assay buffer, ADP probe, ADP converter, and ADP developer) and incubation for 30 min at room temperature. After dilution with 900 μl of DDW, absorbance at 570 nm was measured.
RESULTS
DosS and DosT Contain ATP Binding Domains without ATP Lid Motifs or F Boxes
For insight into the DosS ABD and DosT ABD, their amino acid sequences were compared with those of conceptually translated ABDs of other known HK genes from M. tuberculosis H37Rv. This sequence alignment revealed that the domain sequences can be divided into two groups, one containing both the conserved F box and the ATP lid, known to be involved in ATP binding, and the other without either of these motifs (Fig. 1B). The former group belongs to the OmpR family and includes the gene products of PrrB, TrcS, TcrY, PhoR, SenX3, MprB, mtrB, KdpD, and Rv0600c, whereas DosS, DosT, NarS, and PdtaS belong to the latter group. Among the HKs of M. tuberculosis, the crystal structure of the PrrB ABD has been determined (16), and other OmpR family members also contain a conserved N box, F box, and three G (G1, G2, and G3) boxes involved in ATP binding. ABDs belonging to this group share key residues in each of the conserved boxes. The ATP lid motif, which connects the F and G2 boxes, consists of 11–14 residues and is absent from the second group of ABDs, including those of DosS and DosT. Although the ABDs of these kinases have conserved N and G boxes, they also lack an F box, and their G1 and G2 boxes are directly connected by 8–15 residues. DosS, DosT, and NarS belong to the NarL family, whereas PdtaS is considered to be a member of the AmiR family. DesK, a NarL family HK from B. subtilis, which also lacks the ATP lid motif, has been well characterized structurally (29). A region connecting its G1 and G2 boxes functions as an ATP lid and contains two phenylalanine residues to retain an ATP molecule in a similar manner to an F box. Compared with the DesK ABD, DosS ABD and DosT ABD are three residues shorter, and DosS has only one phenylalanine, whereas DosT has none in this region of the molecule (Fig. 1B).
Crystal Structures of ATP Binding Domains from DosS and DosT
The observation that DosS and DosT proteins apparently lack the F box and ATP lid regions suggests they may possess a previously undescribed ATP binding mechanism. To explore this possibility, we first determined the crystal structure of the DosS ABD at a resolution of 1.8 Å (Table 1). The crystal of the DosS ABD contained two almost identical ABD molecules in an asymmetric unit. Each DosS ABD molecule consisted of three α helices and a five stranded β-sheet with a strand order of 1-2-3-5-4 (Fig. 2A). The helices are all located on one side of the sheet, resulting in a globular shape revealing an α/β sandwich fold. A search for homologous proteins using DALI revealed structural similarities of the DosS ABD to ABDs of HK, including B. subtilis DesK (PDB code 3GIG), T. maritima CheA (PDB code 1I59), Salmonella enterica PhoQ (PDB code 3CGZ), and M. tuberculosis PrrB (PDB code 1YSR) with Z-scores of 14.7, 12.8, 11.9, and 11.3, respectively. The crystal structure of the DosS ABD clearly showed the absence of an ATP lid motif, as expected from the amino acid sequence alignment, although its overall structure is similar to those of canonical ABDs of HKs.
TABLE 1.
Data collection and refinement statistics
| DosS ABD | DosT ABD | |
|---|---|---|
| Data collection | ||
| X-ray source | PAL 6B | PAL 6B |
| Wavelength (Å) | 0.97908 | 1.1000 |
| Space group | P41212 | P212121 |
| Unit cell parameters (Å) | 53.2 × 53.2 × 184.6 | 50.9 × 65.9 × 72.7 |
| Resolution limit (Å) | 50–1.8 (1.83–1.80)a | 40–1.9 (1.93–1.90)a |
| Total reflections | 571,828 | 125,573 |
| Unique reflections | 25,671 (1,278) | 19,905 (994) |
| Redundancy | 22.3 (20.8) | 6.3 (5.8) |
| Completeness (%) | 99.9 (99.9) | 99.9 (100.0) |
| Rsymb | 0.081 (0.459) | 0.098 (0.434) |
| Average I/σ(I) | 56.0 (8.3) | 21.9 (4.2) |
| Refinement | ||
| Space group | P41212 | P212121 |
| Resolutions (Å) | 46–1.9 | 32–1.9 |
| Reflections (working) | 20,025 | 18,850 |
| Reflections (test) | 1,093 | 1,016 |
| Rworkc | 0.186 | 0.194 |
| Rfreec | 0.236 | 0.232 |
| Number of water molecules | 190 | 144 |
| r.m.s.d. from ideal geometryd | ||
| Bond length (Å) | 0.015 | 0.012 |
| Bond angle | 1.458° | 1.353° |
| Average B factors (Å) | ||
| Molecule A (main/side chain) | 20.5 (19.2/21.9) | 17.8 (16.5/19.2) |
| Molecule B (main/side chain) | 22.8 (21.3/24.4) | 20.0 (19.3/20.8) |
| Waters | 31.4 | 25.3 |
a Numbers in parentheses describe the relevant value for the last resolution shell.
b Rsym = Σ|Ii − 〈I〉|/ΣI where Ii is the intensity of the ith observation and 〈I〉 is the mean intensity of the reflections.
c Rwork = Σ‖Fobs| − |Fcalc‖/Σ|Fobs|, crystallographic Rfactor, and Rfree = Σ‖Fobs| − |Fcalc‖/Σ|Fobs| when all reflections belong to a test set of randomly selected data.
d r.m.s.d., root mean square deviation.
FIGURE 2.

Crystal structures of DosS ABD and DosT ABD. A, ribbon diagram of the structure of the DosS ABD showing an α/β-sandwich fold. Three helices are one side of a mixed β-sheet, with strand order 1-2-3-5-4. Protein regions are colored from blue to red from the N to C terminus. B, superimposition of the DosS ABD (green) on the PhoQ ABD (orange) bound to an ATP analog. The ATP is surrounded by the F box helix and long ATP lid. A loop connecting β3 and α3 in the DosS ABD overlies the ATP binding site. C, A zinc ion found in the DosS ABD is coordinated by His507, Glu537, and Asp529, and a water molecule, which is hydrogen-bonded to the carbonyl oxygen of Val505. This interaction holds the position of the loop connecting β3 and α3 via Glu537. D, ribbon diagram of a DosT ABD dimer from the crystal structure showing domain swapping. The C-terminal parts (from the β3 strand to the C terminus) of two molecules (mol; green and cyan) were swapped. The two DosT ABDs reconstituted by switching C-terminal region are almost identical to one another. E, superimposition of the DosT ABD (blue) on to the PhoQ ABD (orange) bound to ATP. The loop connecting β3 and α3 in the DosT ABD overlies the ATP binding site. F, superimposition of two DosT ABDs. The orientations of the Asp534 side chains in the loops connecting β3 and α3 are opposite when the loop is rotated.
The conformation of an ATP lid surrounding an ATP analog has been described in detail using PhoQ from E. coli (17). Superimposition of the crystal structures of the DosS ABD and the ATP analog-bound PhoQ revealed a marked difference in their configurations in the region between the β3 strand and α3 helix, which form the ATP binding pocket (Fig. 2B). Specifically, a loop connecting the β3 strand and the α3 helix of the DosS ABD appears to be taut, leaving little room for ATP binding, whereas the PhoQ has the F box helix, and a loop connecting the F box and α3 helices extends to form the ATP lid, surrounding the ATP binding site along with G1, G2, and N boxes (Fig. 3).
FIGURE 3.

The position of the structural elements for ATP binding in the ABD. In the structure of ATP-bound PhoQ ABD, ATP is surrounded by N, F, G1, and G2 boxes and the ATP lid wraps the phosphate groups of the ATP. A short loop containing Glu537 between the G1 and G2 boxes occupies the space for ATP binding in the DosS ABD; for ATP to bind, this loop would need to be withdrawn to create sufficient space. Area of the boxes for ATP binding was shown as the dotted surfaces.
A zinc atom was found in the putative ATP binding pocket of the DosS ABD (Fig. 2C) coordinated by Asp529, His509, Glu537, and a water molecule connected to the carbonyl oxygen atom of Val505. This tetrahedral interaction with the zinc ion apparently stabilizes the loop connecting β3 and α3 at the ATP binding site through the Zn-Glu537 interaction because the Glu537 residue is located at the loop. These observations, combined with the fact that the DosS ABD does not have an F box, suggest that the DosS ABD adopts a closed conformation, in which the ATP binding site is buried and covered by the short loop. This, in turn, suggests that ATP binding to the DosS ABD requires conformational changes in the loop region.
To confirm that the closed conformation is due to the short β3-α3 loop of the ABD, the structure of DosT ABD was determined at a resolution of 1.9 Å using a molecular replacement method with the DosS ABD structure (Table 1). In the crystal structure of the DosT ABD, two molecules in an asymmetric unit have a domain swapped (Fig. 2D). The N-terminal region of the α/β sandwich structure of ABD, which consists of two strands (β1 and β2) and two helices (α1 and α2), comes from one molecule, and the C-terminal part (three strands and one helix) of the ABD is derived from a second molecule. A loop connecting the β2 and β3 strands is the switching point. However, monomeric DosT ABD would be functional because it eluted as a monomer in gel-filtration chromatography. The two structures of DosT ABD were almost identical to one another and similar to that of the DosS ABD. The structure of the DosT ABD was also superimposed onto that of PhoQ bound to ATP (Fig. 2E). In DosT, similar to DosS, a loop connecting the β3 strand and α3 helix crosses the putative ATP binding site, which does not have an ATP lid or F box, and the loop folds into the pocket for the adenine moiety of ATP. The fact that DosT ABD does not contain a zinc atom favors the hypothesis that the closed conformation for ATP binding is due to the short ATP lid. When the two structures of DosT ABD were superimposed, a predominant difference was the orientation of the loop connecting β3 and α3 (Fig. 2F). Asp534, a negatively charged residue in the loop corresponding to Glu537 of DosS, faces toward the ATP binding site and has a hydrogen bond to Asn500 to stabilize the structure of the fold, whereas in the second structure, the loop is rotated and the side chain of Asp534 faces toward the solvent.
The DosS ABD Requires Specific Conformational Change to Allow ATP Binding
The crystal structures of the DosS ABD indicate that it is not suitable for ATP binding. Because the DosS KC dimer has autophosphorylation activity, the conformation of the ABD may be expected to change to an open conformation to permit ATP binding in the presence of HisKA in the KC dimer. To confirm the ability of the ABD to bind ATP in solution, the kinase activities of the ABD and KC were examined in the presence of additional HisKA. As expected, the KC was autophosphorylated, whereas the ABD was not (Fig. 4A). In the presence of additional HisKA, KC phosphorylated the additional HisKA, whereas the ABD was unable to phosphorylate. Thus, although the ABD alone does not possess kinase activity, the ABD can bind ATP and phosphorylate HisKA in the context of the KC. This implies that the free ABD is not able to bind ATP as suggested by the crystal structure but that the ABD is functional when connected to HisKA in the KC dimer.
FIGURE 4.
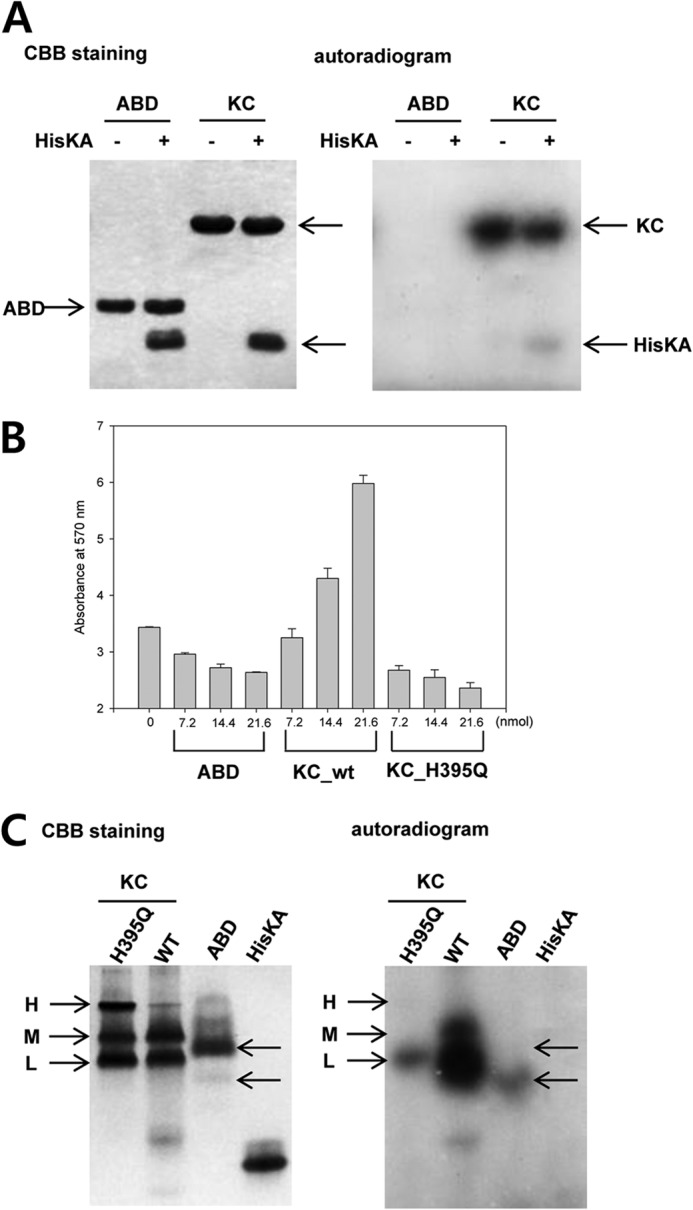
Autophosphorylation and ATP binding of DosS. A, the kinase activity of the DosS KC and ABD were assayed in the presence of additional HisKA. KC phosphorylated not only its own HisKA but also free HisKA, whereas the ABD did not. B, ATP hydrolysis activity was measured by colorimetric measurement of ADP. KC activity increased the concentration of ADP due to autophosphorylation, whereas the ABD and the H395Q mutant did not. C, ATP binding in native PAGE. The H395Q KC mutant and the ABD are radioactive due to the binding of radiolabeled ATP. The H395Q KC mutant migrates as at least three distinctive bands (H, M, and L) by native PAGE and only the L band demonstrates interaction with ATP. The majority of the ABD does not bind ATP, whereas a minor band was radioactive. Proteins were stained with Coomassie Brilliant Blue (CBB).
If the ABD alone has intrinsic ATPase activity, it could hydrolyze ATP, rather than phosphorylate HisKA. To rule out this possibility ADP generation was monitored for the KC and ABD. A H395Q mutant of KC, which lacked the phosphate-accepting histidine, was also employed to test for intrinsic ATPase activity of KC. This mutant was able to bind ATP but not to transfer a phosphate group to the histidine. Wild-type KC produced increased levels of ADP, whereas the H395Q mutant and the ABD alone did not (Fig. 4B). An increase in protein concentration appeared to hamper the ADP-dependent color reaction, leading to a decrease in absorbance.
The ABD alone, therefore, does not have ATPase activity and cannot phosphorylate HisKA. We next confirmed that the ABD cannot bind ATP, as suggested by its crystal structure. We mixed radiolabeled ATP with the ABD, HisKA, KC, and the H395Q mutant of KC and performed native PAGE (Fig. 4C). Wild-type KC showed a strong radioactive signal due to autophosphorylation. The mutant H395Q, which lacks the phosphate-accepting histidine, also demonstrated radioactivity in one of three major bands resolved by native PAGE; this represents ATP binding in the H395Q mutant, rather than autophosphorylation. Unlike HisKA, which has no ATP binding function, the ABD did have a radioactive signal; however, the major ABD band was not radioactive, whereas a minor, faster migrating conformation did contain radioactivity, suggesting ATP binding. These results indicate that the majority of the ABD is not able to bind ATP; however, the domain is able to bind ATP after undergoing conformational change to a form which migrates more quickly.
The Conformation of KC Required for ATP Binding
In the previous experiment, an interesting observation was that KC H395Q migrated as at least three distinctive bands: high, middle, and low (H, M, or L) by native PAGE, whereas the ABD showed one major band (Fig. 4C). These three distinctive bands may represent three different conformations. Because KC was obtained as a single peak by gel filtration during purification steps, these forms were considered to be conformations of KC dimers. KC consists of an ABD and the HisKA region, of which the HisKA region is important for dimerization of KC. The three bands may, therefore, represent three different configurations of the two globular ABDs with respect to the helix bundle of HisKAs in a KC dimer.
As shown in Fig. 4C, among the three bands of the H395Q mutant, only the L-form bound ATP. This implies that the ABDs in the other KC forms (H and M) are in a closed conformation, whereas a particular orientation of the ABD relative to the HisKA domain, as present in the L-form, induces a change of the ABD to an open conformation. Wild-type KC migrated as two major bands (M and L) and the L-form was autophosphorylated. Although a small amount of radioactivity was also observed in the M band, it appears that the L-form alters, becoming the M-form, after an autophosphorylation event.
To identify differences between the three bands, possible conformational changes were monitored by native PAGE. After treatment with a reducing agent (β-mercaptoethanol), the M band was diminished, whereas the L band increased (Fig. 5A), suggesting that the M-form has a disulfide bridge. There is only one cysteine, Cys524, in DosS KC; therefore, the single possibility for disulfide bond formation would be between two Cys524 residues in a KC dimer. In this case, it would be expected that a dimer-size band would be observed after separation by SDS-PAGE in the absence of reducing agent, due to the presence of the disulfide bond. Indeed, such a dimer-sized band was identified in both wild-type and H395Q mutant forms of KC (Fig. 5B); however, when Cys524 was mutated to a serine, the C524S mutant did not migrate as a dimer-sized band in the absence of dithiothreitol (Fig. 5B). Investigation of autophosphorylation of wild-type and C524S mutant KC revealed that both migrated as radioactive monomeric bands in SDS-PAGE, whereas the H395Q mutant does not (Fig. 5B). These results indicate that mutation of Cys524 to a serine does not affect autophosphorylation activity. The wild-type dimer-sized band, with a disulfide bridge between two Cys524 residues, is not phosphorylated (Fig. 5B). Thus, Cys524 does not appear to have a critical role in autophosphorylation, but the disulfide bridge via Cys524 does affect this process.
FIGURE 5.

ATP binding ability depends on the conformation of the KC. A, three distinctive bands (H, M, and L) of the DosS KC were detected by native PAGE. The M band is diminished by β-mercaptoenthanol, suggesting that it is an oxidized form. B, a band representing a dimer of DosS KC was detected by SDS-PAGE in the absence of reducing agent. The C524S mutant did not form a dimer while H395Q did (upper panel). Autophosphorylation was detected in the monomer band of wild-type KC and the C524S mutant, but not in the dimer band of wild-type KC or the H395Q mutant (lower panel). C, a schematic diagram for a KC dimer. ABDs are tethered to HisKA dimer with flexibility of positioning. The M form has a disulfide bridge between two ABDs far away from the ATP phospho-accepting histidine in HisKA, whereas the L form has a position of ATP binding site closely to the histidine.
The M-form, a dimeric KC with a disulfide bridge, does not undergo autophosphorylation, possibly due to the position of the ATP binding site with respect to the phosphate-accepting histidine. As revealed by the crystal structure of the DosS ABD, Cys524 is located in the β3 strand of the ABD β-sheet and its side chain faces toward the solvent on the opposite side of the molecule to the three α helices. To complete a disulfide bond involving Cys524, two ABDs would need to be orientated with their β-sheets in a back-to-back position (Fig. 5C). In this configuration, molecular handcuffs are snapped onto the ABD, preventing ATP from accessing the phosphate-accepting histidine of HisKA.
Ionic Interaction between the ABD and the HisKA Domain May Be Necessary for Autophosphorylation
In the crystal structures, the ABD had a closed conformation. A proper arrangement of the ABD relative to the HisKA domain, as in the L-form of the KC dimer, is required for both autophosphorylation and ATP binding. These data suggest that an interaction between the HisKA domain and the ABD activates ATP binding of the ABD in a KC dimer, allowing autophosphorylation. How does the HisKA domain open the ATP binding site of the ABD? Based on the crystal structure, the loop connecting β3-α3 in the ABD would need to be withdrawn from the putative ATP binding site (see Fig. 3). In this loop region, glutamate (Glu537) and aspartate (Asp534) residues are located in the DosS ABD and DosT ABD, respectively. In the DosT ABD structures, this negatively charged residue can be rotated to face outside of the binding pocket (Fig. 2F). If a positively charged residue can be located close to this site in the loop, it may attract the loop out of the ATP binding site by an ionic interaction.
Amino acid sequence alignment between DosS and DesK demonstrated high sequence similarity in the KC region, suggesting similar structures for these two molecules (Fig. 6A). The spatial arrangement of the DosS ABD with respect to the HisKA dimer was modeled based on the structure of DesK (Fig. 6B). In this model, it is possible that the ATP binding site of the ABD locates close to His395 in the DosS HisKA domain. In this configuration, a positively charged residue, Arg440, in HisKA near H395 would then be in proximity to Glu537 in the loop of DosS ABD. The arginine residue could potentially then interact with the glutamic residue and play a role in drawing Glu537 away from the ATP binding site.
FIGURE 6.

Prediction of ABD orientation with respect to HisKA in DosS KC. A, the amino acid sequence of the DosS KC is aligned to that of DesK. The secondary structures (blue helices and yellow strands) of DesK KC and DosS ABDs are displayed at the top and bottom of the aligned sequences, respectively. Conserved residues are indicated by an asterisk between the two sequences. Key residues for the interaction between the ABD and HisKA are indicated by triangles. B, the possible position of the ABD with respect to HisKA in the DosS KC is modeled based on the crystal structure (PBD code 3GIE) and a docked model (29) of the DesK KC. When the ATP binding site of the ABD is located close to His395 in the HisKA domain, and the loop containing Glu537 is extended in a similar manner to that of the DesK ABD, Glu537 in the DosS ABD may interact with Arg440 close to His395 in the HisKA domain. Interprotomer interaction is suggested with a dashed line connecting ABD and HisKA. Conserved (asterisks) and similar (colons) residues between two sequences are marked, and arrowheads indicate the residues shown in B.
To investigate whether Arg440 can approach Glu537 to form such an ionic interaction, both Arg440 and Glu537 residues of DosS KC were mutated to cysteine, as if the two residues can be closely located in KC, they could then potentially form a disulfide bond under oxidized conditions. As shown in Fig. 5B, an intermolecular disulfide bond between two Cys524 residues in a wild-type KC dimer can be detected by SDS-PAGE separation in the absence of a reducing agent. To avoid unexpected disulfide bridges, we introduced the mutations at Arg440 and Glu537 into the C524S mutant. We purified wild-type KC protein and four mutant forms, C524S, C524S/R440C, C524S/E537C, and C524S/R440C/E537C. All purified proteins migrated as a single monomeric band by SDS-PAGE in the presence of dithiothreitol (Fig. 7A). A band migrating at the expected size of a dimer, representing an intermolecular disulfide bond, was detected in WT but not in C524S mutant protein preparations. Using SDS-PAGE in the absence of a reducing agent, E537C also contains a dimer band, similar to wild-type, whereas the C524S/R440C mutant does not. This suggests that intermolecular interaction between two Cys537 residues is possible. The mutant C524S/R440C/E537C showed three bands in addition to those observed in the C524S/E537C mutant, which had both monomer and dimer bands. These extra bands suggest intermolecular interactions between R440C and E537C because Cys440-Cys440 interactions were undetectable. Moreover, double Cys537-Cys440 interactions are also possible in a single dimer.
FIGURE 7.
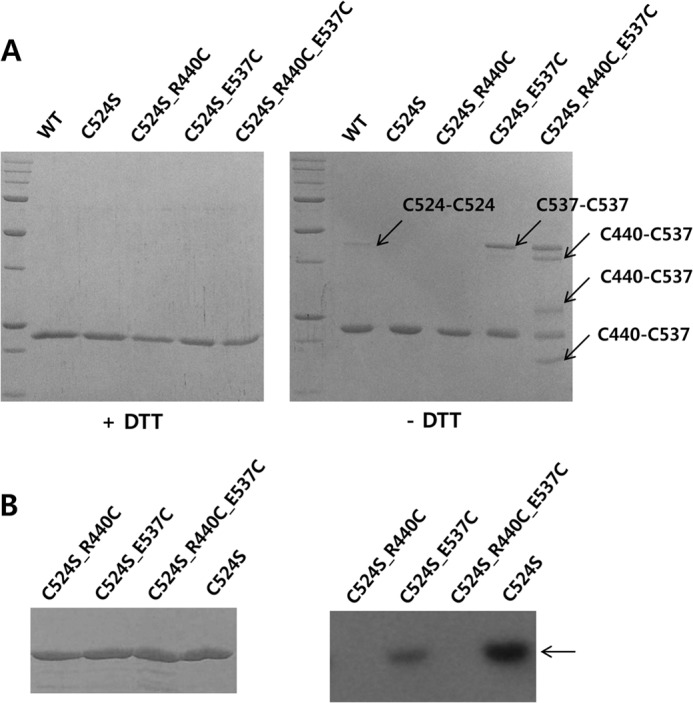
Arg440 and Glu537 can be in close proximity in the KC and these residues are essential for autophosphorylation. A, mutation of Arg440 and Glu537 to cysteines induced disulfide bond formation between the two residues. Although wild-type KC and four mutants (C524S, C524S/R440C, C524S/E537C, and C524S/R440C/E537C) were shown as a single band by SDS-PAGE in the presence of dithiothreitol, dimer-sized bands were detected in WT, C524S/E537C, and C524S/R440C/E537C in the absence of the reducing agent. The presence of multiple bands in the C524S/R440C/E537C mutant indicates various interactions between Cys440 and Cys537 in a KC dimer. B, mutation of Arg440 or Glu537 leads to a loss of the autophosphorylation activity compared the C524S mutant; activity of the R440C mutant was completely ablated.
If the interaction between Glu537 and Arg440 plays a role in assisting ATP binding for autophosphorylation, elimination of these charged residues could result in a loss of autophosphorylation activity. To investigate this, the four mutants were subjected to an autophosphorylation assay. The majority of radioactivity was absent in mutant C524S/E537C compared with the C524S mutant, whereas C524S/R440C and C524S/R440C/E537C mutants completely lost their autophosphorylation activity (Fig. 7B).
DISCUSSION
The current work utilized a combination of crystal structure determination, structural modeling, and site-directed mutagenesis to address fundamental questions underlying the DosS mechanism of action such as how signal detection in the sensor core is communicated to the KC and how this process controls the activity of the KC. The crystal structures of DosS ABD and DosT ABD revealed their stable inactive conformation with a closed ATP binding site. Structural modeling and functional analyses of DosS mutants suggest that a conformational change required for the ATP binding is triggered by the interaction of the ABD with the HisKA region.
The first structure of a KC to be revealed was that of HK853 of T. maritima. The structure showed two globular ABDs, which were linked to a dimer of DHp domains (15). The three distinct conformational states elucidated for the DesK KC clearly demonstrate that the ABD can be positioned differently with respect to the HisKA domain dimer (14). Two ABDs tether to a dimer of HisKA and movement of the ABD has certain flexibility. Considering the possibility of this type of flexibility, the DosS ABD may also have different preferred positions with respect to HisKA (Fig. 5C). In fact, native-PAGE revealed the presence of at least three distinct bands (H, M, and L) for the DosS KC, which apparently represents different conformational states with the ABD in various positions. Among the three major conformations of the KC, autophosphorylation is only detected in the L-form. In the M-form, an intermolecular disulfide bridging using Cys524 hampers autophosphorylation; however, the M-form also failed to bind ATP, as demonstrated by the H395Q mutant (Fig. 4C), indicating that the ATP binding site is not open in the M conformation. The L-form was the only conformation to exhibit detectable ATP binding ability. It indicates that the proper positioning of the ABD with respect to HisKA in the KC is necessary not only for kinase activity but also for exposure of the ATP binding site.
Comparison of the ATP binding sites within the crystal structures of DosS and E. coli PhoQ shows that the loop connecting the β3 and α3 of DosS occupies the ATP binding site. If the β3-α3 loop was sufficiently removed from this site in the DosS ABD, it is possible that ATP may be able to bind because there is sufficient space and N and G boxes are available. As shown in the DosT structure, the β3-α3 loop has a degree of flexibility and Asp534, corresponding to Glu537 of DosS, in the loop can be rotated to face the opposite side. Although in DosS the side chain of Glu537 was found to face into the ATP binding site, it could rotate and participate in an interaction, if a positively charged residue were in close proximity. We proposed that HisKA is responsible for opening of the ATP binding site. There is a positively charged residue, Arg440, close to His395 in the DosS HisKA. Trajtenberg et al. (29) showed the interaction between Gly334 of ABD and Gly192 of HisKA using the disulfide cross-linking assay. The same authors also reported that Glu342 frequently interacts to Arg228 or Arg235 in the docked structures (29). Accordingly, we hypothesized that Arg440 and Glu537 of DosS KC can make a key interaction under these circumstances. This arginine residue is conserved in the DosT HisKA, where Asp534 of DosT corresponds structurally to Glu537 of DosS. When Arg440 and Glu537 were mutated to cysteines, proteins with disulfide bridges were detected. The ability of R440C and E537C to form a disulfide bond indicates that the two residues can be sufficiently closely located to interact ionically. When the ABD approaches the HisKA domain at an orientation where His395 and the ATP binding site are in close proximity, Arg440 can extend to Glu537. The interaction between these two residues will trigger the movement of Glu537 out of the ATP binding site, resulting in extension of the loop to hold the ATP as shown in ATP-bound DesK structure. The ABD will then bind ATP, and His395 is able to accept the phosphate group at the correct position.
If interaction of these two residues plays the role described above, elimination of this charged residue would be expected to hamper autophosphorylation of KC; accordingly, mutation at Arg440 or Glu537 reduces autophosphorylation levels. Unlike the expectation that R440C and E537C mutants behave similarly with respect to autophosphorylation, R440C completely lost its activity, whereas the E537C mutant retained residual activity. One possible explanation for this is that, in the Glu537-containing short loop of DosS, another negatively charged residue, Asp538, interacts with Arg440, instead of Glu537. Alternatively, the marginal activity of the E537C mutant could occur if the mutation caused the loop to withdraw from the ATP binding site, allowing basal autophosphorylation activity of KC to occur in the absence of ionic interaction. In this case, the complete loss of activity in the R440C mutant implies an involvement of Arg440 in another mechanism important for autophosphorylation. A catalytic regulation mechanism with interhelical rearrangements in the HisKA coiled-coil domain was proposed for DesK (14); similarly, a helical rotation in parallel coiled-coils of the HAMP domain relays an input signal in a cog-wheel model (30). In DosS, rotational control of the helices in the HisKA dimer may also be important for the phosphorylation reaction. If Arg440 plays a role in interaction between helices during helix rotation, a mutation at Arg440 could abolish autophosphorylation activity.
To the best of our knowledge, nucleotide-free ABD structures have not been reported in the HK family lacking a full-size ATP lid motif although DesK ABD showed the nucleotide bound structures (14, 29). However, in this study, we showed that the ABDs of DosS and DosT have a closed conformation with regard to ATP binding and that the DosS KC fails both to undergo autophosphorylation and to bind ATP, as a result of the closed conformation of the ABD. To bind ATP, it is necessary for the DosS ABD to undergo a conformational change. Thus, the binding of ATP at the ABD can provide a point of control for regulation of KC activity. Furthermore, the fact that the closed conformation of DosS makes ATP binding possible only when the ABD extends toward HisKA, indicating that ATP binding occurs when the position of the ABD and HisKA are suitable for autophosphorylation. This mimics the ATP lid function of guiding of the ATP to the histidine in HisKA. Protection of ATP by surrounding the phosphate group is suggested as another role for the ATP lid. Closing the ATP binding site until the autophosphorylation reaction is imminent would be another way to protect ATP from unnecessary interactions and would be beneficial for a HK without full-length ATP lid. Previously, autophosphorylation has been thought to proceed stepwise, including one step where ATP binding triggers the proper positioning of the long ATP lid for interaction with the HisKA domain. Here, we propose another mechanism, where proper positioning of a short ATP lid for interaction with the HisKA domain triggers ATP binding and DosS autophosphorylation.
This work was supported by National Research Foundation of Korea Grants 2009-0077493 and 2011-0015987 by the Korean government (Ministry of Education, Science, and Technology).
The atomic coordinates and structure factors (codes 3ZXO and 3ZXQ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HK
- histidine kinase
- DHp
- dimerization and histidine phosphor accepting
- RR
- response regulator
- KC
- kinase core
- ABD
- ATP binding domain
- SeMet
- selenomethionine
- Ni-NTA
- nickel-nitrilotriacetic acid
- PDB
- Protein Data Bank
- H
- high
- M
- middle
- L
- low.
REFERENCES
- 1. Parrish N. M., Dick J. D., Bishai W. R. (1998) Mechanisms of latency in Mycobacterium tuberculosis. Trends Microbiol. 6, 107–112 [DOI] [PubMed] [Google Scholar]
- 2. Wayne L. G., Sohaskey C. D. (2001) Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55, 139–163 [DOI] [PubMed] [Google Scholar]
- 3. Gao R., Stock A. M. (2009) Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63, 133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stewart R. C. (2010) Protein histidine kinases: assembly of active sites and their regulation in signaling pathways. Curr. Opin. Microbiol. 13, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tucker P. A., Nowak E., Morth J. P. (2007) Two-component systems of Mycobacterium tuberculosis: structure-based approaches. Methods Enzymol. 423, 479–501 [DOI] [PubMed] [Google Scholar]
- 6. Roberts D. M., Liao R. P., Wisedchaisri G., Hol W. G., Sherman D. R. (2004) Two sensor kinases contribute to the hypoxic response of Mycobacterium tuberculosis. J. Biol. Chem. 279, 23082–23087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saini D. K., Malhotra V., Tyagi J. S. (2004) Cross talk between DevS sensor kinase homologue, Rv2027c, and DevR response regulator of Mycobacterium tuberculosis. FEBS Lett. 565, 75–80 [DOI] [PubMed] [Google Scholar]
- 8. Kumar A., Toledo J. C., Patel R. P., Lancaster J. R., Jr., Steyn A. J. (2007) Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc. Natl. Acad. Sci. U.S.A. 104, 11568–11573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shiloh M. U., Manzanillo P., Cox J. S. (2008) Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe 3, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sardiwal S., Kendall S. L., Movahedzadeh F., Rison S. C., Stoker N. G., Djordjevic S. (2005) A GAF domain in the hypoxia/NO-inducible Mycobacterium tuberculosis DosS protein binds haem. J. Mol. Biol. 353, 929–936 [DOI] [PubMed] [Google Scholar]
- 11. Cho H. Y., Cho H. J., Kim Y. M., Oh J. I., Kang B. S. (2009) Structural insight into the heme-based redox sensing by DosS from Mycobacterium tuberculosis. J. Biol. Chem. 284, 13057–13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Podust L. M., Ioanoviciu A., Ortiz de Montellano P. R. (2008) 2.3 A X-ray structure of the heme-bound GAF domain of sensory histidine kinase DosT of Mycobacterium tuberculosis. Biochemistry 47, 12523–12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee J. M., Cho H. Y., Cho H. J., Ko I. J., Park S. W., Baik H. S., Oh J. H., Eom C. Y., Kim Y. M., Kang B. S., Oh J. I. (2008) O2- and NO-sensing mechanism through the DevSR two-component system in Mycobacterium smegmatis. J. Bacteriol. 190, 6795–6804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Albanesi D., Martín M., Trajtenberg F., Mansilla M. C., Haouz A., Alzari P. M., de Mendoza D., Buschiazzo A. (2009) Structural plasticity and catalysis regulation of a thermosensor histidine kinase. Proc. Natl. Acad. Sci. U.S.A. 106, 16185–16190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marina A., Waldburger C. D., Hendrickson W. A. (2005) Structure of the entire cytoplasmic portion of a sensor histidine-kinase protein. EMBO J. 24, 4247–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nowak E., Panjikar S., Morth J. P., Jordanova R., Svergun D. I., Tucker P. A. (2006) Structural and functional aspects of the sensor histidine kinase PrrB from Mycobacterium tuberculosis. Structure 14, 275–285 [DOI] [PubMed] [Google Scholar]
- 17. Marina A., Mott C., Auyzenberg A., Hendrickson W. A. (2001) Structural and mutational analysis of the PhoQ histidine kinase catalytic domain. Insight into the reaction mechanism. J. Biol. Chem. 276, 41182–41190 [DOI] [PubMed] [Google Scholar]
- 18. Casino P., Rubio V., Marina A. (2009) Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139, 325–336 [DOI] [PubMed] [Google Scholar]
- 19. Cho H. Y., Cho H. J., Kim M. H., Kang B. S. (2011) Blockage of the channel to heme by the E87 side chain in the GAF domain of Mycobacterium tuberculosis DosS confers the unique sensitivity of DosS to oxygen. FEBS Lett. 585, 1873–1878 [DOI] [PubMed] [Google Scholar]
- 20. Sheffield P., Garrard S., Derewenda Z. (1999) Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15, 34–39 [DOI] [PubMed] [Google Scholar]
- 21. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 22. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Terwilliger T. C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 26. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 27. Navaza J. (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr. Sect. A 50, 157–163 [Google Scholar]
- 28. Berman H., Henrick K., Nakamura H. (2003) Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 10, 980. [DOI] [PubMed] [Google Scholar]
- 29. Trajtenberg F., Graña M., Ruétalo N., Botti H., Buschiazzo A. (2010) Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase. J. Biol. Chem. 285, 24892–24903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hulko M., Berndt F., Gruber M., Linder J. U., Truffault V., Schultz A., Martin J., Schultz J. E., Lupas A. N., Coles M. (2006) The HAMP domain structure implies helix rotation in transmembrane signaling. Cell 126, 929–940 [DOI] [PubMed] [Google Scholar]