

Background: The role of miRNAs in mediating stroke-induced neurogenesis remains largely unknown.
Results: The miR17-92 cluster regulated ischemia-induced neural progenitor cell proliferation, and activation of the Shh pathway up-regulated miR17-92 cluster expression.
Conclusion: The miR17-92 cluster plays an important role in mediating adult neural progenitor cell proliferation.
Significance: The present study provides molecular mechanisms underlying miR17-92 cluster in mediating stroke-induced neurogenesis.
Keywords: Apoptosis, MicroRNA, Neural Stem Cell, Proliferation, Stroke, Sonic Hedgehog, Subventricular Zone (SVZ)
Abstract
The role of microRNAs (miRNAs) in mediating adult neurogenesis after stroke has not been extensively studied. The present study investigated the function of the miR17-92 cluster in adult neural progenitor cells after experimental stroke. We found that stroke substantially up-regulated miR17-92 cluster expression in neural progenitor cells of the adult mouse. Overexpression of the miR17-92 cluster either in cultured ischemic neural progenitor cells or in the subventricular zone (SVZ) of ischemic animals significantly increased cell proliferation, whereas inhibition of individual members of the miR17-92 cluster, miR-18a and miR-19a, suppressed cell proliferation and increased cell death. The miR17-92 cluster mediated PTEN (phosphatase and tensin homolog) expression, which is a predicted target of the miR17-92 cluster. Addition of Sonic hedgehog (Shh) protein up-regulated miR17-92 expression and elevated c-Myc protein in ischemic neural progenitor cells, whereas blockade of the Shh signaling pathway down-regulated miR17-92 cluster expression and reduced c-Myc levels. Overexpression of c-Myc up-regulated miR17-92 cluster expression. Intraventricular infusion of Shh and a Shh receptor inhibitor, cyclopamine, to ischemic animals further elevated and suppressed, respectively, miR17-92 cluster expression in the SVZ. These data indicate that the miR17-92 cluster plays an important role in mediating neural progenitor cell function and that the Shh signaling pathway is involved in up-regulating miR17-92 cluster expression.
Introduction
Stroke induces neurogenesis and neuroblasts generated in the SVZ2 migrate to the ischemic boundary region, which participate in the brain repair process (1–4). Molecular mechanisms underlying stroke-induced neurogenesis are not fully known. MicroRNAs (miRNAs), small noncoding RNAs, regulate neural stem cell function. Inhibition of miRNA biogenesis by ablation of Dicer, an endoribonuclease that cleaves double-stranded RNA and pre-miRNA into short double-stranded RNA, in neural stem cells during development results in reduction of neural stem cells, abnormal neuronal differentiation, and a thin cortical wall of the mouse brain (5). Multiple miRNAs including miR-124a mediate adult neurogenesis (6–13). Using miRNA array, we previously found that stroke robustly changes miRNA profiles in neural progenitor cells. Of interest, is that members of the miR17-92 cluster are increased in ischemic SVZ neural progenitor cells on the array, suggesting that this cluster could play a role in stroke-induced neurogenesis (11). The miR17-92 cluster comprises a cluster of six miRNAs on chromosome 13 that is transcribed as a single polycistronic unit (14). Despite the wealth of information about the biological effects of the miR17-92 cluster in cancer cells (15), the effect of this cluster on regulation of neural progenitor cells after stroke is unknown.
Sonic hedgehog (Shh), a secreted glycoprotein, is critical for embryonic tissue induction and patterning (16–20). The Shh pathway plays an important role in adult neurogenesis under physiological conditions (21). Stroke up-regulates Shh expression, and blockage of the Shh signaling pathway suppresses ischemia -induced neural progenitor cell proliferation (19, 21). Recent studies indicate that the Shh signaling pathway along with the miR17-92 cluster mediate the development of medulloblastomas (22, 23). However, little is known about the effect of activation of the Shh signaling pathway on miR17-92 cluster expression and the biological function of the miR17-92 cluster in ischemic neural progenitor cells.
The present study investigated the effect of altering the miR17-92 cluster levels on the proliferation and differentiation of neural progenitor cells and the linkage between the Shh pathway and expression of the miR17-92 cluster. Using an animal model of focal cerebral ischemia and primary neural progenitor cells, we show that the miR17-92 cluster regulated ischemia-induced neural progenitor cell proliferation and that activation of the Shh pathway up-regulated miR17-92 cluster expression.
EXPERIMENTAL PROCEDURES
All experimental procedures were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Henry Ford Hospital.
Model of Middle Cerebral Artery Occlusion (MCAO)
Male C57BL/6J mice (3–4 months) were employed in this study. The right MCA was permanently occluded by placement of an embolus at the origin of the right MCA as described previously (24). In this model, MCAO evokes a peak increase of neurogenesis 7 days after stroke (3, 25). Therefore, all mice were sacrificed 7 days after MCAO.
SVZ Cell Culture
SVZ neural progenitor cells were isolated from adult mice, as described previously (26). The cells were plated at a density of 2 × 104 cells/μl in the growth medium, which contains DMEM/F-12 medium (Invitrogen), 20 ng/ml EGF (R&D Systems, Minneapolis, MN), and basic FGF (R&D Systems). DMEM/F-12 medium contains l-glutamine (2 mmol/liter), glucose (0.6%), putrescine (9.6 mg/ml), insulin (0.025 mg/ml), progesterone (6.3 ng/ml), apotransferrin (0.1 mg/ml), and sodium selenite (5.2 ng/ml). The generated SVZ neurospheres (primary spheres) were passaged by mechanical dissociation and reseeded as single cells at a density of 20 cells/μl. Cultured SVZ neural progenitor cells used in the experiments were less than 3 passages to avoid the likely genetic variation of progeny.
Laser Capture Microdissection (LCM)
LCM was performed according to our published protocol (26, 27). Briefly, frozen brain coronal sections (8 μm) stored at −80 °C were immediately immersed in acetone for 2 min of fixation and air-dried for 30s. After a brief rinse with 0.1% diethylpyrocarbonate-treated phosphate buffered saline, sections were stained with propidium iodide dye (1:5000 dilution, Sigma Aldrich) for 5 min and rinsed with phosphate-buffered saline twice. Sections were then air-dried under laminar flow for 10 min and immediately used for LCM. Dense SVZ cells on sections stained by propidium iodide were readily distinct from the ependymal cells that have cilia along the lateral wall of the lateral ventricle and from the adjacent striatal cells. In the non-ischemic mouse, the dorsal and ventral SVZ of the lateral wall was defined as a 20–30-μm-wide zone approximately of two to three cell bodies immediately adjacent to ependymal cells, whereas in the ischemic mouse, the SVZ was expanded to a 60–80-μm-wide zone (28). Propidium iodide-positive cells within the SVZ were dissected with a Leica AS LMD System (Leica Microsystems, Inc, Buffalo Grove, IL). The excised cells fell into a collection tube under gravity, ensuring contamination-free processing and minimizing sample damage. Eppendorf tubes containing 20 μl of lysis buffer were stored at −80 °C before miRNA isolation. Approximately 1,000 cells were isolated in the SVZ from each animal.
Implantation of an Osmotic Pump
An Alzet micro-osmotic pump was implanted in the animal brain for 7 days starting 1 day after MCAO. The intraventricular catheter with osmotic pump (1 μl/hr, Alzet, Cupertino, CA) was placed into the right lateral ventricle (ipsilateral to ischemia). The catheter placement was 0.8 mm lateral and 0 mm caudal from the bregma and 2.2 mm deep. The pump was loaded with either 5 μmol/liter recombinant mouse Shh (rmShh, catalog no. 461-SH-025, R&D Systems) or control vehicle (0.1% BSA in PBS).
To examine whether inhibition of the Shh pathway affects neural progenitor cell proliferation, cyclopamine (Millipore, Philadelphia, PA), a potent inhibitor of Smoothen (Smo), was intraventricularly infused at a dose of 0.11 μg/h into the lateral ventricle of the ischemic hemisphere via the osmotic pump for 28 days starting 1 day after MCAO. Ischemic animals infused with vehicle were used as a control group. For labeling proliferating cells, BrdU (100 mg/kg) was administered (i.p.) daily for 7 consecutive days starting at 24 h after stroke. All animals were sacrificed 35 days after MCAO.
Plasmid DNA Transfection
The primary miR17-92 cluster (786 bp) was amplified by PCR using the following primers: forward, 5′-CGGAATTCGTCAGGATAATGTCAAAGTGCTTACA and reverse, CGGGTACCACCAAACTCAACAGGCCG, with EcoRI and KpnI at 5′-ends. pCAG-miR17-92 cluster plasmid was constructed by cloning a primary miR17-92 cluster fragment into the EcoRI and KpnI sites in the pCAG-GFP vector. The recombinant DNA protocol follows the National Institutes of Health guidelines. pCAG-miR17-92 plasmid (2 μg) was introduced into neural progenitor cells by means of electroporation using a NucleofectorTM kit (Lonza, Walkersville, MD), an empty pCAG-GFP vector was used as a negative control. Briefly, DNA plasmids were mixed with 100 μl of Nucleofector solution, and cell-DNA mixtures were transferred into a cuvette and electroporated using program A33. In addition, to examine the effect of c-Myc on miR17-92 cluster expression, neural progenitor cells were transfected with pCNDA3.0 vector carrying c-Myc (Addgene, Cambridge, MA) or an empty vector. Total RNAs or proteins were extracted at 24 or 72 h after nucleofection for the following experiment.
miRNA and siRNA Electroporation in Vitro
miRNA hairpin inhibitors against miR-18a (mature sequence, UAAGGUGCAUCUAGUGCAGAUAG), miR-19a (mature sequence, UGUGCAAAUCUAUGCAAAACUGA), and their mimics and cel-miR-67 (mature sequence, UCACAACCUCCUAGAAAGAGUAGA) were purchased from Dharmacon (Chicago, IL). cel-miR-67 was used as the negative control for miRNA inhibitor and mimics. For attenuation of endogenous PTEN, SVZ neural progenitor cells were transfected with Silencer® Select siRNA-PTEN (s132220, Invitrogen), and a Silencer® Select Negative Control (Invitrogen, 4390843) was employed as a control. siRNA or miRNA oligonucleotides (200 pmol) were mixed with 100 μl of nucleofector solution and were introduced into cultured neural progenitor cells using a NucleofectorTM kit as described above.
Quantification of Mature miRNAs by Real-time qRT-PCR
Total RNAs (10 ng) were reverse transcribed using a TaqMan® MicroRNA Reverse Transcription (RT) kit (Applied Biosystems, Carlsbad, CA). Each RT reaction contained 1× stem-loop RT-specific primer, 1× reaction buffer, 0.25 mm each of dNTPs, 3.33 units/μl Multiscribe RT enzyme and 0.25 U/μl RNase inhibitor. The 15-μl reactions were incubated for 30 min at 16 °C, 30 min at 42 °C, and 5 min at 85 °C and then held at 4 °C. The PCR reaction was performed using a standard TaqMan® PCR kit protocol (Applied Biosystems). Briefly, following the RT step, 1.33 μl of the RT reaction were combined with 1 μl of a TaqMan microRNA assay (20×; forward primer, reverse primer, and probe) and 17.67 μl of TaqMan® Universal PCR Master Mix, No AmpErase® UNG in a 20-μl final volume. The reactions were incubated at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The expression of miRNAs was normalized against the expression of U6 snRNA as an endogenous normalization control. All assays were performed in triplicate and were calculated on the basis of the ΔΔCt method. The n-fold change in miRNAs expression was determined according to the method of 2−ΔΔCT.
Neurosphere Assay
A neurosphere assay was employed to investigate the effect of miRNAs on SVZ neural progenitor cells. The assay has been widely used by us and others (7, 11, 19, 26) as a valuable tool for investigating the biology of neural progenitor cells. The SVZ neural progenitor cells were transfected with miRNA mimics or inhibitors of miR-18a and miR-19a and incubated in growth medium in the presence of EGF and basic FGF for 24 h to allow the cells to recover. Then single cells at a density of 10 cells/μl were plated directly onto laminin-coated glass coverslips and incubated for an additional 48 h to allow cell growth. Immediately after the termination of incubation, cell apoptosis was analyzed with TUNEL staining. To analyze cell proliferation, BrdU (30 μg/ml, Sigma Aldrich), the thymidine analog that is incorporated into the DNA of dividing cells during S-phase, was added 18 h before the termination of incubation. BrdU-positive cells were measured (see below for quantification). To examine the SVZ cell differentiation, neurospheres were plated directly onto laminin-coated glass coverslips in DMEM-F-12 medium containing 2% FBS, which is referred to as differentiation medium. Every 4 days, one-half of the medium was replaced with fresh medium. Incubation was terminated 10 days after plating, and immunostaining for neuronal and astrocyte markers was performed for evaluation of cell differentiation.
Generation of Lentivirus and Injection of Lentivirus
High-titer vesicular stomatitis virus-pseudotyped lentivirus were produced in 293TN cells by transient transfection of the transfer vector pre-miR17-92 cluster in pcopGFP lentivector (System Biosciences, Mountain View, CA) and pPACK packaging mixture, and concentrated by PEG-it virus precipitation solution as described in the manual. The titers of pseudoviral particles were determined by fluorescence real time PCR and were 1 × 109 to 1 × 1010 transducing units/ml. Five μl of vector concentrate was injected by a 33G Hamilton syringe (0.2 μl/min) into the SVZ at stereotactic coordinates (mm from bregma): anterior-posterior = 0, mediolateral = +0.8 cm, and dorsoventral = 2.2 cm from the skull surface.
Immunocytochemistry and Quantification
Immunofluorescent staining was performed on cultured cells. The following primary antibodies were used in the present study: mouse anti-BrdU (1:1,00; Roche Applied Science), mouse anti-β-III tubulin (TuJ-1, 1:500; Covance the Development Services Company), rabbit anti-glial fibrillary acidic protein (1:500; Dako Cytomation California, Inc., Carpinteria, CA), rabbit anti-NG2 (1:500; Abcam, Cambridge, MA), and chicken anti-GFP (Aves Labs, Tigard, OR). Cultured cells were fixed in 4% paraformaldehyde for 20 min at room temperature. Nonspecific binding sites were blocked with phosphate-buffered saline with 1% bovine serum albumin for 1 h at room temperature. The cells were then incubated with the primary antibodies listed above and with CY3-conjugated secondary antibodies. Nuclei were counterstained with DAPI (1:10,000, Vector Laboratories, Burlingame, CA). Apoptosis was assayed with TUNEL staining according to manufacturer's instructions (Millipore).
The number of BrdU-positive cells and total DAPI nuclei was counted under a 20× objective (IX71; Olympus Optical, Tokyo, Japan), and the percentage of BrdU/DAPI was determined. For all measurements, we counted at least 500 cells from three wells/group (n = 3 individual cultured cells).
ChIP
A ChIP assay was performed using the Magna ChIPTM A kit (Upstate, Charlottesville, VA). SVZ cells were cross-linked with 1% formaldehyde and sonicated to an average length of 200–500 bp. The chromatin solutions were precleared with the addition of protein G beads for 2 h at 4 °C. The precleared chromatins were incubated with Myc antibody (2 μg, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and normal IgG serum as a negative control overnight. The antibody/chromatin mixtures were precipitated with protein A/G magnetic beads, and magnetic beads were pelleted with a DynaMagTM-Spin (Invitrogen). The beads were sequentially washed with ChIP wash buffer. Cross-linkings were reversed by adding 4 μl of 5 m NaCl and incubating at 65 °C overnight. DNAs were purified by phenol/chloroform extraction and ethanol precipitation. The real-time PCR primers (forward primer, 5′-CCTTGTGCGACATGTGCTG, and reverse primer, 5′-GTTTCCCCAACTGCTGTGAT (202 bp) were used to amplify primary miR17-92 cluster promoter region flanking the Myc-binding site. Binding activities were calculated as percentage of pre-immunoprecipitated input DNA.
SDS-PAGE and Western Blot
Cells were lysed in radioimmune precipitation assay buffer containing 50 mm Tris-HCl, pH 8.0, with 150 mm sodium chloride, 1.0% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate. Lysate was sonicated and then centrifuged for 10 min at 12,000 rpm to remove cell debris. Protein concentrations were determined using a BCA assay (Thermo Scientific, Waltham, MA). Equal amounts of proteins were then separated by SDS-PAGE and transferred to a nitrocellulose membrane. Membrane was probed with an appropriate primary antibody and a secondary antibody conjugated to horseradish peroxidase. The following antibodies were utilized: β-actin (1:10,000 dilution, Abcam), cleaved caspase-3 (1:500 dilution, Cell Signaling, Danvers, MA), PTEN (phosphatase and tensin homolog; 1:500 dilution, Santa Cruz Biotechnology), c-Myc and N-Myc (1:500 dilution, Santa Cruz Biotechnology), Fas ligand (FasL, 1:500 dilution, Abcam). Proteins were visualized by enhanced chemiluminescence (Thermo Fisher Scientific, Rockford, IL).
Statistical Analysis
The data are presented as mean ± S.E. Independent sample t test was used for two-group comparisons from the non-MCAO and MCAO samples. One-way analysis of variance followed by a Student's Newman-Keuls test was performed for multiple sample analysis. A value of p < 0.05 was taken as significant.
RESULTS
The miR17-92 Cluster Up-regulated by Stroke Promotes the Proliferation and Survival of SVZ Neural Progenitor Cells
Using microarray, we previously demonstrated that stroke up-regulated miR17-92 expression in neural progenitor cells (11). To verify microarray findings, single neural progenitor cells in the SVZ were isolated from animals subjected to 7 days of MCAO by means of LCM (Fig. 1A), and levels of individual members of the miR17-92 cluster in these cells were measured by quantitative real-time RT-PCR. Compared with the cells isolated from non-ischemic SVZ, the cells from the ischemic SVZ had substantial increases in levels of miR-18a, miR-19a, and miR-19b (Fig. 1B), which is consistent with published microarray findings (11). To further confirm the in vivo findings, we examined miR17-92 cluster expression in cultured neural progenitor cells harvested from the SVZ of mice subjected to 7 days of stroke and found a significant elevation of individual members including miR-18a, miR-19a, miR-19b and miR-92a of the miR17-92 cluster (Fig. 1C). These data indicate that stroke up-regulates miR17-92 cluster expression in neural progenitor cells.
FIGURE 1.

Quantification of miR17-92 cluster and its paralogs in SVZ neural progenitor cells after stroke. Panel A shows the SVZ neural progenitor cells captured before and after laser microdissection. Panels B and C show qRT-PCR data of individual members of the miR17-92 cluster in single neural progenitor cell population captured by laser microdissection (B) and in primary cultured neural progenitor cells harvested from the SVZ (C), respectively. Panel D shows qRT-PCR data of miR106b-25 components in primary cultured neural progenitor cells. The fold change of miRNAs was normalized against the expression of U6 snRNA as an endogenous normalization control. Scale bar, 50 μm. LV, lateral ventricle; CC, corpus callosum; Str, striatum.
There are two miR17-92 cluster analogs, miR-106a-363 and miR-106b-25 (14). To examine whether stroke alters expression of these two clusters, levels of individual members of the two clusters were measured in ischemic neural progenitor cells using real-time RT-PCR. In contrast with the up-regulation of the miR17-92 cluster, we found that stroke substantially decreased the expression of miR-106a, miR-106b, miR-25, miR-93, and miR-363 (Fig. 1D). Another member of the miR-106a-363 cluster, miR-19b-2, was poorly expressed as indicated by a high CT value (∼35). We therefore, focused on the miR17-92 cluster for the following experiments.
Aforementioned data indicate that miR-18a and miR-19a were highly up-regulated among individual components of the miR17-92 cluster after stroke (Fig. 1B). Stroke increases neural progenitor cell proliferation and promotes progenitor cell differentiation into neuroblasts (1–4). To examine whether up-regulation of these two members affects cell proliferation and differentiation, neural progenitor cells harvested from the ischemic SVZ were transfected with miR-18a and miR-19a inhibitors, and in vitro cell proliferation and differentiation were measured. Quantitative RT-PCR analysis showed that transfection of miR-18a and miR-19a inhibitors almost completely abolished endogenous levels of miR-18a (Fig. 2A) and miR-19a (Fig. 2A), respectively, indicating the efficacy of ablation of endogenous miR-18a and miR-19a in ischemic neural progenitor cells. Analysis of the number of BrdU-positive cells showed that attenuation of endogenous miR-18a and miR-19a by the inhibitors substantially decreased the percentage of BrdU-positive cells (Fig. 2B). Blockade of endogenous miR-19a significantly increased the number of apoptotic cells, i.e. TUNEL-positive cells, whereas inhibition of miR-18a increased apoptotic cell death but did not reach a statistical significance (Fig. 2, C and D).
FIGURE 2.

The effects of individual miR-18a and miR-19a on the proliferation and survival of ischemic neural progenitor cells. Panel A demonstrates the introduction of miR-18a and miR-19a inhibitors significantly decreased the expression of miR-18a and miR-19a in neural progenitor cells. Panels B and C show quantitative data of BrdU-positive (B) and TUNEL-positive (C) cells, respectively, after treatment with miR-18 inhibitor, miR-19a inhibitor, or cel-miR-67 (control). Panel D shows representative immunostaining images of TUNEL-positive cells. Panel E shows that delivery of miR-18a and miR-19a mimics dramatically increased the expression of endogenous miR-18a and miR-19a. Panels F and G show quantitative data of the number of BrdU-positive cells (F) and representative immunofluorescent images of BrdU-positive cells (G) after treatment with miR-18a mimic, miR-19a mimics, or cel-miR-67 (control). *, p < 0.05; #, p < 0.01. Scale bar, 20 μm.
To examine whether forced increases in miR-18a and miR-19a levels further augment ischemia-induced cell proliferation, miR-18a and miR-19a mimics were delivered into ischemic neural progenitor cells. Compared with mimic controls, delivering the two mimics elevated miR-18a and miR-19a levels by 389- and 236-fold, respectively (Fig. 2E). Augmentation of miR-18a and miR-19a levels significantly increased the number of BrdU-positive cells (Fig. 2, F and G). In addition, ischemic progenitor cells were transfected with a pCAG-GFP vector carrying primary miR17-92 cluster or an empty vector (Fig. 3, A and B). Overexpression of miR17-92 resulted in 4.3- and 6.5-fold increases in miR-18a and miR-19a levels, respectively, compared with levels in neural progenitor cells transfected with an empty vector (Fig. 3C). Other members of the miR17-92 cluster, including miR-17, miR-19b, miR-20, and miR-92, were also up-regulated after transfection (Fig. 3C). Neural progenitor cells with overexpression of primary miR17-92 cluster exhibited a significant increase in proliferation measured by the BrdU-positive cells (Fig. 3, D and E). Overexpression of the miR17-92 cluster significantly reduced the number of glial fibrillary acidic protein (Fig. 3, F and G), but not Tuj1- and NG2-positive cells (Fig. 3G). The effect of the miR-18a and miR-19a mimics and overexpression of the miR17-92 cluster on apoptotic cells were not detected because only few TUNEL-positive cells were found in control group (data not shown). To further examine the effect of overexpression of the miR17-92 cluster on neural progenitor cells in vivo, we intraventricularly injected the lentiviral vector carrying miR17-92 cluster precursor and GFP into the lateral ventricle of the ischemic hemisphere 1 day prior to stroke. Compared with ischemic animals treated with the lentiviral vector carrying only GFP, ischemic animals that received the lentiviral vector carrying miR17-92 cluster and GFP exhibited a substantial increase of neural progenitor cells measured by the number of GFP+/SOX2+ cells in the ischemic SVZ (Fig. 3, H and I), which is in line with in vitro data. Collectively, these in vitro and in vivo data suggest that either exogenous miR-18a and miR-19a or overexpression of the miR17-92 cluster enhance ischemia-increased neural progenitor cell proliferation.
FIGURE 3.

The biological effect of miR17-92 cluster on the neurogenesis. Panels A and B show that many of neural progenitor cells (A, bright field) exhibited GFP signals (B, fluorescent field) 24 h after transfection with the pCAG-miR17-92 plasmid. Panel C shows qRT-PCR data of individual miR17-92 in the neural progenitor cells after transfection with the pCAG-miR17-92 cluster (miR17-92) or the empty vector (control). Panels D and E show representative immunofluorescent images of BrdU-positive cells (D) and quantitative data of the number of BrdU-positive cells (E) after transfection with the pCAG-miR17-92 cluster (miR17-92) or the empty vector (control). F panels are microphotographs showing glial fibrillary acidic protein (GFAP; red) immunoreactive cells and GFP (green). Panel G shows the quantification data derived from transfected SVZ precursors after miR17-92 overexpression. Panels H and I demonstrate that the intraventricular infusion of the lentiviral vector carrying miR17-92 cluster and GFP increased the number of GFP+/SOX2+ cells in the SVZ (I, miR17-92, arrows) compared with the infusion of the lentiviral vector carrying only GFP (I; control, arrows). LV, lateral ventricle. Scale bar, 20 μm.
Using Targetscan, miRanda, and Pictar, we found that PTEN is one of the predicted targets of miR-18a and miR19a (Fig. 4A). PTEN is well known to regulate cell proliferation and survival (29–35). Accordingly, we examined whether elevation and attenuation of miR-18a and miR-19a alter PTEN protein levels in neural progenitor cells. Western blot analysis revealed that miR-18a and miR-19a mimics substantially reduced PTEN levels (Fig. 4B), whereas inhibitors of miR-18a and miR-19a elevated PTEN levels (Fig. 4C). These data suggest that the both miR-18a and miR19a target PTEN. Interestingly, although Fas ligand, an inducer of apoptosis (36), is not a putative target of miR-18a and miR-19a, we found that introduction of the mimics of miR-18a and miR-19a suppressed Fas ligand protein (Fig. 4B), whereas their inhibitors elevated Fas ligand levels in ischemic neural progenitor cells (Fig. 4C). To examine the effect of PTEN on neural progenitor cells, we transfected ischemic neural progenitor cells with a siRNA-PTEN or a scrambled control siRNA. Western blot analysis confirmed that siRNA-PTEN substantially decreased endogenous PTEN levels in ischemic neural progenitor cells 48 h after the transfection (Fig. 4D). Attenuation of PTEN by siRNA-PTEN considerably reduced cleaved caspase-3 proteins (Fig. 4D) and the number of TUNEL-positive cells (Fig. 4, E and F) and greatly increased the number of BrdU-positive cells (Fig. 4, G and H), compared with the cells transfected with the scrambled siRNA. These results are consistent with the effect of miR-18a and miR-19a overexpression on the neural progenitor cells.
FIGURE 4.
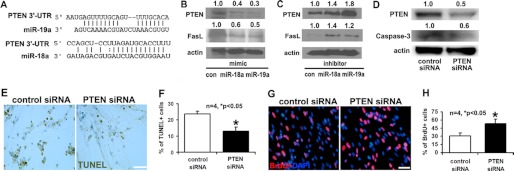
Panel A shows sequences of miR-18a and miR-19a binding sites in the 3′-UTRs of PTEN are predicted based on Targetscan software (version 5.1). Western blots (B and C) show protein levels of PTEN and Fas ligand in neural progenitor cells after transfection with miR-18a and miR-19a mimics (B) or miR-18a and miR-19a inhibitors (C). Control is cel-miR-67 (con). Panel D shows that protein levels of PTEN and caspase-3 were decreased in neural progenitor cells transfected with siRNA against PTEN, compared with those transfected with scrambled siRNAs. Intensity of the bands, normalized to actin and relative to the control lane, is shown above the images. Panels E and G show immunostaining of TUNEL and BrdU on neural progenitor cells transfected with PTEN or scrambled siRNAs. Panels F and H show the quantitative data of TUNEL- and BrdU-immunoreactive cells, respectively. *, p < 0.05. Scale bar, 20 μm.
Shh Signaling Regulates miR17-92 Cluster Expression
The Shh signaling pathway regulates neurogenesis in the adult SVZ under non-ischemic and ischemic conditions (16, 17, 19, 21). To examine the effect of Shh on miR17-92 cluster expression in neural progenitor cells, primary adult SVZ neural progenitor cells were treated with rmShh (100 ng/ml) or transfected with siRNA against Shh (siRNA-Shh) for 24 h. Transfection of siRNA-Shh reduced endogenous mRNA levels of Shh by 80% compared with cells transfected with control scrambled siRNA. Quantitative real-time RT-PCR analysis revealed that rmShh substantially increased levels of individual members of the miR17-92 cluster (Fig. 5A), whereas attenuation of endogenous Shh in ischemic neural progenitor cells by siRNA-Shh substantially decreased levels of miR-18a, miR-19a, and miR-92a compared with levels in ischemic neural progenitor cells transfected with scrambled siRNA (Fig. 5B). Shh binds to its transmembrane receptor Ptch that results in the activation of the transmembrane protein Smo and subsequently activates the Gli transcription factors (37–39). To examine whether the Shh signaling pathway regulates miR17-92 cluster expression, the ischemic neural progenitor cells were incubated with cyclopamine (5 μm), a specific inhibitor of the Smo, in the presence of rmShh, and levels of individual members of the miR17-92 cluster were measured. Real-time RT-PCR analysis showed that cyclopamine suppressed rmShh-increased levels of miR-18a, miR-19a, miR-19b, and miR-92a (Fig. 5A). The effect of Shh on the miR17-92 cluster appears specific because rmShh treatment in the presence or absence of cyclopamine did not significantly affect the expression of miR-106a-363 and miR-106b-25 components on neural progenitor cells (Fig. 5D). To examine whether exogenous Shh regulates miR17-92 cluster expression in ischemic brain, rmShh or vehicle BSA was intraventricularly infused to ischemic mice for 7 consecutive days starting 1 day after stroke via an osmotic mini-pump. Single SVZ cells in these mice were isolated with LCM and levels of the miR17-92 cluster in SVZ cells were analyzed. Infusion of rmShh substantially increased levels of miR-18a, miR-19a, and miR-92a compared with treatment with BSA (Fig. 5C). In contrast, intraventricular infusion of cyclopamine suppressed ischemia-up-regulated miR-18a, miR-19a, miR-19b, and miR-92a in SVZ cells (Fig. 5E) and ischemia-increased BrdU-positive cells (Fig. 5, F and G) compared with the infusion of vehicle. These in vitro and in vivo data indicate that the Shh signaling pathway mediates miR17-92 cluster expression in neural progenitor cells.
FIGURE 5.

The effect of the Shh signaling pathway on the expression of miR17-92 cluster in ischemic neural progenitor cells. Panels A–C show qRT-PCR data of individual members of the miR17-92 cluster (A to C) in primary cultured neural progenitor cells treated with rmShh (A, Shh), rmShh with cyclopamine (A, Shh+CPM), the cultured cells transfected with siRNA-Shh (B) or SVZ cells after intraventricular infusion of rmShh (C). rmShh (100 ng/ml) and cyclopamine treatment did not significantly affect the expression of miR106a-363 and miR106b-25 components on neural progenitor cells (D). Control for panel B is scrambled siRNA and control for panel C is BSA. * and #, p < 0.05 versus control and Shh, respectively. Panel E shows qRT-PCR data of individual members of the miR17-92 cluster in the ischemic SVZ of animals received intraventracular infusion of cyclopamine (E, ischemia+CPM, n = 8) or vehicle (E, ischemia+vehicle, n = 4) and non-ischemia SVZ (E, n = 4). Representative microscopic images show BrdU-positive cells obtained from brain coronal sections of animals that received infusion of cyclopamine (F, ischemia+CPM, arrows) or vehicle (F, ischemia+vehicle, arrows) and the non-ischemia (F, arrows) SVZ. Panel G shows quantitative data of BrdU-positive cells in the vehicle and cyclopamine groups. Scale bar, 50 μm. LV, lateral ventricle.
Myc is a downstream target of Shh (40–42). C- and N-Myc regulate miR17-92 cluster expression in HeLa cells (43), neuroblastoma (22, 44, 45), and cerebellar granule neuron precursors (22). Whether C- and N-Myc are involved in Shh-mediated miR17-92 cluster expression in adult neural progenitor cells is unknown. SVZ tissues were isolated from 7 day ischemic animals and ischemic animals treated with rmShh and protein levels of Myc were assayed by means of Western blot. We found that stroke significantly increased levels of c-Myc and N-Myc (Fig. 6, A and B), and intraventricular infusion of rmShh to ischemic mice further increased c-Myc protein levels (Fig. 6C). These in vivo data suggest that Shh targets Myc in neural progenitor cells. To confirm the in vivo observation, ischemic neural progenitor cells were treated with rmShh in the presence or absence of cyclopamine and Myc proteins were measured. Incubation of neural progenitor cells with rmShh considerably increased c-Myc levels, whereas cyclopamine abolished rmShh-augmented c-Myc (Fig. 6D). These in vivo and in vitro data indicate that the Shh signaling pathway regulates c-Myc proteins in neural progenitor cells. We then examined the effect of c-Myc on miR17-92 cluster expression. Ischemic neural progenitor cells were transfected with pCNDA vector carrying c-Myc or an empty vector and expression of the miR17-92 cluster was measured. Western blot analysis confirmed that transfection of pCDNA-c-Myc plasmid increased protein levels of c-Myc (Fig. 6E). Elevation of c-Myc resulted in considerable increases in levels of individual members of the miR17-92 cluster (Fig. 6F). These data indicate that c-Myc activates transcription of the miR17-92 cluster in neural progenitor cells, which is consistent with published findings in fibroblasts (46). Using TRANSFAC software (BIOBASE Corp., Beverly, MA), we found that there are putative c-Myc binding sites in the promoter of primary miR17-92 cluster (Fig. 6G). To examine whether c-Myc binds to the miR17-92 cluster promoter, ChIP was performed. Proteins isolated from normal and ischemic SVZ neural progenitor cells were incubated with antibodies against c-Myc. The immune complexes were precipitated with magnetic protein A/G beads. The recovered DNA from the input, unbound, and bound fractions were determined by PCR, which amplified the c-Myc motif in the miR17-92 promoter sequences. A binding band was detected on ChIP (Fig. 6H), and stroke enhanced the binding of c-Myc to the promoter of miR17-92 (Fig. 6H), suggesting that Myc is involved in miR17-92-promoted neurogenesis. These results suggest that Shh could target c-Myc to regulate miR17-92 cluster expression in neural progenitor cells.
FIGURE 6.

The effect of the Shh signaling pathway on the Myc in ischemic neural progenitor cells. Western blots show levels of c-Myc (A and C) and N-Myc (B) in SVZ tissues after stroke and after intraventricular infusion of rmShh (C) to ischemic animals. Panel D is a Western blot showing c-Myc protein levels in cultured neural progenitor cells treated with rmShh (Shh) or rmShh with cyclopamine (Shh+cyclopamine). A Western blot (E) shows c-Myc levels after transfection with pDNA-c-Myc. RT-PCR data (F) show individual members of the miR17-92 cluster in cultured neural progenitor cells transfected with pCNDA3.0 vector carrying c-Myc or an empty vector (control). Panel G shows c-Myc consensus factor-specific matrix analyzed by TRANSFAC (TRANScription FACtor database). The ChIP assay (H) reveals binding of c-Myc to primary miR17-92 cluster promoter sequence under non-ischemic (non-MCAO) and ischemic (MCAO) conditions. Anti-rabbit IgG (IgG) was employed as a negative control.
DISCUSSION
There are few reports on the biological functions of miRNAs in adult neural progenitor cells after stroke. In the present study, we found that stroke substantially up-regulated the miR17-92 cluster in SVZ neural progenitor cells. Furthermore, we demonstrate that the miR17-92 cluster up-regulated by stroke increased neural progenitor cell proliferation and survival and that the Shh signaling pathway mediated miR17-92 cluster expression. Taken together, these data provide evidence for biological function of the miR17-92 cluster in ischemic neural progenitor cells.
The miR17-92 cluster comprises a cluster of seven miRNAs on chromosome 13 that are transcribed as a single polycistronic unit (47). During development, the miR17-92 cluster regulates neural progenitor cell proliferation and oligodendrogenesis (23, 48). Clinical relevance of the miR17-92 cluster has recently been implicated in patients with Feingold syndrome, an autosomal dominant syndrome, and germ line deletion of the miR17-92 cluster in the patients causes microcephaly and skeletal abnormalities (49). We previously demonstrated that stroke alters miRNA profiles in SVZ neural progenitor cells (11). The present study showed that stroke robustly up-regulated members of the miR17-92 cluster, miR-18a, miR-19a, miR-19b, and miR-92a, in SVZ neural progenitor cells. Attenuation of endogenous miR-18a and miR-19a levels elevated by stroke suppressed neural progenitor cell proliferation and increased progenitor cell death, whereas addition of exogenous miR-18a and miR-19a further enhanced stroke-induced progenitor cell proliferation, indicating that these two miRNAs mediate stroke-induced neurogenesis by augmentation cell proliferation and survival. Others have reported that the miR17-92 cluster regulates oligodendrogenesis during development (48). Using conditional knock-out of the miR17-92 cluster in CNPase-expressing oligodendrocytes, Budde et al. (48) show that ablation of the miR17-92 cluster substantially reduces oligodendrocyte progenitor cell proliferation and survival at postnatal day 0. Our data showed that alteration of miR-18a and miR-19a levels inversely change PTEN protein levels and that the effect of attenuation of endogenous PTEN by siRNA-PTEN on ischemic neural progenitor cells mimics the effect of elevation of miR-18a and miR-19a on the progenitor cells, suggesting that PTEN is likely a target of miR-18a and miR-19a in mediating neural progenitor cell proliferation, which is consistent with published studies showing that the miR17-92 cluster represses PTEN level on oligodendrocytes (48) and oncogenic cells (14, 50, 51). PTEN negatively regulates embryonic neural stem cell proliferation and survival (29–35). Following a focal cerebral ischemia, PTEN null progenitors give rise to greater numbers of neuroblasts that migrate to peri-infarct cortex (29). Interestingly, we also found that FasL levels were negatively regulated by miR-18a and miR-19a, although FasL is not a predicted target of these two miRNAs by the miRNA target algorithms. Studies in Pten+/− mice and others have shown that Fas/FasL is involved in PTEN-mediated cell survival (52). Thus, the effect of miR-18a and miR-19a on FasL expression observed in the present study is likely indirectly affected by alteration of PTEN levels. Although PTEN is a featured target of miR18a and miR-19a, it is likely the miR17-92 cluster targets other potential genes in the neural progenitor cells, which coordinately regulate neurogenesis. In addition, contrary to the up-regulation of the miR17-92 cluster, we found that several components of its paralogs, miR-106b-25 and miR-106a-363 clusters, were decreased in ischemic neural progenitor cells, suggesting different roles of miR-106b-25, miR-106a-363 clusters in stroke-induced neurogenesis.
The Shh signaling pathway mediates adult neurogenesis under physiological and stroke conditions (16–19, 21). Our previous study demonstrated that the Shh pathway mediates carbamylated erythropoietin-enhanced proliferation of adult neural progenitor cells (19). The present study indicates that endogenous Shh in neural progenitor cells and exogenous Shh regulate miR17–19 cluster expression in neural progenitor cells and that administration of exogenous Shh to ischemic animals further up-regulated miR17-92 cluster in SVZ neural progenitor cells, whereas blockage of the Shh pathway suppressed ischemia-up-regulated miR17-92 cluster and ischemia-increased neural progenitor cell proliferation. These in vitro and in vivo data strongly suggest that the miR17-92 cluster likely mediates Shh-induced neural progenitor cell proliferation. Our findings are consistent with reports by others showing that the Shh signaling pathway up-regulates miR17-92 expression (22, 23). Myc is an effector of the Shh signaling pathway, and Myc activates miR17-92 cluster expression by binding to the promoter of the primary miR17-92 cluster (40–45). We found that Shh increased Myc levels, which was associated with up-regulation of the miR17-92 cluster. Collectively, these data suggest that the Shh signaling pathway up-regulates miR17-92 cluster expression at least in part through activation of Myc in neural progenitor cells.
In summary, the present study suggests that the miR17-92 cluster mediates adult neural progenitor cell proliferation and survival and that the Shh signaling pathway up-regulates miR17-92 cluster expression. Our data raise the possibility that augmentation of the miR17-92 cluster could amplify endogenous neurogenesis in ischemic brain.
This work was supported by National Institutes of Health Grants RO1 AG037506 (to M. C.) and RO1 NS075156 (to Z. G. Z.) and American Heart Association Scientist Development Grant 10SDG2790012 (to X. S. L.).
- SVZ
- subventricular zone
- miRNA
- microRNA
- Shh
- Sonic hedgehog
- MCAO
- middle cerebral artery occlusion
- LCM
- laser capture microdissection
- rmShh
- recombinant mouse Shh
- qRT-PCR
- quantitative RT-PCR
- FasL
- Fas ligand.
REFERENCES
- 1. Parent J. M., Vexler Z. S., Gong C., Derugin N., Ferriero D. M. (2002) Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 52, 802–813 [DOI] [PubMed] [Google Scholar]
- 2. Yamashita T., Ninomiya M., Hernández Acosta P., García-Verdugo J. M., Sunabori T., Sakaguchi M., Adachi K., Kojima T., Hirota Y., Kawase T., Araki N., Abe K., Okano H., Sawamoto K. (2006) Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 26, 6627–6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang R., Zhang Z., Wang L., Wang Y., Gousev A., Zhang L., Ho K. L., Morshead C., Chopp M. (2004) Activated neural stem cells contribute to stroke-induced neurogenesis and neuroblast migration toward the infarct boundary in adult rats. J. Cereb. Blood Flow. Metab. 24, 441–448 [DOI] [PubMed] [Google Scholar]
- 4. Zhang R. L., Zhang Z. G., Chopp M. (2005) Neurogenesis in the adult ischemic brain: generation, migration, survival, and restorative therapy. Neuroscientist 11, 408–416 [DOI] [PubMed] [Google Scholar]
- 5. De Pietri Tonelli D., Pulvers J. N., Haffner C., Murchison E. P., Hannon G. J., Huttner W. B. (2008) miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 135, 3911–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brett J. O., Renault V. M., Rafalski V. A., Webb A. E., Brunet A. (2011) The microRNA cluster miR-106b∼25 regulates adult neural stem/progenitor cell proliferation and neuronal differentiation. Aging 3, 108–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng L. C., Pastrana E., Tavazoie M., Doetsch F. (2009) miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 12, 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Denli A. M., Cao X., Gage F. H. (2009) miR-9 and TLX: chasing tails in neural stem cells. Nat. Struct. Mol. Biol. 16, 346–347 [DOI] [PubMed] [Google Scholar]
- 9. Liu C., Teng Z. Q., Santistevan N. J., Szulwach K. E., Guo W., Jin P., Zhao X. (2010) Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell 6, 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu C., Zhao X. (2009) MicroRNAs in adult and embryonic neurogenesis. Neuromolecular Med. 11, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X. S., Chopp M., Zhang R. L., Tao T., Wang X. L., Kassis H., Hozeska-Solgot A., Zhang L., Chen C., Zhang Z. G. (2011) MicroRNA profiling in subventricular zone after stroke: MiR-124a regulates proliferation of neural progenitor cells through Notch signaling pathway. PLoS One 6, e23461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Magill S. T., Cambronne X. A., Luikart B. W., Lioy D. T., Leighton B. H., Westbrook G. L., Mandel G., Goodman R. H. (2010) microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc. Natl. Acad. Sci. U.S.A. 107, 20382–20387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szulwach K. E., Li X., Smrt R. D., Li Y., Luo Y., Lin L., Santistevan N. J., Li W., Zhao X., Jin P. (2010) Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 189, 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiao C., Srinivasan L., Calado D. P., Patterson H. C., Zhang B., Wang J., Henderson J. M., Kutok J. L., Rajewsky K. (2008) Lymphoproliferative disease and autoimmunity in mice with increased miR-17–92 expression in lymphocytes. Nat. Immunol. 9, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cho W. C. (2007) OncomiRs: the discovery and progress of microRNAs in cancers. Mol. Cancer 6, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lai K., Kaspar B. K., Gage F. H., Schaffer D. V. (2003) Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat. Neurosci. 6, 21–27 [DOI] [PubMed] [Google Scholar]
- 17. Marti E., Bovolenta P. (2002) Sonic hedgehog in CNS development: one signal, multiple outputs. Trends Neurosci. 25, 89–96 [DOI] [PubMed] [Google Scholar]
- 18. Rowitch D. H., S-Jacques B., Lee S. M., Flax J. D., Snyder E. Y., McMahon A. P. (1999) Sonic hedgehog regulates proliferation and inhibits differentiation of CNS precursor cells. J. Neurosci. 19, 8954–8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang L., Zhang Z. G., Gregg S. R., Zhang R. L., Jiao Z., LeTourneau Y., Liu X., Feng Y., Gerwien J., Torup L., Leist M., Noguchi C. T., Chen Z. Y., Chopp M. (2007) The Sonic hedgehog pathway mediates carbamylated erythropoietin-enhanced proliferation and differentiation of adult neural progenitor cells. J. Biol. Chem. 282, 32462–32470 [DOI] [PubMed] [Google Scholar]
- 20. Zhu G., Mehler M. F., Zhao J., Yu Yung S., Kessler J. A. (1999) Sonic hedgehog and BMP2 exert opposing actions on proliferation and differentiation of embryonic neural progenitor cells. Dev. Biol. 215, 118–129 [DOI] [PubMed] [Google Scholar]
- 21. Sims J. R., Lee S. W., Topalkara K., Qiu J., Xu J., Zhou Z., Moskowitz M. A. (2009) Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke 40, 3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Northcott P. A., Fernandez-L A., Hagan J. P., Ellison D. W., Grajkowska W., Gillespie Y., Grundy R., Van Meter T., Rutka J. T., Croce C. M., Kenney A. M., Taylor M. D. (2009) The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer Res. 69, 3249–3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uziel T., Karginov F. V., Xie S., Parker J. S., Wang Y. D., Gajjar A., He L., Ellison D., Gilbertson R. J., Hannon G., Roussel M. F. (2009) The miR-17∼92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc. Natl. Acad. Sci. U.S.A. 106, 2812–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Z., Chopp M., Zhang R. L., Goussev A. (1997) A mouse model of embolic focal cerebral ischemia. J. Cereb. Blood Flow Metab. 17, 1081–1088 [DOI] [PubMed] [Google Scholar]
- 25. Zhang R. L., Zhang Z. G., Zhang L., Chopp M. (2001) Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience 105, 33–41 [DOI] [PubMed] [Google Scholar]
- 26. Liu X. S., Chopp M., Zhang R. L., Hozeska-Solgot A., Gregg S. C., Buller B., Lu M., Zhang Z. G. (2009) Angiopoietin 2 mediates the differentiation and migration of neural progenitor cells in the subventricular zone after stroke. J. Biol. Chem. 284, 22680–22689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu X. S., Zhang Z. G., Zhang R. L., Gregg S., Morris D. C., Wang Y., Chopp M. (2007) Stroke induces gene profile changes associated with neurogenesis and angiogenesis in adult subventricular zone progenitor cells. J. Cereb. Blood Flow Metab. 27, 564–574 [DOI] [PubMed] [Google Scholar]
- 28. Zhang R. L., Zhang Z. G., Lu M., Wang Y., Yang J. J., Chopp M. (2006) Reduction of the cell cycle length by decreasing G1 phase and cell cycle reentry expand neuronal progenitor cells in the subventricular zone of adult rat after stroke. J. Cereb. Blood Flow Metab. 26, 857–863 [DOI] [PubMed] [Google Scholar]
- 29. Gregorian C., Nakashima J., Le Belle J., Ohab J., Kim R., Liu A., Smith K. B., Groszer M., Garcia A. D., Sofroniew M. V., Carmichael S. T., Kornblum H. I., Liu X., Wu H. (2009) Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J. Neurosci. 29, 1874–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Groszer M., Erickson R., Scripture-Adams D. D., Dougherty J. D., Le Belle J., Zack J. A., Geschwind D. H., Liu X., Kornblum H. I., Wu H. (2006) PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc. Natl. Acad. Sci. U.S.A. 103, 111–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Groszer M., Erickson R., Scripture-Adams D. D., Lesche R., Trumpp A., Zack J. A., Kornblum H. I., Liu X., Wu H. (2001) Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294, 2186–2189 [DOI] [PubMed] [Google Scholar]
- 32. Li L., Liu F., Ross A. H. (2003) PTEN regulation of neural development and CNS stem cells. J. Cell. Biochem. 88, 24–28 [DOI] [PubMed] [Google Scholar]
- 33. Li L., Liu F., Salmonsen R. A., Turner T. K., Litofsky N. S., Di Cristofano A., Pandolfi P. P., Jones S. N., Recht L. D., Ross A. H. (2002) PTEN in neural precursor cells: regulation of migration, apoptosis, and proliferation. Mol. Cell Neurosci. 20, 21–29 [DOI] [PubMed] [Google Scholar]
- 34. Otaegi G., Yusta-Boyo M. J., Vergaño-Vera E., Méndez-Gómez H. R., Carrera A. C., Abad J. L., González M., de la Rosa E. J., Vicario-Abejón C., de Pablo F. (2006) Modulation of the PI 3-kinase-Akt signalling pathway by IGF-I and PTEN regulates the differentiation of neural stem/precursor cells. J. Cell Sci. 119, 2739–2748 [DOI] [PubMed] [Google Scholar]
- 35. Zheng H., Ying H., Yan H., Kimmelman A. C., Hiller D. J., Chen A. J., Perry S. R., Tonon G., Chu G. C., Ding Z., Stommel J. M., Dunn K. L., Wiedemeyer R., You M. J., Brennan C., Wang Y. A., Ligon K. L., Wong W. H., Chin L., DePinho R. A. (2008) p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455, 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miwa K., Asano M., Horai R., Iwakura Y., Nagata S., Suda T. (1998) Caspase 1-independent IL-1β release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med. 4, 1287–1292 [DOI] [PubMed] [Google Scholar]
- 37. Ahn S., Joyner A. L. (2005) In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature 437, 894–897 [DOI] [PubMed] [Google Scholar]
- 38. Ingham P. W., McMahon A. P. (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087 [DOI] [PubMed] [Google Scholar]
- 39. Ruiz i Altaba A., Sánchez P., Dahmane N. (2002) Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat. Rev. Cancer 2, 361–372 [DOI] [PubMed] [Google Scholar]
- 40. Hatton B. A., Knoepfler P. S., Kenney A. M., Rowitch D. H., de Alborán I. M., Olson J. M., Eisenman R. N. (2006) N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 66, 8655–8661 [DOI] [PubMed] [Google Scholar]
- 41. Kenney A. M., Cole M. D., Rowitch D. H. (2003) Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 130, 15–28 [DOI] [PubMed] [Google Scholar]
- 42. Kenney A. M., Widlund H. R., Rowitch D. H. (2004) Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 131, 217–228 [DOI] [PubMed] [Google Scholar]
- 43. O'Donnell K. A., Wentzel E. A., Zeller K. I., Dang C. V., Mendell J. T. (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843 [DOI] [PubMed] [Google Scholar]
- 44. Lovén J., Zinin N., Wahlström T., Müller I., Brodin P., Fredlund E., Ribacke U., Pivarcsi A., Påhlman S., Henriksson M. (2010) MycN-regulated microRNAs repress estrogen receptor-α (ESR1) expression and neuronal differentiation in human neuroblastoma. Proc. Natl. Acad. Sci. U.S.A. 107, 1553–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schulte J. H., Horn S., Otto T., Samans B., Heukamp L. C., Eilers U. C., Krause M., Astrahantseff K., Klein-Hitpass L., Buettner R., Schramm A., Christiansen H., Eilers M., Eggert A., Berwanger B. (2008) MycN regulates oncogenic MicroRNAs in neuroblastoma. Int. J. Cancer 122, 699–704 [DOI] [PubMed] [Google Scholar]
- 46. Hong L., Lai M., Chen M., Xie C., Liao R., Kang Y. J., Xiao C., Hu W. Y., Han J., Sun P. (2010) The miR-17–92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. 70, 8547–8557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hayashita Y., Osada H., Tatematsu Y., Yamada H., Yanagisawa K., Tomida S., Yatabe Y., Kawahara K., Sekido Y., Takahashi T. (2005) A polycistronic microRNA cluster, miR-17–92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 65, 9628–9632 [DOI] [PubMed] [Google Scholar]
- 48. Budde H., Schmitt S., Fitzner D., Opitz L., Salinas-Riester G., Simons M. (2010) Control of oligodendroglial cell number by the miR-17–92 cluster. Development 137, 2127–2132 [DOI] [PubMed] [Google Scholar]
- 49. de Pontual L., Yao E., Callier P., Faivre L., Drouin V., Cariou S., Van Haeringen A., Geneviève D., Goldenberg A., Oufadem M., Manouvrier S., Munnich A., Vidigal J. A., Vekemans M., Lyonnet S., Henrion-Caude A., Ventura A., Amiel J. (2011) Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat. Genet. 43, 1026–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Olive V., Bennett M. J., Walker J. C., Ma C., Jiang I., Cordon-Cardo C., Li Q. J., Lowe S. W., Hannon G. J., He L. (2009) miR-19 is a key oncogenic component of mir-17–92. Genes Dev. 23, 2839–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takakura S., Mitsutake N., Nakashima M., Namba H., Saenko V. A., Rogounovitch T. I., Nakazawa Y., Hayashi T., Ohtsuru A., Yamashita S. (2008) Oncogenic role of miR-17–92 cluster in anaplastic thyroid cancer cells. Cancer Sci 99, 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Di Cristofano A., Kotsi P., Peng Y. F., Cordon-Cardo C., Elkon K. B., Pandolfi P. P. (1999) Impaired Fas response and autoimmunity in Pten+/− mice. Science 285, 2122–2125 [DOI] [PubMed] [Google Scholar]