

Background: MarP is a serine protease critical for pH homeostasis in Mycobacterium tuberculosis.
Results: MarP is localized in the periplasm, and its substrate specificity was uncovered using synthetic peptides.
Conclusion: The periplasmic location of MarP is essential for function and the substrate profile shows high selectivity at multiple subsites.
Significance: Understanding the MarP proteolytic pathway may lead to the development of novel anti-tuberculosis chemotherapeutics.
Keywords: Mycobacterium tuberculosis, Peptide Biosynthesis, Peptides, pH Regulation, Serine Protease, Stress Response, Multiplex Substrate Profiling, Positional-scanning Synthetic Combinatorial Library
Abstract
The transmembrane serine protease MarP is important for pH homeostasis in Mycobacterium tuberculosis (Mtb). Previous structural studies revealed that MarP contains a chymotrypsin fold and a disulfide bond that stabilizes the protease active site in the substrate-bound conformation. Here, we determined that MarP is located in the Mtb periplasm and showed that this localization is essential for function. Using the recombinant protease domain of MarP, we identified its substrate specificity using two independent assays: positional-scanning synthetic combinatorial library profiling and multiplex substrate profiling by mass spectrometry. These methods revealed that MarP prefers bulky residues at P4, tryptophan or leucine at P2, arginine or hydrophobic residues at P1, and alanine or asparagine at P1′. Guided by these data, we designed fluorogenic peptide substrates and characterized the kinetic properties of MarP. Finally, we tested the impact of mutating MarP cysteine residues on the peptidolytic activity of recombinant MarP and its ability to complement phenotypes of Mtb ΔMarP. Taken together, our studies provide insight into the enzymatic properties of MarP, its substrate preference, and the importance of its transmembrane helices and disulfide bond.
Introduction
Tuberculosis is the second highest cause of death from a communicable disease, killing ∼1.5 million people annually (1). This can in part be attributed to the multiple mechanisms Mycobacterium tuberculosis (Mtb)4 has evolved to resist bactericidal host defenses, including acid and oxidative stresses (2–5). Although Mtb can stall phagosome-lysosome fusion (6–8), activation of macrophages with the T-cell-derived cytokine IFN-γ turns the relatively neutral (pH ∼ 6.2) environment of the Mtb-containing phagosome into the much more hostile, acidic environment of the phagolysosome with a pH as low as 4.5 (9–13). Mtb uses mechanisms of pH homeostasis to persist within host cells and maintain a near neutral intrabacterial pH despite the acidic phagolysosomal pH surrounding the bacteria (3).
Mechanisms of pH homeostasis have been well characterized in several Gram-negative and Gram-positive bacteria (14, 15), but those active in Mtb remain elusive. A small number of proteins has been implicated in the mycobacterial response to low pH including: MgtC, a predicted magnesium transporter (16); OmpATB, an outer membrane protein (17); PhoPR, a two component regulator (18); aprABC, a gene locus regulated by PhoPR in response to acid (19); and Rv3671c, a transmembrane serine protease (3, 20). A mutant with a transposon insertion in Rv3671c failed to maintain its neutral intrabacterial pH in acidic conditions both in vitro and in IFN-γ activated macrophages and lost viability (3). pH homeostasis was dependent on Rv3671c proteolytic activity, because a catalytically inactive mutant (Rv3671c-S343A) failed to rescue the acid sensitivity of the transposon mutant, whereas the wild-type enzyme fully restored the phenotypes of the mutant. Furthermore, disruption of Rv3671c significantly attenuated Mtb in both the acute and chronic phases in a mouse model of Mtb infection. A better mechanistic understanding of the Rv3671c protease, which we named MarP (mycobacterial acid resistance protease), may aid in the discovery of new inhibitors that target pH homeostasis.
MarP is highly conserved in mycobacteria (21). Computational predictions of its secondary structure reveal four transmembrane helices in the N-terminal region connected by a linker region to the C-terminal protease domain (22). The predicted topology places the protease domain of MarP outside of the cytoplasm, in the periplasmic compartment. The presence of an outer membrane and, hence, a periplasm in mycobacteria, was recently demonstrated by cryo-electron microscopy studies (23, 24). Structural and biochemical analyses demonstrated that the C-terminal domain of MarP is a chymotrypsin-like serine protease, with a classic catalytic triad composed of His-235, Asp-264, and Ser-343 (20). Recombinant MarP lacking its transmembrane domain readily cleaved β-casein. Furthermore, a reversible disulfide bond between Cys-214 and Cys-395 regulated proteolysis in vitro by stabilizing the active site in a catalytically active conformation. A construct of the protease containing a part of the linker that connects the N-terminal transmembrane helices with the C-terminal protease domain underwent autoproteolysis within the linker at several sites, but the major cleavage (↓) occurred after Leu-160 in the sequence PKRL↓SALL (20). The biological significance of this autocleavage site is unknown, as the truncated form of the native protease has not been observed in Mtb lysates.
The closest characterized homologues of MarP are the HtrA (high temperature requirement A) serine proteases, whose protease domains exhibit ∼20% sequence identity with that of MarP. HtrA-family proteases are periplasmic serine proteases characterized by at least one C-terminal PDZ protein-protein interaction domain and an association with the cytoplasmic membrane (25, 26). They function as both proteases and oligomeric chaperones depending on the molecular signal (27). Mtb contains three HtrA proteases (28, 29). One of them, HtrA2 (PepD) was shown to be important for Mtb stress response and for virulence in the mouse model of infection (29, 30). However, MarP differs from these proteases because it lacks a PDZ domain, contains more than one predicted transmembrane helix (31), and exists as a monomer (3, 20). Despite knowledge about the structure of the protease domain and functional role of MarP in pH homeostasis, the localization of the protease domain, the function of the transmembrane helices, and the substrate preference of the protease remain unclear.
In this study we investigated the importance of the transmembrane helices in MarP-mediated acid resistance and established the periplasmic location of the protease domain experimentally. We used two distinct substrate profiling methods (32, 33) to determine the peptide sequence cleavage specificity of the enzyme. Knowledge of the preferred cleavage sequences enabled us to design fluorogenic substrates and characterize the proteolytic activity of MarP. MarP was active over a broad pH range and sensitive to serine protease inhibitors and reducing reagents. This work will facilitate future studies to identify in vivo substrates and elucidate the MarP proteolytic pathway.
EXPERIMENTAL PROCEDURES
Protease Constructs
MarP_179–397 and MarP_179–397-S343A were purified as previously described (20). MarP_179–397-C214A and MarP_179–397-C214A-C395A were constructed by mutating Cys-214 and Cys-395 to alanine using the QuikChange site-directed mutagenesis kit (Stratagene). PCR products were cloned into the pET28b vector including a C-terminal His6 tag and purified as previously described (20). MarP complementation plasmids were created using Gateway Cloning Technology (Invitrogen). For MarP-TM1234, full-length marP was amplified from the chromosome. To generate MarP-TM12, the MarP protease domain at Leu-119 was fused to Val-52 of the second MarP transmembrane helix using overlap PCR. Similarly, the MarP protease domain at Leu-119 was fused to Val-102 of TM helix three to create MarP-TM123. A C-terminal FLAG tag was introduced to all constructs by PCR, and all genes were cloned behind the Hsp60 promoter on plasmids integrating into the Mtb chromosome. In MarP-C214A and MarP-C214A/C395A, cysteine residues were mutated to alanine using the QuikChange site-directed mutagenesis kit (Stratagene). Both constructs were expressed under the control of the Hsp60 promoter from plasmids that integrate into the chromosome. ΔMarP was electroporated, and transformants were selected for on 7H10 plates with kanamycin (20 μg/ml). Expression of each protein was confirmed by immunoblotting with FLAG-tag-specific antibodies (Sigma) or MarP-specific antibodies.
Bacterial Strains and Media
Mtb (H37Rv) strains were grown at 37 °C in Middlebrook 7H9 liquid medium (Difco) containing 0.2% glycerol, 0.5% BSA, 0.2% dextrose, 0.085% NaCl and 0.05% Tween80 or on Middlebrook 7H10 agar plates containing 10% oleic acid albumin dextrose catalase supplement (BD Biosciences) and 0.5% glycerol. Hygromycin B (50 μg/ml) and kanamycin (20 μg/ml) were included when selection was required. Phosphate citrate buffer contained 200 mm sodium phosphate, 100 mm citric acid, and 0.02% Tyloxapol. A knock-out strain lacking MarP (ΔMarP) with the entire marP gene replaced by a hygromycin cassette was previously created and confirmed by Southern blot (data not shown).
Immunoblotting
Mtb lysates were prepared and fractionated as described (3), and 60–80 μg of cytosolic fractions and an equal volume of membrane and cell envelope fraction based on total volume of these fractions were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Antibodies and dilutions used are as follows: 1:1,000 anti-alkaline phosphatase (Sigma), 1:2,000 anti-MarP; 1:10,000 anti-PrcB (proteasome β subunit); 1:3,000 anti-FLAG (Sigma).
Alkaline Phosphatase Fusions and Activity
MarP fragments were amplified using PCR and cloned into pMB111 using the BamHI restriction sites to create in-frame fusions to alkaline phosphatase (PhoA). The pMB111 plasmid (a gift from Miriam Braunstein) contains Escherichia coli PhoA lacking a signal sequence. Plasmid pMB124 (gift from Miriam Braunstein) contains PhoA fused to the signal sequence of the Mtb secreted antigen, antigen 85B (34), and served as a positive control. Plasmids were electroporated into Mycobacterium smegmatis mc2155 and plated on LB agar plates containing 60 μg/ml 5-bromo-4-chloro-3-indolyl phosphate, 0.2% glucose, and 0.05% Tween80.
Mtb Susceptibility to Low pH and Hydrogen Peroxide
Early to mid-log phase cultures were washed with PBS containing 0.05% Tween 80 and centrifuged at 800 rpm for 12 min. Single-cell suspensions were inoculated into stress media at an A600 = 0.05. Colony forming units (cfu) were determined by plating serial dilutions of the suspensions on 7H10 agar plates. Mtb was exposed to 8 mm H2O2 in 7H9 medium for 4 h at 37 °C. To measure low pH susceptibility, Mtb was inoculated into phosphate citrate buffer at pH 4.5 and incubated at 37 °C for 3 and 6 days.
The Complete Diverse Positional Scanning Synthetic Combinatorial Library (PS-SCL)
PS-SCL consists of four sublibraries of fluorogenic substrates with the general structure acetyl-P4-P3-P2-P1-amino-4-carbamoyl-methylcoumarin. One position is fixed in each sublibrary, whereas the remaining positions contain an equimolar mixture of all amino acids (32). 2 μm MarP_179–397 was assayed with the library in 50 mm Tris-HCl, pH 7.4, 500 mm NaCl, 0.01% Triton X-100 for 1 h at 37 °C. The enzyme mixture was preincubated for 15 min at 37 °C before adding it to the PS-SCL substrates. All reactions were run in triplicate at 37 °C in 96-well Microfluor-1 Black “U” bottom plates (Dynex Technologies, Chantilly, VA) on a SpectraMax Gemini fluorescence spectrometer (Molecular Devices) with the excitation and emission wavelengths set at 380 and 460 nm, respectively.
Peptide Cleavage Site Identification by Multiplex Substrate Profiling (MSP)-Mass Spectrometry
MarP (250 nm) was profiled using the MSP-MS assay as described (33). Each peptide pool was incubated at room temperature in 50 mm Tris-HCl, pH 7.4, 500 mm NaCl, and aliquots were removed and acid-quenched to pH 3 or less with formic acid (4% final) after 60, 240, and 1200 min. A control sample without enzyme was prepared under identical conditions and quenched at the first and last time point of the assay to account for non-enzymatic degradation of the substrates. Cleaved bonds derived from non-enzymatic degradation were subtracted from the MarP cleavage sites. Cleavage site identification was performed using peptide sequencing by mass spectrometry. Samples containing 1–3 μg of total peptide (calculated as 60 μl of enzyme reaction containing peptide pools at 500 nm) were desalted using C18 zip tips (Millipore) and rehydrated in 0.1% formic acid. The peptide mixture containing 0.1 μg of peptide was injected onto the column.
For LC-MS/MS, a LTQ-FT mass spectrometer (Thermo) equipped with a 10,000 p.s.i. system nanoACUITY (Waters) UPLC instrument for reverse phase chromatography with a C18 column (BEH130, 1.7-μm bead size, 100 μm × 100 mm) was used with standard operating conditions as previously reported (33). Mass spectrometry peak lists were generated using in-house software called PAVA, and Protein Prospector software Version 5.10.0 (35) was used to search a database containing 620 14-mer sequences. This database contains the sequences of the 124 14-mer synthetic peptides, concatenated with 4 different copies of randomized sequences for the same 124 entries to create a final database of 620 sequences for estimation of false discovery rate (33). For database searching, peptide sequences were matched with no enzyme specificity requirement, and potential modifications included oxidation of Trp, Pro, and Phe and N-terminal pyroGlu from Gln. Protein Prospector score thresholds were selected to be as follows: a minimum protein score of 22, a minimum peptide score of 15, and maximum expectation values of 0.01 for “protein” and 0.05 for peptide matches. This resulted in a peptide false discovery rate of <0.2%. Cleavage site data were extracted from Protein Prospector using an in-house script called “MSP extractor” software (33). iceLogo software was used to generate substrate specificity profiles for amino acids at ±4 positions on both sides of the identified scissile bond (36). Raw mass spectrometry data can be obtained from the ProteomeCommons Tranche network.
Assay of Enzymatic Activity
Fluorogenic peptides were designed based on preferred substrate sequences from the PS-SCL and MSP-MS assays. Trp-Lys-Leu-Leu-AMC was synthesized by Anaspec (Anaspec, Freemont, CA), whereas 7-methoxycoumarin-4-acetic acid (MCA)-PSLIAKWV-K-2,4-dinitrophenol (DNP) was synthesized in-house using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) synthesis. To carry out steady-state kinetic measurements, 250 nm MarP_179–397 was assayed with 0–250 μm WKLL-AMC, whereas 2 μm MarP_179–397 was assayed with 0–160 μm MCA-PSLIAKWV-K(DNP). Quadruplicate assays were run in 50 mm Tris-HCl, pH 7.4, 500 mm NaCl, 0.01% Triton X-100 at 37 °C for 1 h. The enzyme was preincubated in the above reaction buffer at 37 °C for 5 min before adding substrate. Michaelis-Menten kinetic parameters were determined by non-linear curve-fitting of raw data to the Michaelis-Menten equation using Prism software (GraphPad, Inc.). In addition, two fluorogenic substrates, MCA-PKRLSALL-K(DNP) and MCA-SALLNTSG-K(DNP), corresponding to the autocleavage site were synthesized in-house. These substrates were incubated with 2 μm MarP for 48 h. Samples were desalted as outlined previously, mixed with Alpha-CHC Matrix (Agilent) and subjected to MALDI-TOF MS analysis on a Voyager DE Pro BioSpectrometry work station (Applied Biosystems).
The effect of pH on enzymatic activity was determined in different buffers in the pH range from 4.5 to 11.0 (50 mm MES, pH 4.5–6.5; 50 mm Tris-HCl, pH 7.0–9.0; 50 mm CAPS, pH 9.0–11.0) with 500 mm NaCl, 0.01% Triton X-100, and 50 μm WKLL-AMC as substrate. Fluorescence of the reaction mixture without the enzyme in each of the above buffers was monitored to control for peptide hydrolysis.
Enzyme inhibition was characterized with 750 nm MarP_179–397 in 50 mm Tris-HCl, pH 7.4, 500 mm NaCl, 0.01% Triton X-100, and the following inhibitors: 20 μm pepstatin A, 1 mm phenylmethylsulfonyl fluoride (PMSF), 2 μm E-64, 10 mm EDTA, 20 mm sodium fluoride, 20 μm fluorophosphonate-rhodamine (FP-Rho, a gift from Dr. Benjamin Cravatt, Scripps Research Institute), 2 mm DTT (Sigma). The reactions were initiated by the addition of the enzyme and inhibitor mixture to 50 μm WKLL-AMC in the final reaction volume of 200 μl. All samples were run in triplicate. For the IC50 determination of PMSF and FP-Rho, 250 nm MarP_179–397 was incubated with 0–1200 μm PMSF and 0–20 μm FP-Rho for 36 min at room temperature. The reactions were initiated by the addition of the enzyme and inhibitor mixture to 50 μm WKLL-AMC in the final reaction volume of 200 μl. All samples were run in triplicate. The IC50 was calculated by non-linear curve-fitting using Prism Software (Graphpad, Inc).
Modeling of Peptides Bound to the MarP Protease Domain
The models were generated based on the crystal structure of MarP in the active state (20) PDB ID 3K6Y) using Coot (37). The backbone conformation of the modeled peptide was the same as that of the bound peptide in the crystal structure. The residues of the peptide were mutated from those in the crystal structure to a given sequence, and the side chain rotamers of Lys, Tyr, Trp that do not clash sterically with the protease were chosen among most energetically preferred ones, as defined in Coot. Side chain torsion angles were manually adjusted slightly whenever needed to avoid minor steric clashes. The figure was created in PyMOL (Schrödinger, LLC) with the surface electrostatic potential generated by the APBS plug-in (38).
RESULTS
Localization of the MarP Protease Domain
MarP is predicted to contain four transmembrane (TM) helices by the TransMembrane server, TMHMM (22), with both the N terminus and C-terminal protease domain located in the periplasm (Fig. 1A). The TM helices are predicted to consist of amino acids 5–23 (TM1), 30–52 (TM2), 62–84 (TM3), and 98–119 (TM4). TM4 and the protease domain are connected by a 58-residue-long linker region (20). We tested these predictions experimentally. PhoA from E. coli has been used to determine the topology of membrane proteins (39) including those in mycobacterial species (34, 40). PhoA is active only when located in the oxidizing, extracytoplasmic environment and can thus be used as a periplasmic reporter. We created four PhoA fusions at different residues of MarP, resulting in constructs containing different numbers of the predicted transmembrane helices. Two fusions, after Leu-123 and Leu-119, were C-terminal of all four predicted transmembrane helices, which places PhoA in the periplasm. Two fusions, at Val-102 and Pro-95, were made C-terminal of the first three helices, which localizes PhoA in the cytosol (Fig. 1A). M. smegmatis clones expressing Leu-123 and Leu-119 fusions demonstrated alkaline phosphatase activity, whereas those at Val-102 and Pro-95 were inactive (Fig. 1B). The plasmid containing PhoA lacking a signal sequence served as a negative control, and a plasmid expressing PhoA fused to the signal sequence of the secreted antigen 85B served as the positive control (34). Immunoblotting with a PhoA-specific antibody verified that all fusion proteins were expressed (Fig. 1C). These experiments confirm the topology predictions and indicate that MarP activity occurs in the periplasm.
FIGURE 1.

Membrane topology of the protease MarP. A, shown is a model of MarP topology. Black circles indicate the location of the PhoA fusions that were created at residues Leu-123, Leu-119, Val-102, and Pro-95. B, alkaline phosphatase activity of M. smegmatis strains expressing MarP-PhoA fusion proteins are shown. Strains were plated on LB agar containing the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate. Plasmid pMB124 (positive control) expresses PhoA fused to the signal sequence of Mtb antigen 85B, and pMB111 (negative control) expresses PhoA without a signal sequence; MarP-Leu-123, MarP-Leu-11, MarP-Val-102, and MarP-Pro-95 express PhoA fused to the respective residues of MarP. C, shown is an immunoblot of M. smegmatis lysates probed with a PhoA-specific monoclonal antibody (α-PhoA). An immunoblot for the proteasome subunit B (α-PrcB) serves as loading control.
Importance of the Transmembrane Helices
MarP proteolysis is important for resistance to both acid and oxidative stress; however, the role of the transmembrane helices in these functions has not been explored. To address this, we created two Mtb strains in the background of a MarP knock-out strain (ΔMarP). In ΔMarP the entire marP gene is replaced with a hygromycin cassette (data not shown). MarP-TM12 expressed MarP lacking transmembrane helices 3 and 4, placing the protease domain in the periplasm (Fig. 2A, upper panel). MarP-TM123 expressed MarP lacking transmembrane domain 4, so that the protease domain is located in the cytoplasm (Fig. 2A, lower panel). MarP-TM1234 contained full-length MarP. To test the importance of the transmembrane helices for pH homeostasis, we exposed the bacteria to phosphate-citrate buffer, pH 4.5, and measured viability (Fig. 2B). As predicted, the ΔMarP strain was hypersusceptible to acid; its cfu decreased 30- and 300-fold compared with that of wild-type (WT) Mtb after 3 and 6 days, respectively. MarP-TM1234 and MarP-TM12 were significantly more acid-resistant and displayed survival rates similar to WT Mtb, whereas MarP-TM123 phenocopied ΔMarP. A similar trend was observed for susceptibility to H2O2 stress. Mtb lacking MarP was previously shown to be hypersusceptible to hydrogen peroxide (20). Cfu of MarP-TM123 and ΔMarP were 10-fold lower than those of WT after exposure to 8 mm H2O2 for 4 h, whereas MarP-TM1234 and MarP-TM12 survived like WT (Fig. 2C). All MarP proteins were expressed and were of expected sizes (Fig. 2D), demonstrating that the lack of complementation with MarP-TM123 was not due to lack of protein expression. These data indicate that transmembrane helices 3 and 4 are dispensable for resistance to acid and oxidative stress, whereas the periplasmic localization of the protease is essential for these protective functions.
FIGURE 2.

Role of the transmembrane helices. A, shown is schematic representation of MarP truncated constructs. E and C correspond to extracellular and cytosolic, respectively. B, sensitivity of WT (H37Rv), ΔMarP, and complemented strains to acid stress are shown. Mtb strains were incubated in phosphate citrate buffer, pH 4.5, and cfu were quantified on day 3 and day 6; data are the means of ± S.D. of triplicate cultures. C, survival of WT, ΔMarP, and complemented strains after exposure to 8 mm H2O2 for 4 h is shown; data are the means of ± S.D. of triplicate cultures. D, shown is an immunoblot to determine expression of MarP constructs. Mtb lysates were fractionated into soluble (s) and insoluble (i) fractions by centrifugation, separated by SDS-PAGE, and transferred to a membrane. FLAG-tagged proteins were detected by immunoblot with anti-FLAG antibody. An immunoblot for dihydrolipoamide S-acyltransferase (α-DlaT) serves as a loading control.
Determination of Substrate Specificity
We previously purified two recombinant MarP proteins without their transmembrane domains: MarP_142–397 and MarP_179–397 (20). The longer construct, MarP_142–397, underwent autocleavage at its N terminus, in the linker region. Liquid chromatography-mass spectrometry (LC/MS/MS) revealed that autocleavage occurred preferentially between Leu-160 and Ser-161 in the sequence PKRLSALL; however, additional cleavage positions were also observed in this region (20). We generated two internally quenched fluorescent peptides based on the autocleavage site sequences of MarP, namely PKRLSALL and SALLNTSG. Each peptide contained an MCA group on the N terminus and a 2,4-dinitrophenol group coupled to a C-terminal lysine. In a kinetic assay, no cleavage of these peptides was detected. However, MALDI-TOF MS analysis revealed that PKRLSALL was partially hydrolyzed between Leu and Ser and between Ser and Ala after 48 h of incubation with 2 μm MarP. SALLNTSG was not cleaved under these conditions (supplemental Fig. 1, A–D).
To uncover the substrate specificity of MarP, the enzyme was assayed with two distinct substrate-profiling methods. The complete diverse PS-SCL has been previously used to profile the P1 to P4 substrate specificity of more than 85 proteases, most of which are serine and cysteine proteases (32, 41). The PS-SCL consists of tetrapeptide substrates containing a fixed amino acid at a specific position linked to a fluorogenic 7-amino-4-carbamoylmethylcoumarin group on the C terminus. The unfixed positions contain equimolar mixtures of all natural amino acids, except cysteine, and norleucine is included to increase the information content (20, 32). In the PS-SCL assay, MarP specificity was dominated by leucine in the P2 position, and it displayed a 12.5-fold or higher preference over tryptophan, norleucine (n), and all other amino acids (Fig. 3). Cleavage also occurred when leucine and, to a lesser extent, methionine, proline, and arginine were present in the P1 position. Little preference was evident in the P3 position, whereas the P4 position was selective for bulky hydrophobic residues such as tryptophan, tyrosine, and phenylalanine as well as histidine.
FIGURE 3.
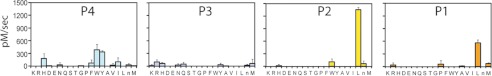
Substrate specificity of MarP determined using the complete diverse positional scanning synthetic combinatorial library. Results from P1, P2, P3, and P4 diverse positional scanning libraries are shown. The y axis represents the substrate cleavage rates in picomolar concentrations of fluorophore released per second. The x axis indicates the amino acid held constant at each position, designated by the one-letter code (with n representing norleucine). The amino acids are grouped based upon the chemical characteristics of the side chain residues (acid, basic, polar, aromatic, and aliphatic amino acids). Data are representative of three independent experiments with error bars representing ± S.D.
In addition, the extended substrate specificity of MarP was determined by incubating the enzyme with a mixture of 124 tetradecapeptides. These substrates were designed to contain a unique dipeptide at each terminus (positions 1–2 and 13–14) to profile exo-acting proteases and a central decapeptide sequence (positions 3–12) consisting of all combinations of neighbor and near-neighbor amino acid pairs that is used to profile endoproteases. This assay is termed Multiplex Substrate Profiling by Mass Spectrometry and has been used to profile numerous endo- and exo-acting proteases (33, 42). After the addition of a protease to mixtures of these peptides, the proteolytic reaction is quenched at various time intervals, and cleaved bonds are identified by peptide sequencing using mass spectrometry.
MarP was assayed with the peptide library, and the activity was quenched using concentrated acid to pH < 3 after incubation for 60, 240, and 1200 min. Cleavage (↓) of four peptides was evident after 60 min, namely: APSLI↓AKWVGFEPH, GnYYKRFn↓AHWVGI, AnTDR↓GWYLAIQAV, and AYNnWSLYRnI↓RQE, where n corresponds to norleucine. By 4 h, 8 additional cleavage sites were observed, and by 20 h a total of 25 sites were cleaved (Fig. 4A). An iceLogo plot was generated to illustrate the frequency of amino acids found at the P4 to P4′ positions of all cleaved sequences (n = 25). The P1 position was enriched with norleucine, isoleucine, and arginine, whereas small polar and negatively charged residues were not tolerated at this position (Fig. 4B). The P2 position was enriched with tryptophan and, to a lesser extent leucine and norleucine, whereas bulky hydrophobic residues such as tyrosine, phenylalanine, norleucine, and isoleucine were most often found in the P4 position. The MSP-MS assay also yielded information on the prime-side substrate specificity. The P1′ position was frequently occupied by alanine and asparagine, whereas P4′ was enriched with leucine and histidine. In general, MarP cleaved at sites that were distal from the termini of the peptides with 84% of substrates having amino acids present in the P4 to P3′ positions; however, there was no statistically significant enrichment (p > 0.05) of amino acids in the P3, P2′, or P3′ subsites.
FIGURE 4.

Substrate specificity of MarP determined using multiplex substrate profiling by mass spectrometry. A, a list of all cleavage sites observed over the course of the assay is shown. Only P4 to P4′ residues are shown, and the time at which the cleavage was first observed is indicated at the top of each list. When cleavage occurs near the N or C termini, then some subsites are not occupied by an amino acid and are represented as a blank space. B, an iceLogo represents amino acids that are most frequently (above the axis) and least frequently (below the axis) observed in each of the 25 cleavage sites. Residues that are highlighted in black text are significantly (p = 0.05) enriched in the MarP subsites relative to the frequency that these same amino acids are found in the peptide library (5.2 ± 0.5%).
Design and Synthesis of Fluorescent MarP Substrates
Two fluorescent substrates were generated to enable enzymatic characterization of MarP. First, a tetrapeptide substrate containing a C-terminal 7-amino-4-methylcoumarin (AMC) group was synthesized. The tetrapeptide sequence (WKLL-AMC) corresponded to the preferred amino acids at the P4, P2, and P1 positions as established by the PS-SCL profiling assay. No obvious preference was evident at the P3 position, so Lys was chosen at P3 to add hydrophilicity to an otherwise hydrophobic peptide sequence. Second, an internally quenched octapeptide, MCA-PSLIAKWV-K(DNP), was synthesized that corresponded to the P4-P4′ amino acids of a substrate observed in the MSP-MS assay at the earliest time point.
Steady-state kinetic studies were performed with each substrate, and kcat/Km values of 14 and 62 m−1s−1 were obtained for WKLL-AMC and MCA-PSLIAKWV-K(DNP), respectively (Table 1; supplemental Fig. 2, A and B). Mass spectrometry analysis of MCA-PSLIAKWV-K(DNP) confirmed that cleavage occurred between Ile and Ala (supplemental Fig. 2, C and D) as was observed for the 14-mer substrate in the MSP-MS assay. To identify key interactions of these synthetic peptides with the protease substrate binding pocket, we performed structural modeling of these substrates bound in the active site of MarP (Fig. 5). The peptides were modeled based on the experimentally observed peptide AVLEPFSR bound in the active site pocket of MarP in the crystal structure of this protease in the active state (20) (PDB ID 3K6Y). In the model of MarP-WKLL-AMC, the P1 leucine is stabilized by hydrophobic interactions with the side chains of Ala-361 and Val-338, which line the S1 pocket. Leucine at P2 interacts with Ile-320 and the catalytic His-235. At P3, which had no preferred amino acid, lysine points into the solvent. P4 tryptophan makes hydrophobic interactions with Leu-315, Phe-359, and Phe-370. For MCA-PSLIAKWV-K(DNP), interactions at P1 (isoleucine), P2 (leucine), and P3(serine) with the protease are similar to those described above. Proline at P4 makes nonpolar contacts with Phe-370, Leu-315, and Phe-359. On the other side of the scissile bond, alanine at P1′ contacts Leu-218, lysine at P2′ likely forms a salt bridge with Glu-219, and tryptophan at P3′ is sandwiched by stacking between Lys-216 and Gln-340. The rest of the peptide chains on this side of the scissile bond as well as the fluorophores are highly solvent-exposed, and therefore, their specific interactions with the protease are difficult to predict. In summary, substrate profiling using PS-SCL and MSP-MS identified two peptide substrates that were cleaved by MarP and bound in the active site consistent with our previous structural analysis.
TABLE 1.
Kinetic parameters of MarP_179–397
Protease activity assays were performed as described under “Experimental Procedures.”
| Substrate | Kma | Vmaxa | kcat | kcat/Km |
|---|---|---|---|---|
| μm | pm s−1 | s−1 | m−1 s−1 | |
| WKLL-AMC | 42.62 ± 4.4 | 145.6 ± 3.5 | 5.8 × 10−4 ± 1.4 × 10−5 | 13.7 ± 1.1 |
| AMC-PSLIAKWV-K(DNP) | 6.5 ± 0.6 | 806.5 ± 18.3 | 4.0 × 10−4 ± 7.3 × 10−5 | 62.0 ± 12.4 |
a Parameters were determined by non-linear curve fitting of the data to the Michaelis-Menten equation.
FIGURE 5.

Models of the fluorogenic peptides bound in the active site of MarP. The fluorescent substrates WKLL-AMC (A) and MCA-PSLIAKWV-K(DNP) (B) (shown as sticks) are modeled in the MarP active site according to their observed cleavage positions. The fluorescent MCA is highly solvent-exposed and could not be modeled accurately. Positively charged, negatively charged, and neutral regions of the protein surface are shown in blue, red, and white, respectively. Side chains and the AMC group are labeled.
Enzymatic Characterization of MarP
Because MarP-mediated proteolysis was important for resistance to acid stress, we were interested in the pH optimum of the protease. Fluorescence of free AMC was unaffected by pH (supplemental Fig. 3); therefore, a pH titration was performed using WKLL-AMC. MarP displayed optimal activity at pH 9.0 but retained >95% activity between pH 7.4 and pH 10 (supplemental Fig. 3B).
We next determined the activity of protease inhibitors against MarP using the WKLL-AMC substrate (Table 2). The serine protease inhibitors PMSF (1 mm) and FP-Rho (20 μm) inhibited catalysis, resulting in 100 and 94.8% inhibition, respectively. The calculated IC50 values for PMSF and FP-Rho were 284 and 4.5 μm, respectively (Fig. 6, A and B). 20 μm pepstatin A, which targets aspartic acid proteases, partially inhibited MarP activity. We have previously shown that MarP autocleavage activity is significantly reduced as a result of disruption of the intramolecular disulfide bond between Cys-214 and Cys-395 by reducing agents and mutagenesis of the two Cys residues (20). To examine if this disulfide is also important for cleavage of a peptide substrate, we tested the effect of 2 mm DTT on catalysis and observed 97.5% inhibition of protease activity. Finally, we tested the effect of complete disruption of the disulfide bond by mutagenesis on the peptide cleavage activity by MarP. We generated recombinant MarP with a single C214A mutation and double C214A/C395A mutations. Neither mutant protease cleaved WKLL-AMC (supplemental Fig. 4), which confirmed that the disulfide bond is an important structural element required for MarP activity in vitro.
TABLE 2.
Effect of protease inhibitors on MarP_179–397 mediated hydrolysis of WKLL-AMC
Inhibition of protease activity was determined as described under “Experimental Procedures.” Samples were run in triplicate. nd, not determined.
| Drug | Concentration | Target | % Inhibition | IC50 |
|---|---|---|---|---|
| μm | ||||
| Pepstatin A | 20 μm | Aspartic acid proteases | 26.4 | nd |
| PMSF | 1 mm | Serine proteases | 100 | 284 ± 14 |
| E-64 | 2 μm | Cysteine proteases | 23.8 | nd |
| EDTA | 10 mm | Metalloproteases | 8.0 | nd |
| Sodium fluoride | 20 mm | Serine/threonine and acidic phosphatases | 12.2 | nd |
| Fluorophosphonate-rhodamine | 20 μm | Serine proteases | 94.8 | 4.48 ± 0.23 |
| DTT | 2 mm | Reduces disulfides | 97.5 | nd |
FIGURE 6.
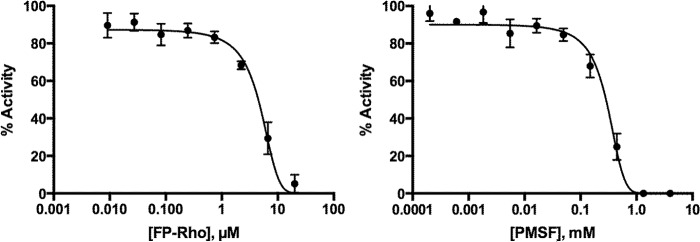
IC50 curves of FP-Rho and PMSF. The IC50 curves were run in triplicate with 250 nm MarP_179–397 and 50 μm WKLL-AMC for 36 min at room temperature. Inhibitor concentrations were varied from 0 to 20 μm FP-Rho and 0–1200 μm PMSF in 3-fold dilutions. Activities were normalized to uninhibited. The calculated IC50 for FP-Rho and PMSF were 4.5 and 284 μm, respectively.
Role of the MarP Disulfide Bond in Vivo
The role of the disulfide bond in regulating MarP protease in vitro demonstrated here and previous work (20, 29) suggested that the disulfide bond (Fig. 7A) may have an important function in vivo. To test this hypothesis, we transformed Mtb ΔMarP with MarP-C214A and MarP-C214A/C395A. Expression of the mutant proteases was confirmed by immunoblot with anti-MarP antiserum (data not shown). Surprisingly, both mutant proteases complemented the ΔMarP mutant and phenocopied WT Mtb with respect to survival after exposure to acid (Fig. 7B) and H2O2 (Fig. 7C) stresses, whereas cfu of ΔMarP were significantly reduced. Thus, the disulfide bond, although important for in vitro catalysis, appears to be dispensable for MarP-mediated resistance to low pH or H2O2.
FIGURE 7.

Importance of the MarP disulfide bond for survival of Mtb exposed to acid and oxidative stress. A, a schematic representation of the MarP protease domain (PDB ID 3K6Y) shows the location of the disulfide bond between Cys-214 and Cys-395 (purple). The active site residues are shown in pink. The disulfide bonds links the N-terminal domain (green) with the C-terminal domain (yellow) of the protease. B, shown is survival of Mtb in low pH. Mtb strains were plated after 3 (gray bars) and 6 (black bars) days of exposure to phosphate citrate buffer, pH 4.5, and cfu were quantified. Data are the means ± S.D. of triplicate cultures. C, survival of Mtb after exposure to hydrogen peroxide is shown. Mtb strains were plated after exposure to 8 mm H2O2 for 4 h (black bars). Data are means ± S.D. of triplicate cultures.
DISCUSSION
Proteolysis is an important aspect of bacterial pathogenesis, with proteases contributing to quality control processes and regulatory pathways. Several proteases have been shown to play an important role in the pathogenesis of tuberculosis including Rip1, a protease important for regulation of cell envelope composition and virulence (43, 44), the mycobacterial proteasome (45), HtrA2 (29), the ClpP1P2 complex (46), the serine protease MycP1 (47), and MarP (3). The periplasmic location of MarP is consistent with its role in resistance against extracellular stresses; its substrates must be present in the extracytoplasmic space and may include periplasmic proteins or proteins that are part of the cell envelope. In this study we determined that MarP was optimally active at pH 9, consistent with our previous observation that autoproteolysis increased at alkaline pH (20). This pH profile suggests that MarP can cleave its substrates independently of acid stress. The pH of Mtb periplasm is unknown, and whether it is actively controlled when the bacteria are exposed to acid has not been investigated. E. coli is unable to control the pH of its periplasm (48), and recent work demonstrated that the pH optimum of the E. coli HtrA protease, DegQ, is pH 5.5, consistent with its role in degrading misfolded proteins during acid stress (49). In contrast, MarP may play a different function in the periplasmic space and is perhaps involved in cell envelope maintenance, which when disrupted results in hypersusceptibility to acid and oxidative stress. This hypothesis is further supported by the increased susceptibility of Mtb lacking MarP to lipophilic antibiotics and detergent (3).
Previously, we reported that recombinant MarP underwent autoproteolysis at its N-terminal linker region. However, MarP did not efficiently cleave two fluorescent peptides containing the autocleavage sequence (PKRLSALL and SALLNTSG). In addition, we were unable to detect truncated MarP in Mtb lysates, suggesting that autocleavage may be observed only at high concentrations of the recombinant enzyme. Alternatively the absence of the transmembrane domain results in an exposed linker region that is prone to proteolysis. Using two independent substrate-profiling methods, we uncovered the specificity of MarP. Both methods determined that the enzyme preferred bulky residues at P4 and hydrophobic residues at P2 and P1; however, the exact residues often differed between assays, and the dominant preference for leucine at P2 in the PS-SCL was not as evident in the MSP-MS assay. The differences observed between methods may be due to the size of the peptide substrates (14-mer versus 4-mer) or contribution of the prime side residues to non-prime side specificity. In particular, the MSP-MS assay revealed that MarP prefers alanine and asparagine in the S1′ pocket, both of which are dissimilar to the bulky fluorescent acetyl-P4-P3-P2-P1-amino-4-carbamoyl-methylcoumarin moiety present in the PS-SCL library.
To make substrates for enzymatic characterization of MarP, we generated a fluorescent tetrapeptide (WKLL-AMC) based on the preferred residues identified from the PS-SCL assay. In addition we synthesized an internally quenched octapeptide substrate based on the P4 to P4′ residues of a robustly cleaved 14-mer peptide, namely APSLI↓AKWVGFEPH. This octapeptide sequence was chosen over others because Leu was present in the P2 position and isoleucine and alanine were frequently found in the P1 and P1′ sites of the other substrates in the MSP-MS assay.
Using these substrates, we noted that the steady-state kinetics of MarP was significantly lower than those of other well characterized serine proteases, which generally cleave their substrates with kcat/Km values of >10,000 m−1s−1 (50). MarP lacks a PDZ domain that regulates protease activity upon binding to substrates in HtrA family proteases. However, the C-terminal region of MarP may also be involved in protein-protein interactions or form an exosite (20) regulating protease activity. Exosites that enhance enzymatic activity upon binding to a preferred ligand have been found in other proteases (51). In addition, it is possible that the transmembrane domain or full-length linker region, which is absent from the recombinant enzyme, are important for substrate recognition. Finally, we cannot rule out that the optimal peptide substrate for MarP has not been identified, because structural features of protein substrates that are not close to the cleavage site may also dictate the specificity of the protease. Notwithstanding, the substrate specificity profiles obtained in this study will help predict extracytoplasmic proteins as potential native MarP substrates. Additional rigorous studies will need to be performed to determine which of these proteins are true substrates of the membrane-anchored protease.
Proteins in the periplasmic space can encounter harsh conditions due to their proximity to the extracellular environment. To tolerate these conditions, many secreted proteins contain disulfide bonds that provide extra stability to their tertiary structures (52, 53). Disulfide bonds are introduced into periplasmic proteins by a dedicated disulfide bond-forming system, the DSB system (54). Mtb encodes a DSB system with vitamin K epoxide reductase (VKOR) instead of DsbB promoting disulfide bond formation (55, 56). In E. coli, the HtrA protease is stabilized by a disulfide bond in the N-terminal region whose formation depends of the DsbA oxidase (57, 58). In contrast to the above systems, the disulfide bond of MarP is not required per se for structural stability of the protease-fold as a whole; instead, the disulfide constrains the conformation of the protease in the active state (20). We tested the importance of the MarP disulfide bond for proteolysis in vitro and for complementation of the MarP mutant phenotypes. Surprisingly, the cysteine mutant proteins fully complemented acid and H2O2 hypersusceptibility of ΔMarP. In contrast, the in vitro experiments determined that the proteases lacking the disulfide were inactive. However, autocleavage of these mutant proteases was not completely abolished (20) indicating that, although important, the disulfide bond is not essential for MarP-mediated proteolysis. Together these data suggest that the structural support provided by the disulfide bond can be compensated for when the protease is expressed in the Mtb periplasm at conditions tested here. This could be due to its attachment to the transmembrane domain, which is missing in the recombinant protease, or afforded by interacting proteins including protease substrate(s). Finally, we may not have identified the condition in which the disulfide becomes essential when MarP is expressed in Mtb.
In summary, we confirmed that MarP is a periplasmic protease, determined the substrate specificity profile of recombinant MarP, and characterized its enzymatic properties. Catalytic inactivation of MarP by mutagenesis yielded an acid and peroxide sensitive Mtb strain, which may be mimicked chemically by inhibitors directed against the active site of the protease. To facilitate the screening of MarP inhibitors we have used the substrate profiling data to develop sensitive fluorogenic peptide substrates. Further understanding of the mechanism by which MarP protects Mtb from acid and oxidative stress and the development of MarP selective inhibitors may lead to the discovery of novel anti-TB chemotherapeutics.
Acknowledgments
We thank Dr. B. Cravatt (Scripps Research Institute, La Jolla, CA) for fluorophosphonate-rhodamine. Mass spectrometry analysis was performed in the Bio-Organic Biomedical Mass Spectrometry Resource at the University of California, San Francisco (A. L. Burlingame, Director), which is supported by the Biomedical Technology Research Centers program of the NIGMS, National Institutes of Health, Grant 8P41GM103481.
This work was supported, in whole or in part, by National Institutes of Health Grants AI08172 (to S. E.) and P50 GM082250 (to C. S. C.). This work was also supported by the Sandler Center (to C. S. C.).

This article contains supplemental Figs. 1–4.
- Mtb
- M. tuberculosis
- AMC
- 7-amino-4-methylcoumarin
- DNP
- 2,4-dinitrophenol
- FP-Rho
- fluorophosphonate-rhodamine
- MCA
- 7-methoxycoumarin-4-acetic acid
- MSP-MS
- multiplex substrate profiling by mass spectrometry
- PS-SCL
- positional scanning synthetic combinatorial library
- PhoA
- alkaline phosphatase
- TM
- transmembrane.
REFERENCES
- 1. WHO (2011) Global Tuberculosis Control, http://www.who.int/tb/publications/global_report/en/index.html
- 2. Sherman D. R., Sabo P. J., Hickey M. J., Arain T. M., Mahairas G. G., Yuan Y., Barry C. E., 3rd, Stover C. K. (1995) Disparate responses to oxidative stress in saprophytic and pathogenic mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 92, 6625–6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vandal O. H., Pierini L. M., Schnappinger D., Nathan C. F., Ehrt S. (2008) A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 14, 849–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vandal O. H., Roberts J. A., Odaira T., Schnappinger D., Nathan C. F., Ehrt S. (2009) Acid-susceptible mutants of Mycobacterium tuberculosis share hypersusceptibility to cell wall and oxidative stress and to the host environment. J. Bacteriol. 191, 625–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ehrt S., Schnappinger D. (2009) Mycobacterial survival strategies in the phagosome. Defence against host stresses. Cell. Microbiol. 11, 1170–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Armstrong J. A., Hart P. D. (1971) Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 134, 713–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clemens D. L., Horwitz M. A. (1995) Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J. Exp. Med. 181, 257–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sturgill-Koszycki S., Schlesinger P. H., Chakraborty P., Haddix P. L., Collins H. L., Fok A. K., Allen R. D., Gluck S. L., Heuser J., Russell D. G. (1994) Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263, 678–681 [DOI] [PubMed] [Google Scholar]
- 9. Ohkuma S., Poole B. (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. U.S.A. 75, 3327–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaible U. E., Sturgill-Koszycki S., Schlesinger P. H., Russell D. G. (1998) Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 160, 1290–1296 [PubMed] [Google Scholar]
- 11. Via L. E., Fratti R. A., McFalone M., Pagan-Ramos E., Deretic D., Deretic V. (1998) Effects of cytokines on mycobacterial phagosome maturation. J. Cell Sci. 111, 897–905 [DOI] [PubMed] [Google Scholar]
- 12. MacMicking J. D. (2003) Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science 302, 654–659 [DOI] [PubMed] [Google Scholar]
- 13. Huynh K. K., Grinstein S. (2007) Regulation of vacuolar pH and its modulation by some microbial species. Microbiol. Mol. Biol. Rev. 71, 452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foster J. W. (1999) When protons attack. Microbial strategies of acid adaptation. Curr. Opin. Microbiol. 2, 170–174 [DOI] [PubMed] [Google Scholar]
- 15. Cotter P. D., Hill C. (2003) Surviving the acid test. Responses of gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 67, 429–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buchmeier N., Blanc-Potard A., Ehrt S., Piddington D., Riley L., Groisman E. A. (2000) A parallel intraphagosomal survival strategy shared by Mycobacterium tuberculosis and Salmonella enterica. Mol. Microbiol. 35, 1375–1382 [DOI] [PubMed] [Google Scholar]
- 17. Song H., Huff J., Janik K., Walter K., Keller C., Ehlers S., Bossmann S. H., Niederweis M. (2011) Expression of the ompATb operon accelerates ammonia secretion and adaptation of Mycobacterium tuberculosis to acidic environments. Mol. Microbiol. 80, 900–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rohde K. H., Abramovitch R. B., Russell D. G. (2007) Mycobacterium tuberculosis invasion of macrophages. Linking bacterial gene expression to environmental cues. Cell Host Microbe 2, 352–364 [DOI] [PubMed] [Google Scholar]
- 19. Abramovitch R. B., Rohde K. H., Hsu F.-F., Russell D. G. (2011) aprABC. A Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol. Microbiol. 80, 678–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biswas T., Small J., Vandal O., Odaira T., Deng H., Ehrt S., Tsodikov O. V. (2010) Structural insight into serine protease Rv3671c that protects M. tuberculosis from oxidative and acidic stress. Structure 18, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ribeiro-Guimarães M. L., Pessolani M. C. (2007) Comparative genomics of mycobacterial proteases. Microb. Pathog. 43, 173–178 [DOI] [PubMed] [Google Scholar]
- 22. Sonnhammer E. L., von Heijne G., Krogh A. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182 [PubMed] [Google Scholar]
- 23. Hoffmann C., Leis A., Niederweis M., Plitzko J. M., Engelhardt H. (2008) Disclosure of the mycobacterial outer membrane. Cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U.S.A. 105, 3963–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zuber B., Chami M., Houssin C., Dubochet J., Griffiths G., Daffé M. (2008) Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 190, 5672–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clausen T., Southan C., Ehrmann M. (2002) The HtrA family of proteases. Implications for protein composition and cell fate. Mol. Cell 10, 443–455 [DOI] [PubMed] [Google Scholar]
- 26. Skorko-Glonek J., Zurawa-Janicka D., Koper T., Jarzab M., Figaj D., Glaza P., Lipinska B. (2013) HtrA protease family as therapeutic targets. Curr. Pharm. Des. 19, 977–1009 [DOI] [PubMed] [Google Scholar]
- 27. Krojer T., Garrido-Franco M., Huber R., Ehrmann M., Clausen T. (2002) Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature 416, 455–459 [DOI] [PubMed] [Google Scholar]
- 28. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 29. Mohamedmohaideen N. N., Palaninathan S. K., Morin P. M., Williams B. J., Braunstein M., Tichy S. E., Locker J., Russell D. H., Jacobs W. R., Jr., Sacchettini J. C. (2008) Structure and function of the virulence-associated High Temperature Requirement A of Mycobacterium tuberculosis. Biochemistry 47, 6092–6102 [DOI] [PubMed] [Google Scholar]
- 30. White M. J., He H., Penoske R. M., Twining S. S., Zahrt T. C. (2010) PepD participates in the mycobacterial stress response mediated through MprAB and SigE. J. Bacteriol. 192, 1498–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim D. Y., Kim K. K. (2005) Structure and function of HtrA family proteins, the key players in protein quality control. J. Biochem. Mol. Biol. 38, 266–274 [DOI] [PubMed] [Google Scholar]
- 32. Choe Y., Leonetti F., Greenbaum D. C., Lecaille F., Bogyo M., Brömme D., Ellman J. A., Craik C. S. (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 281, 12824–12832 [DOI] [PubMed] [Google Scholar]
- 33. O'Donoghue A. J., Eroy-Reveles A. A., Knudsen G. M., Ingram J., Zhou M., Statnekov J. B., Greninger A. L., Hostetter D. R., Qu G., Maltby D. A., Anderson M. O., Derisi J. L., McKerrow J. H., Burlingame A. L., Craik C. S. (2012) Global identification of peptidase specificity by multiplex substrate profiling. Nat. Methods 9, 1095–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braunstein M., Griffin T. J., IV, Kriakov J. I., Friedman S. T., Grindley N. D., Jacobs W. R., Jr. (2000) Identification of genes encoding exported Mycobacterium tuberculosis proteins using a Tn552′phoA in vitro transposition system. J. Bacteriol. 182, 2732–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chalkley R. J., Baker P. R., Medzihradszky K. F., Lynn A. J., Burlingame A. L. (2008) In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell. Proteomics 7, 2386–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Unni S., Huang Y., Hanson R. M., Tobias M., Krishnan S., Li W. W., Nielsen J. E., Baker N. A. (2011) Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. Comput. Chem. 32, 1488–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manoil C., Beckwith J. (1986) A genetic approach to analyzing membrane protein topology. Science 233, 1403–1408 [DOI] [PubMed] [Google Scholar]
- 40. Goldman B. S., Beck D. L., Monika E. M., Kranz R. G. (1998) Transmembrane heme delivery systems. Proc. Natl. Acad. Sci. U.S.A. 95, 5003–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harris J. L., Backes B. J., Leonetti F., Mahrus S., Ellman J. A., Craik C. S. (2000) Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. U.S.A. 97, 7754–7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sojka D., Franta Z., Frantová H., Bartosová P., Horn M., Váchová J., O'Donoghue A. J., Eroy-Reveles A. A., Craik C. S., Knudsen G. M., Caffrey C. R., McKerrow J. H., Mares M., Kopácek P. (2012) Characterization of gut-associated cathepsin D hemoglobinase from tick Ixodes ricinus (IrCD1). J. Biol. Chem. 287, 21152–21163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Makinoshima H., Glickman M. S. (2005) Regulation of Mycobacterium tuberculosis cell envelope composition and virulence by intramembrane proteolysis. Nature 436, 406–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sklar J. G., Makinoshima H., Schneider J. S., Glickman M. S. (2010) M. tuberculosis intramembrane protease Rip1 controls transcription through three anti-sigma factor substrates. Mol. Microbiol. 77, 605–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gandotra S., Schnappinger D., Monteleone M., Hillen W., Ehrt S. (2007) In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat. Med. 13, 1515–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raju R. M., Unnikrishnan M., Rubin D. H., Krishnamoorthy V., Kandror O., Akopian T. N., Goldberg A. L., Rubin E. J. (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 8, e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ohol Y. M., Goetz D. H., Chan K., Shiloh M. U., Craik C. S., Cox J. S. (2010) Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe 7, 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilks J. C., Slonczewski J. L. (2007) pH of the cytoplasm and periplasm of Escherichia coli. Rapid measurement by green fluorescent protein fluorimetry. J. Bacteriol. 189, 5601–5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sawa J., Malet H., Krojer T., Canellas F., Ehrmann M., Clausen T. (2011) Molecular adaptation of the DegQ protease to exert protein quality control in the bacterial cell envelope. J. Biol. Chem. 286, 30680–30690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hedstrom L. (2002) Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4524 [DOI] [PubMed] [Google Scholar]
- 51. Jabaiah A. M., Getz J. A., Witkowski W. A., Hardy J. A., Daugherty P. S. (2012) Identification of protease exosite-interacting peptides that enhance substrate cleavage kinetics. Biol. Chem. 393, 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Darby N., Creighton T. E. (1995) Disulfide Bonds in Protein Folding and Stability. pp. 219–252, Humana Press Inc., Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 53. Wedemeyer W. J., Welker E., Narayan M., Scheraga H. A. (2000) Disulfide bonds and protein folding. Biochemistry 39, 7032. [DOI] [PubMed] [Google Scholar]
- 54. Ito K., Inaba K. (2008) The disulfide bond formation (Dsb) system. Curr. Opin. Struct. Biol. 18, 450–458 [DOI] [PubMed] [Google Scholar]
- 55. Dutton R. J., Boyd D., Berkmen M., Beckwith J. (2008) Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dutton R. J., Wayman A., Wei J. R., Rubin E. J., Beckwith J., Boyd D. (2010) Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc. Natl. Acad. Sci. U.S.A. 107, 297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Skórko-Glonek J., Sobiecka-Szkatuła A., Lipińska B. (2006) Characterization of disulfide exchange between DsbA and HtrA proteins from Escherichia coli. Acta Biochim. Pol. 53, 585–589 [PubMed] [Google Scholar]
- 58. Skórko-Glonek J., Zurawa D., Tanfani F., Scirè A., Wawrzynów A., Narkiewicz J., Bertoli E., Lipińska B. (2003) The N-terminal region of HtrA heat shock protease from Escherichia coli is essential for stabilization of HtrA primary structure and maintaining of its oligomeric structure. Biochim. Biophys. Acta 1649, 171–182 [DOI] [PubMed] [Google Scholar]