

Background: Endocytosis can involve dimerization and membrane association of the protein endophilin.
Results: We found subnanomolar affinity for endophilin N-BAR dimerization. Membrane dissociation is substantially slower than association, and membrane-bound protein density-dependent.
Conclusion: Endophilin binds membranes as dimers that subsequently oligomerize.
Significance: Our findings illuminate the membrane binding mechanism of endophilin, which is important in understanding the regulation of membrane trafficking events.
Keywords: Endocytosis, Kinetics, Membrane Biophysics, Membrane Trafficking, Protein Self-assembly, Membrane Curvature, Protein Dimerization, Protein-Membrane Interactions
Abstract
The recruitment to plasma membrane invaginations of the protein endophilin is a temporally regulated step in clathrin-mediated endocytosis. Endophilin is believed to sense or stabilize membrane curvature, which in turn likely depends on the dimeric structure of the protein. The dynamic nature of the membrane association and dimerization of endophilin is thus functionally important and is illuminated herein. Using subunit exchange Förster resonance energy transfer (FRET), we determine dimer dissociation kinetics and find a dimerization equilibrium constant orders of magnitude lower than previously published values. We characterize N-BAR domain membrane association kinetics under conditions where the dimeric species predominates, by stopped flow, observing prominent electrostatic sensitivity of membrane interaction kinetics. Relative to membrane binding, we find that protein monomer/dimer species equilibrate with far slower kinetics. Complementary optical microscopy studies reveal strikingly slow membrane dissociation and an increase of dissociation rate constant for a construct lacking the amphipathic segment helix 0 (H0). We attribute the slow dissociation kinetics to higher-order protein oligomerization on the membrane. We incorporate our findings into a kinetic scheme for endophilin N-BAR membrane binding and find a significant separation of time scales for endophilin membrane binding and subsequent oligomerization. This separation may facilitate the regulation of membrane trafficking phenomena.
Introduction
Clathrin-mediated endocytosis represents a prominent mode of cellular regulation and uptake of external material (1). In this dynamic process, assembly of clathrin at the plasma membrane packages nascent vesicles for intracellular trafficking of receptors and cargo. Endophilin functions as an accessory protein in clathrin-mediated endocytosis and additional membrane-trafficking processes (2–6). The localization of endophilin to endocytic sites at the plasma membrane has been shown by live-cell fluorescence imaging to mark pivotal points in the endocytic process (7–9). Membrane association of endophilin (10) is achieved by means of its N-BAR4 domain (11), facilitating membrane recruitment of additional proteins through its Src homology 3 (SH3) domain (4, 6, 12–15).
Endophilin-membrane complexes exhibit membrane insertion of hydrophobic segments (16, 17), electrostatic interactions (10, 16, 18), and intricate surface ordering of protein subunits (10, 19–23). In particular, two amphipathic segments, designated as helix 0 (H0) (residues 1–22) and the helix 1 insert (H1I) (residues 59–87), form helices upon membrane association that insert into the bilayer (16, 17). This membrane insertion is important for functional membrane remodeling by endophilin (6, 11, 18). An additional aspect of the membrane-remodeling mechanism arises from the shape that endophilin presents to the membrane (11, 24, 25). Importantly, the crescent shape of endophilin depends on its dimeric structure (26, 27).
These aspects of endophilin function are well appreciated structurally because of both experiment (10, 16–18, 20, 21, 23, 28) and computation (19, 29, 30). However, beyond qualitative kinetic characterization by surface plasmon resonance (SPR) (28), little is known about the timescales and mechanisms determining the kinetics of endophilin-membrane interactions.
To fully elucidate the mechanisms of endophilin function, kinetic characterization of its membrane association is essential. Toward this end, we hypothesized that endophilin associates with membranes as a dimer in an effective two-step process leading to oligomerization even in the absence of pronounced membrane deformation.
We assess this mechanistic hypothesis as follows. The evaluation of N-BAR dimerization in solution serves as a prerequisite of our mechanistic analysis of membrane binding. We employ stopped-flow and microscopy-based techniques for kinetic studies. We find evidence for an important contribution of protein oligomerization on the membrane in the mechanism. We arrive at a kinetic model that is fitted to the experimental data. Finally, we discuss the physiological relevance of the separation of time scales for N-BAR membrane interactions observed here.
EXPERIMENTAL PROCEDURES
Materials
Lipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG), l-α-phosphatidylinositol (4)- and (4,5)-bis-phosphate (PI(4)P and PI(4,5)P2; brain, ammonium salts) were obtained from Avanti Polar Lipids (Alabaster, AL). Folch Fraction I was from Sigma. 2-(3-(Diphenylhexatrienyl)propanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine (DPH-PC), Pacific BlueTM (PB) C5-maleimide, Alexa Fluor® 488 (AF-488) C5-maleimide, and Texas Red-1,2-dihexadecanoyl-sn-glycero-3 phosphoethanolamine (triethylammonium salt) were from Invitrogen/Life Technologies. Casein was from Fisher Scientific. Tris(2-carboxyethyl)phosphine was from Pierce/Thermo Fisher Scientific. All commercial reagents were used without further purification.
Membrane Preparation
Extruded vesicles were prepared by standard techniques (31). Chloroform solutions of lipid stock were gently evaporated, by using compressed air, from a round-bottom flask, which was subsequently evacuated. Vesicle compositions are represented as the mole fraction DOPG, with DOPC comprising the remaining portion. DPH-PC was included at 0.7 mol % for stopped-flow Förster resonance energy transfer (FRET) studies. Lipids were hydrated at 2 mg/ml with 20 mm HEPES buffer with indicated NaCl concentrations, pH 7.4, under bath sonication at 40 °C for 10–15 min. Lipid dispersions were extruded 13 times through single polycarbonate membranes (Whatman/GE Healthcare). Dynamic light scattering consistently revealed an average hydrodynamic radius of 75 ± 15–30 nm for all lipid compositions utilized in stopped-flow studies. For tubulation assays, 400-nm pore sizes were used. Phosphate concentrations of extruded preparations were determined by standard analysis (32). Giant unilamellar vesicles (GUVs) were prepared by electroformation (33) as described (34).
Plasmids and Protein Preparation
A plasmid encoding rat endophilin A1, kindly provided by P. De Camilli, served as the template for generation of a single Cys construct comprising residues 1–247 with C108A/E241C substitutions (N-BAR_C241), ΔNH (BAR) (residues 33–247 with and without the A66W substitution), and N-BAR F10W. We employed sequential QuikChange (Stratagene) reactions for mutagenesis. A plasmid encoding residues 1–247 of human endophilin A1 with the A66W mutation in the pGEX6p vector (GE Healthcare) was kindly provided by N. Mochizuki (24) and served as the template for N-BAR_C234 (with A66W/C108S/Q234C substitutions). Relative to rat, human N-BAR differs by an M133I substitution. A plasmid encoding residues 1–247 of rat endophilin A1 with the C108S/A247C substitutions (3) (N-BAR_C247) was kindly provided by R. Langen. All sequences were verified by DNA sequencing.
GST fusion proteins were purified from the soluble portion of bacterial lysates (BL21(DE3) RIL CodonPlus, Stratagene) using glutathione affinity. Fusion proteins were cleaved at the GST moiety using PreScission protease or thrombin. BAR domains were purified further by ion exchange with a linear NaCl gradient and size exclusion chromatography (SEC) (Superdex200 (GE Healthcare)), concentrated, aliquoted, and flash-frozen in liquid nitrogen. Purity was assessed by SDS-PAGE and analytical SEC. For measurements with thawed samples, we used ultracentrifugation supernatant portions. Concentrations were determined by Bradford analysis using bovine serum albumin (Thermo Fisher Scientific) as a standard (35) and absorption at 280 nm. Concentrations indicated refer to total N-BAR in terms of monomeric units. Labeling was conducted at introduced Cys residues for N-BAR_C241 and N-BAR_C247 or endogenous Cys108 for ΔNH (BAR) (both with and without the A66W substitution). 4-fold excess of maleimide dye reagent was used via a DMSO solution for reaction at 4 °C, in 150 mm NaCl, 20 mm HEPES, 1 mm tris(2-carboxyethyl)phosphine, pH 7.4 solution (HNTcep buffer). Commonly, glutamate/arginine/HEPES, pH 7.4, was added to promote protein solubility during labeling (36). Reactions were quenched with excess dithiothreitol (DTT), and excess dye reagent was removed via three 5-ml HiTrap desalting columns (GE Healthcare) connected in series.
Liposome Tubulation Assay
Endophilin N-BAR variants were incubated with 400-nm membrane-extruded vesicles at room temperature for 30 min. Samples were applied to carbon/Formvar-supported copper grids (Electron Microscopy Sciences, Hatfield, PA). After a 5-min incubation, excess material was removed by blotting on filter paper. Grids were then stained with 2% (w/v) uranyl acetate for 2 min, washed in buffer, and air-dried at room temperature. Grids were observed with a JEM 1011 transmission electron microscope (JEOL) with the accelerating voltage set to 100 kV.
Protein Cross-linking
Site-specific irreversible cross-linking by reaction of Cys at residue 241 (Cα-Cα distance, 38 Å) was conducted with Bis-MAL-dPEG®11 (Quanta BioDesign, Powell, OH; linker length, 30 Å). Reactions proceeded for 4 h at 4 °C with 20 μm protein in HNTcep buffer with equimolar cross-linker added via a DMSO solution. Reactions were quenched using 2-fold excess DTT, and unreacted reagent was removed using a HiTrap desalting (GE Healthcare) column achieving buffer exchange for stopped flow and SEC.
Size Exclusion Chromatography
N-BAR samples in HNTcep buffer were run at 4 °C over a Superdex 200 (GE Healthcare) column pre-equilibrated with the same buffer. The retention volume at peak absorption value (V) was used to calculate the distribution coefficient according to
 |
where the column void V0 and total volume Vt were determined using blue dextran (GE Healthcare) and acetone, respectively.
Fluorescence Correlation Spectroscopy (FCS)
Measurements were carried out at 20 °C in glass-bottomed chambers (Nunc, Rochester, NY) passivated with a polylysine-conjugated polyethylene glycol solution. The concentration of labeled protein (N-BAR_C247-AF-488) was held constant (between 7 and 17 nm) for each titration. The total protein concentration was varied from 40 nm to 40 μm via the addition of unlabeled protein. The concentration of unlabeled protein was determined by absorption, and the concentration of labeled protein was determined by FCS. For measurements following a 24-h incubation, DTT was refreshed.
Details of the instrument used for FCS measurements have been described (37). Laser power was adjusted to 5.3–5.6 microwatts (prior to entering the microscope).
For each concentration of protein, 30 traces of 10-s duration were measured. The curves were averaged and analyzed as representing a single diffusing species in three dimensions. The ratio of radial to axial dimensions of the observation volume was determined from measurements of a solution of free AF-488 and was fixed for the protein measurements.
Fluorescence Anisotropy Decay
Experiments were carried out at the University of Pennsylvania Ultrafast Optical Processes Laboratory using time-correlated single photon counting instrumentation described in Ref. 38, at ambient temperature with N-BAR_C241-PB in HNTcep buffer with 0.05 mm phenylmethylsulfonyl fluoride, using 405 nm excitation. Samples were prepared to desired concentrations at 4 °C 18 h before measurements. For 1 μm, a sample prepared 60 h prior to measurements yielded results indistinguishable from the 18-h sample (data not shown).
Fluorescence Spectroscopy
Measurements employed a Cary Eclipse fluorometer with a Peltier-controlled temperature block. Donor (PB) and acceptor (AF-488) N-BAR_C241 conjugates (D and A, respectively)5 were examined with a fluorophore ratio of 1:2, chosen to optimize FRET. For kinetic studies, a temperature of 27 °C was selected to accelerate subunit exchange relative to 22 °C. D and A samples in HNTcep buffer were adjusted to identical concentrations and separately preincubated at the experimental temperature for a duration equal to that of the measurement. Protein adsorption to cuvette walls was <5%, as confirmed by excitation spectra (not shown) in the acceptor emission peak region (519 nm), at the start and end of kinetic monitoring. To achieve this, quartz cuvettes were treated with casein solution for 1 h and washed once with sample buffer. The casein solution comprised the soluble portion of a 2.5 mg/ml suspension in sample buffer following sedimentation.
The dimer dissociation rate kdissn6 was determined from fluorescence emission time courses monitoring donor quenching relative to the initial time point (right after mixing D and A solutions) by single-exponential fitting (39). The reaction scheme for subunit exchange yields the rate of heterodimer (DA) formation
 |
where kassn is the dimerization rate constant for association of monomers. In this mixing experiment, the concentration of monomers is unchanged. Hence, the second term in Equation 2 is constant, and a single exponential with time constant kdissn results as the solution to Equation 2 (40).
For equilibrium measurements, the pre-equilibration period for acceptor-donor mixtures was selected as 12 h at 27 °C, as guided by the kinetics results. Emission measurements at the acceptor wavelength upon excitation of the donor (400 nm) were processed by the following previously reported routine (41, 42). Briefly, acceptor-only and donor-only protein conjugate reference standards enabled accounting for emission arising from donor bleedthrough and acceptor cross-excitation. All spectra were buffer-subtracted. Emission spectra were first corrected for acceptor cross-excitation contribution, as determined by comparing excitation spectra at the acceptor emission wavelength of the standard and sample. Subsequently, the contribution of the donor to measured emission in the acceptor peak region was subtracted, as determined from the donor-only standard equalized to the donor-acceptor mixture sample spectrum at the donor emission wavelength (455 nm). Finally, the values were referenced to that observed at the highest protein concentration used in the titration, yielding relative FRET, F, proportional to the dimer fraction of the sample, and fitting was conducted of
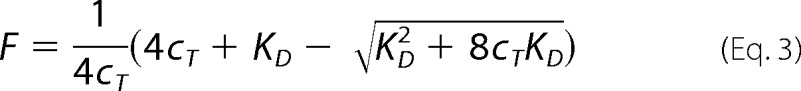 |
where cT is the total protein concentration, and KD is the equilibrium dimerization dissociation constant (41).
Stopped Flow
Measurements were carried out with an Applied Photophysics (Surrey, UK) SX.18MV stopped-flow spectrometer (43) using excitation at 280 nm and collecting emission after passage through a 400-nm long-pass filter. Between two and six traces were averaged for each condition, and 50 μl each of protein and lipid solutions were mixed. For light scattering measurements, we used an incident wavelength of 430 nm, and lipid-only traces were subtracted from protein-lipid mixing traces prior to fitting. Data before the dead time of 2 ms were excluded from fitting. All measurements were conducted at 22.0 ± 0.05 °C, in HEPES, pH 7.4, with varying NaCl concentrations as indicated for particular experiments. For concentration variations analyzed in conjunction with microscopy data (see below), 33 mm NaCl was selected as opposed to physiological levels to improve signal-to-noise ratios.
For global analysis of progress curves incorporating off-rates from microscopy experiments (see “Results”), the reaction scheme (44) detailed below yielded differential equations (Equations 4–7) characterizing chemical species as dimeric N-BAR D, the membrane as L, the intermediate as D*L (where the asterisk is meant to represent an inserted species), and oligomerized species as Dn*L, treated as a dimer of N-BAR dimers represented as D2*L.
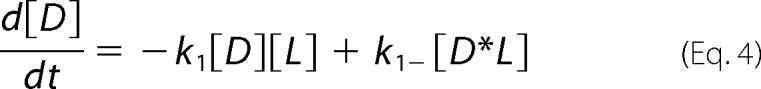 |
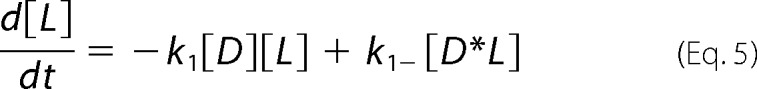 |
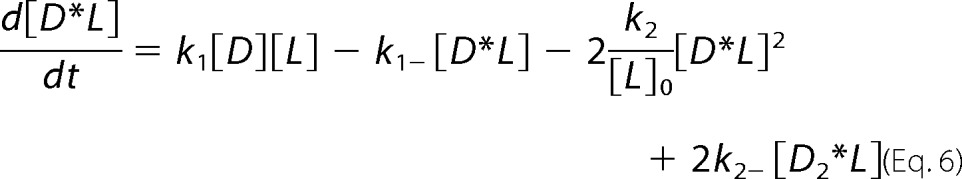 |
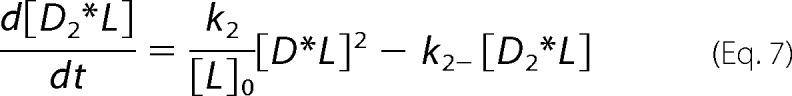 |
Terms for which k2 appears as a coefficient (related to the membrane-bound bimolecular reaction) are divided by [L]0, the initial vesicle concentration in terms of total lipid used in the mixing experiment. This approach considers the concentration of membrane-bound species in terms of a dimensionless density. Equations 4–7 were numerically integrated and fitted to experimental data employing MATLAB (The MathWorks, Natick, MA).
Microscopy-monitored Protein Dissociation
Protein and GUVs containing 25% DOPG were incubated in ∼180 μl of an equimolar solution of sucrose and glucose, pH 7.4, containing HEPES and varying NaCl concentration, with the same osmolarity as the prepared GUV dispersion (measured with an Advanced Instruments Inc. (Norwood, MA) micro-osmometer) in a polypropylene tube for 1 h.
Imaging chambers (2 mm thick) were formed from two coverslips (24 × 40 mm) overhanging a glass microscope slide. The protein-GUV mixture was added to the chamber on a surface pretreated with 2 μl of 2.5 mg/ml casein, 20 mm Tris, and 2 mm EDTA.
Micropipettes were prepared as described (34) and treated with the casein solution applied to the chamber. Transfer capillaries (outer/inner diameters: 1.5 mm/1.12 mm, World Precision Instruments Inc., Sarasota, FL) were cut to desired lengths with a ceramic glass cutter and equipped with a rubber O-ring as a handle.
GUVs were aspirated in micropipettes at constant aspiration pressure (less than 60 pascals). The transfer capillary was manually positioned as to enclose the aspirated GUV. Protein dilution was achieved by transfer of the GUV using a motor-controlled micromanipulator from the protein chamber into an adjacent chamber, composed typically of ∼400 μl of buffer solution, under the protection of the transfer capillary (see Fig. 4A, below). Finally, the transfer capillary was removed to expose the protein-covered GUV.
FIGURE 4.

Dissociation kinetics under nontubulating conditions. A, diagram of dissociation kinetics experiments employing rapid dilution via micropipette-mediated GUV transfer. B and C, electron micrographs of liposomes (composed of 25% DOPG) in the absence of endophilin N-BAR (B) and in the presence of 300 nm N-BAR_C241-AF488 (4 μm lipids, 33 mm NaCl) (C). D, fluorescence micrograph of N-BAR_241-AF-488 bound to a GUV (25% DOPG) aspirated in a micropipette, in 33 mm NaCl, 1 mm DTT following preincubation at 300 nm protein, 4 μm lipids, and identical ionic strength. Scale bar, 5 μm. E, fluorescence (Fl.) intensity record and single-exponential fit (solid line) charting dissociation of N-BAR, as diagrammed in A, with conditions of D. norm., normalized.
The protein dissociation process was monitored by using a confocal fluorescence microscope (34). The protein fluorescence intensity was determined by fitting a Gaussian ring to the GUV contour (excluding the aspirated region) using MATLAB.
RESULTS
Subnanomolar Dimerization Affinity
We first asked the question whether the membrane binding mechanism of endophilin involves a dimerization step. This hypothesis is supported by previous reports of low dimerization affinities for this protein (16, 35, 45, 46).
Because the measurement of affinities requires establishment of monomer/dimer equilibria, we first designed an experiment to accurately assess N-BAR dimerization kinetics using FRET (Fig. 1A). A single Cys N-BAR construct, N-BAR_C241, was separately labeled with donor (PB) and acceptor (AF-488) fluorophores. We showed that this mutant exhibits functionality in liposome tubulation in labeled and unlabeled form (Fig. 1, B–E).
FIGURE 1.

Subunit exchange FRET experimental design. A, schematic in which dimeric N-BAR variants separately labeled with donor (D) and acceptor (A) fluorophores undergo subunit exchange, generating fluorescence changes. B–E, electron micrographs of liposome vesicles (Folch Fraction I) in the absence of endophilin N-BAR (B) and in the presence of N-BAR_C247 (C), N-BAR_C241 (D), or N-BAR_C241-AF-488 (E), revealing N-BAR-induced tubule formation. Protein concentrations were 5–7 μm, and lipid concentration was 0.1 mg/ml, in 20 mm Tris-HCl, 150 mm NaCl, 1 mm DTT, pH 7.4 buffer. Scale bars, 200 nm.
To uncover the equilibration time for dimer dissociation at 27 °C, we monitored FRET (Fig. 2A), characterizing dimer dissociation kinetics for N-BAR_C241. We selected this optimized variant as N-BAR_C247 (see “Experimental Procedures”) exhibited low (although detectable) FRET, hindering quantitative analysis, whereas N-BAR_C234 showed appreciable FRET but poor tubulation functionality (data not shown). We determined a value for kdissn of 1.4 ± 0.3 × 10−4 s−1 at 27 °C (means ± S.E. of five measurements) from fitting FRET time traces such as those shown in Fig. 2B with a model resulting from Equation 2.
FIGURE 2.

Subnanomolar dimerization affinity revealed for N-BAR by FRET. A, emission spectra of a mixture of endophilin N-BAR_C241-PB and N-BAR_C241-AF-488, 2 μm total protein, 27 °C. A representative time series is plotted from ∼30 s after mixing (small dashed line) to 500 min (long dashed line), revealing donor quenching and sensitized acceptor emission (emphasized by arrows). Fl., fluorescence; AU, arbitrary units. B, FRET efficiency EFRET calculated from the example in A as donor quenching relative to the initial spectrum, shown with single-exponential fit, yielding infinite time EFRET = 30% and kdissn = 1.3 × 10−4 s−1. C, fluorescence spectra at 22 °C, donor:acceptor 1:1 of an endophilin N-BAR sample that was simultaneously labeled with PB and AF-488 (black solid line) and a mixture of twice the concentration of free dye compounds (gray dashed line), individually normalized (norm.) to emission maxima. D, fluorescence spectra of a mixture of N-BAR_241-PB and N-BAR_241-AF-488, 0.13 μm total protein, equilibrated for 40 h at 22 °C (black). SDS was added to 1%, and the emission was collected immediately, corrected for dilution, and plotted (gray) relative (rel.) to the maximum of the initial spectrum. PAGE (not shown) revealed that SDS suffices to dissociate N-BAR dimers at 22 °C. E, relative FRET F calculated as described under “Experimental Procedures” (41) for a representative concentration series following a 12-h incubation, with fit of Equation 3 yielding KD of 0.57 nm. See “Results” for average values. The gray lines indicate predictions using a KD of one-half (dotted) and two times (dashed) the best-fit value. F, F as in E extended beyond 10 μm total protein and corrected for the inner filter effect.
We excluded putative “proximity” FRET due to random localization of fluorophores, based on measurements with free dyes in solution (Fig. 2C). Our interpretation of the FRET signal as reporting on protein dimerization (Fig. 1A) is further substantiated by the elimination of FRET upon SDS-induced dimer dissociation (Fig. 2D).
Knowledge of the kinetics determined above allowed us to equilibrate endophilin samples to study dimerization equilibria at 27 °C (Fig. 2, E and F). We determined a KD at 27 °C of 0.41 ± 0.15 nm (mean ± S.E. of three measurements exemplified in Fig. 2E) by fitting FRET data with Equation 3. The determined KD value corresponds to a substantially higher affinity than a value resulting from published measurements at reduced temperature (10 μm) (16). Additional transitions at protein concentrations much higher than KD were not revealed by FRET measurements (Fig. 2F).
Protein cross-linking (Fig. 3, A and B), SEC (Fig. 3, B and C), time-resolved fluorescence anisotropy (Fig. 3D), and FCS measurements (Fig. 3E) are also inconsistent with 10 μm dimerization affinity. SEC of 2 μm N-BAR under conditions of the subunit exchange FRET studies (Fig. 3, B and C) reported a Stokes radius in agreement with that calculated for dimeric N-BAR, using HYDROPRO (47), as opposed to monomeric N-BAR. In support of this reasoning, we note that HYDROPRO analysis outputs a radius of gyration of 35 Å for dimeric N-BAR in agreement with the experimental value of 35.8 Å (48). Although a nearly 2-fold change in rotational correlation time is predicted comparing N-BAR monomer and dimer (using HYDROPRO (47)), indistinguishable values are observed for 1 and 16 μm samples (Fig. 3D). Similarly, the diffusion coefficients of monomeric and dimeric N-BAR are expected to differ, although indistinguishable diffusion times are reported by FCS over a range of concentrations (Fig. 3E). Hence, these data do not report a monomer-dimer transition occurring in the concentration regime investigated. We next aimed to elucidate the N-BAR membrane binding mechanism through measurements of endophilin N-BAR membrane binding and unbinding kinetics.
FIGURE 3.

Demonstration that dimeric N-BAR predominates at low micromolar and submicromolar concentrations. A, denaturing 10% polyacrylamide gel with Coomassie Brilliant Blue staining demonstrating inter-protomer cross-linking of N-BAR_C241 with Bis-MAL-dPEG®11. Molecular mass standard positions are indicated in kDa on the left, along with predicted molecular mass of N-BAR dimer and monomer. B, SEC showing unchanged retention peak after cross-linking (red) relative to N-BAR_C241 at 16 μm (black) and 2 μm N-BAR_C241 (gray). Absorption (Abs.) at 280 nm was plotted individually normalized (norm.) to trace maxima. C, SEC calibration (49) with endophilin N-BAR result (red dashed line) from B when compared with Rs of 28.7 and 34.5 Å for monomer and dimer, respectively (gray and black) determined theoretically via the software HYDROPRO (47). Sqrt, square root. D, representative fluorescence (Fl.) anisotropy decay of N-BAR_241-PB at 1 and 16 μm. Double-exponential fitting (lines) yielded rotational correlation times (± fit error) 0.32 ± 0.05 and 3.04 ± 0.27 ns (1 μm), and 0.36 ± 0.05 and 3.32 ± 0.29 ns (16 μm), whereas a single exponential with time constant 0.22 ns describes free dye data. Similar results were obtained from three consecutive acquisitions using the same protein samples, analyzed with deconvolution of the instrument response function, by FluoFit software (PicoQuant GmbH) (not shown). AU, arbitrary units. E, measured diffusion times by FCS of N-BAR_247-AF-488 as a function of total protein concentration plotted on a log scale, at 20 °C after 5 min of mixing (circles) and then again after 24 h of incubation at 4 °C, for selected concentrations (squares).
Dissociation from Single GUVs Reports Long Membrane Residence Time
We studied dissociation kinetics via a rapid dilution strategy through the transfer of single protein-bound GUVs to a solution devoid of protein (Fig. 4A). To reduce the complexity of kinetic processes, we limited our experiments to conditions where membrane morphology transitions, such as tubulation, are not observed (Fig. 4, B and C).
Considering the electrostatic component of N-BAR/membrane interactions (10, 16, 18), we selected a membrane composition containing 25 mol % of negatively charged lipids that was recently employed in N-BAR studies (50). A fluorescence decay time of 1010 ± 245 s (mean ± S.E. for nine GUVs) was determined (Fig. 4, D and E).
Early Membrane Association Assessed by Stopped Flow
We next extended the membrane binding investigation to shorter timescales by using stopped-flow rapid mixing and FRET between endophilin N-BAR A66W and DPH-PC embedded in vesicle membranes, or alternatively, by monitoring light scattering changes induced by protein binding (Fig. 5A). The kinetic traces are well fitted by a single exponential (Fig. 5B) for a wide range of conditions investigated. We used this approach to compare membrane binding kinetics of cross-linked (irreversibly dimerized) and non-cross-linked endophilin N-BAR domains. Consistent with our finding of subnanomolar dimerization affinity, we determined indistinguishable membrane binding kinetics for cross-linked and non-cross-linked samples under the same experimental conditions (Fig. 5C).
FIGURE 5.

Stopped-flow assessment of early timescale of membrane association. A, FRET between endophilin N-BAR A66W and DPH-PC (alternatively, excess light scattering using unlabeled proteins and membranes) used to follow binding upon rapid mixing of protein with vesicles. B, representative stopped-flow record of light scattering monitoring the process diagrammed in A, using 100% DOPG vesicles, 72 μm total lipid, and 0.4 μm N-BAR_C241 in 100 mm NaCl with 2 mm DTT, and single-exponential fit (gray). AU, arbitrary units. C, single-exponential decay constants from stopped-flow progress curves as in B, with and without cross-linking (Fig. 3, A and B).
Taken on its own, the observation via stopped flow of single-exponential membrane binding kinetics could be consistent with a one-step membrane binding process (51). However, the slow dissociation kinetics observed by microscopy (Fig. 4E) suggest (as further discussed below) the presence of a second step in the membrane binding mechanism.
Kinetic Model
We formulated a kinetic two-step model (see Equations 4–7) as a minimal model consistent with our observations for the endophilin-membrane interaction. This model bridges the time scales characterized by stopped flow and microscopy (Fig. 6A). In this model, dimeric protein components in solution (D in Fig. 6A) represent the membrane-binding species, achieving both association with the lipid membrane (L) and insertion of amphipathic helices effectively in the same step. Our model thus does not include separate steps for association of the protein with the membrane and the insertion of amphipathic segments. This is due to the fact that the stopped-flow data are well described by a single exponential (see e.g. Ref. 52) and that the folding transition associated with membrane insertion is likely not rate-limiting (53). We designate the resultant N-BAR-membrane intermediate species D*L, where the asterisk is meant to represent an inserted species, and −L indicates membrane-bound species. Intermediate D*L subsequently oligomerizes on the membrane, generating the species Dn*L, which is proposed to govern membrane residence time.
FIGURE 6.

Global experimental data analysis to rationalize two prominent timescales of endophilin-membrane interactions under nontubulating conditions. A, scheme as described under “Results.” Amphipathic helices are indicated by outlined circles. Rate constants represent global fitting results from panel D. B and C, electron micrographs of liposomes (55 μm total lipid) in presence of 0.65 μm (B) and 1.3 μm (C) N-BAR A66W. Liposomes in the absence of protein are imaged in Fig. 4B. Scale bars, 200 nm. D, stopped-flow DPH-PC FRET data (black) used for quantitating the proposed model below. Gray lines display global fitting to predictions of the model, as described under “Experimental Procedures,” yielding parameters in A. Each graph shows post-mixing concentrations of 25% DOPG vesicles of 55, 80, 115, and 160 μm total lipid (with increasing signal magnitude) for fixed N-BAR A66W concentrations of 0.65, 0.81, 1.0, and 1.3 μm as indicated in 33 mm NaCl, 1 mm DTT. Protein solutions were prepared to premixing concentrations 24 h prior to measurements. AU, arbitrary units.
To test this kinetic model, we identified a range of protein and vesicle concentrations in which N-BAR is dimeric but does not alter membrane morphology (Fig. 6, B and C) for collection of stopped-flow data (Fig. 6D). We note that this model corresponds to that recently proposed in analysis of the binding mechanism of an amphipathic membrane-binding peptide (44).
To determine the rate constants specified in the model, we employed global fitting of numerically integrated rate equations to the entire data set of Fig. 6D (results in Fig. 6A), treating membrane-bound species (D*L) within a surface density perspective (see “Experimental Procedures”). The oligomeric species was approximated as a dimer of N-BAR dimers (i.e. n = 2 in Fig. 6A). We used the single-exponential decay constant observed in GUV dissociation experiments (see example trace in Fig. 4E; using the acidic lipid content and ionic strength of the stopped-flow series of Fig. 6D) as an estimate of k2− (Fig. 6A). Global fitting with this two-step model yields rate constants for the first step of k1 = 0.11 ± 0.01 μm−1 s−1 and k1− = 36.4 ± 2.4 s−1. These values can be compared with those resulting from fitting a single-step model to the data shown in Fig. 6D; we find k1 = 0.122 ± 0.001 μm−1 s−1 and k1− = 35.1 ± 2.4 s−1, i.e. identical values within fit errors when compared with the two-step model. Consistent with this finding is a large uncertainty of the fit value for k2 of the two-step model; our stopped-flow measurements report on the first membrane binding step (Fig. 6A, k2 not reported) only. These findings are consistent with a large separation in time scales for membrane binding/unbinding of dimeric BAR domain proteins on the one hand and oligomerization/deoligomerization of membrane-bound BAR domain protein on the other hand.
Substantiating the Role of Oligomerization in the Mechanism of N-BAR Membrane Binding
Our hypothesis of oligomerization following membrane binding is supported by the direct observation (via cryo-electron microscopy imaging) of oligomerization of membrane-bound endophilin N-BAR (23). Importantly, endophilin N-BAR domain lattice formation has been observed even in the absence of morphological changes relative to the bare membrane, as is true of our conditions (Figs. 4, B and C, and 6, B and C).
Comparative studies with an endophilin variant lacking the H0 sequence (ΔNH (BAR) (residues 33–247 (16))) further support our incorporation of membrane-mediated oligomerization in the model and exclusion of an explicit step for membrane insertion. Recently published cryo-electron microscopy reconstructions showing intermolecular interactions between helix 0 components (21, 23), site-specific cross-linking studies (21), and recent optical imaging studies (28) document the role of H0 in mediating oligomerization of membrane-bound endophilin N-BAR. Considering these studies, our model predicts that ΔNH (BAR) would exhibit accelerated dissociation relative to N-BAR. Indeed, remarkably accelerated dissociation was observed for the helix deletion construct ΔNH (BAR) relative to N-BAR under the same conditions (Fig. 7, A and B).
FIGURE 7.

N-BAR exhibits density-dependent membrane dissociation and ΔNH (BAR) exhibits accelerated membrane dissociation. A, protein dissociation records, as in Fig. 4, D and E, within a solution without added salt (to increase membrane binding) following preincubation at ∼10 mm NaCl, pH 7.4, and 400 nm protein. GUVs contained 25 mol % DOPG. Lines represent single-exponential fits. For both constructs, the decay constant is orders of magnitude different from the timescale of the unbinding rate constant yielded by analysis of stopped-flow data assuming a single-step binding/association mechanism (data not shown). Fl., fluorescence; norm., normalized. B, dependence of membrane dissociation time of N-BAR and ΔNH (BAR) determined as in A on the initial protein fluorescence (corrected for differing labeling efficiencies of the two proteins) measured on the GUV, proportional to membrane-bound protein density. Protein concentration used for preincubation was varied from 100 to 400 nm for N-BAR and from 100 to 800 nm for ΔNH (BAR), to yield a range of initial membrane-bound protein densities. Other conditions are the same as in A. Lines represent linear fits. Error bars are the errors of single exponential fits to data shown in A. AU, arbitrary units.
In further support of our view on oligomerization being a second step in the membrane binding mechanism, we found a dependence of the dissociation time on the protein density on the membrane (Fig. 7B). Conversely, no density dependence of membrane dissociation rates is expected for a protein that does not oligomerize on the membrane because in that case, protein unbinding will follow a kinetic first-order process. The density dependence is significantly more prominent for N-BAR relative to ΔNH (BAR), consistent with changed N-BAR protein organization at elevated membrane-bound density, leading to slower dissociation.
In sharp contrast to the slow timescale for membrane dissociation of endophilin N-BAR (Figs. 4E and 6A), direct/competitive binding stopped-flow measurements of peripheral proteins not known to oligomerize on the membrane have revealed membrane dissociation rate constants typically between 0.1 and 100 s−1 (see e.g. Refs. 54–57). We note that the single dimer unbinding rate constant (k1− = 36 s−1; Fig. 6A) determined for endophilin does indeed lie within this range. For HIV1-Nef and a myristoylated alanine-rich C kinase substrate (MARCKS) peptide, both known to insert into the bilayer, respective dissociation rate constants of 0.5 s−1 (58) and 1.5 s−1 (59) have been measured. Thus, the alternative hypothesis that a deinsertion process underlies the slow dissociation of N-BAR and the influence of the H0 segment on dissociation kinetics (Fig. 7A) is not consistent with previously measured rate constants. We note in passing that the on-rate for endophilin membrane binding (k1 = 0.1 μm−1 s−1) lies within the range of values typically determined for peripheral membrane proteins (43, 54, 57, 58, 60–63).
Role of Electrostatics in the Association Phase of the Mechanism of N-BAR Membrane Binding
We applied stopped-flow analysis to describe the determinants of the first step in the binding mechanism (Fig. 6A). Promotion of electrostatic interaction strength by varying vesicle acidic lipid (DOPG) content accelerated association and decelerated dissociation (Fig. 8A). This trend is inferred (via the relation kobs = k1− + [L]0k1) from increased slopes and decreased y-intercepts (Fig. 8A) extracted from plots exemplified in Fig. 5C. Increasing ionic strength from 33 to 750 mm NaCl significantly decelerated association, as indicated by similar analysis (Fig. 8B), albeit with a lower influence on dissociation (data not shown).
FIGURE 8.

Prominent role of electrostatics in the first step of the membrane binding mechanism. A, parameters extracted from analysis of titrations as shown in Fig. 5C, for indicated membrane compositions, averaged over 3–7 vesicle preparations (error bars indicate S.E.), with 33 mm NaCl and 0.3 μm N-BAR A66W. B, averaged slope as in A for experiments varying solution ionic strength via the NaCl concentration, with 72% DOPG and 0.3 μm N-BAR A66W (error bars indicate S.D. and S.E. over ≥3 vesicle preparations). C, DPH-PC stopped-flow association analysis in the pseudo-first-order regime, using 0.3 μm N-BAR A66W, 33 mm NaCl, with 25% DOPG and phosphoinositides (PI(4)P and PI(4,5)P2) incorporated to 3 mol %. D, pseudo-first-order parameters extracted from plots exemplified in C. Error bars represent S.D. and S.E. of datasets as in C from ≥3 vesicle preparations. A similar trend was observed using 150 mm NaCl and a background composition of 35% DOPG (data not shown).
The sensitivity of stopped-flow parameters to electrostatic interaction strength is consistent with the assumption that the association step (rather than helix insertion) governs kinetics of processes on this timescale. However, carefully delineating the kinetic contributions of membrane association and amphipathic membrane insertion remains an aim for future study.
Electrostatic influences on kinetics are likely to have implications in the context of a model (64) accounting for the role of endophilin in clathrin-coated vesicle formation (13, 65). In particular, this model attributes a role in vesicle fission to a preference of BAR domain proteins for PI(4,5)P2 over PI(4)P. This affinity difference has been supported by equilibrium lipid binding studies for endophilin (13, 28). We found a marked influence of phosphoinositides on kinetics, although revealing no significant difference comparing PI(4,5)P2 and PI(4)P in association or dissociation (Fig. 8, C and D). Thus, localization of N-BAR proteins to necks of budding vesicles may rely more heavily on membrane curvature than on membrane lipid composition.
DISCUSSION
Our in vitro results document the separation of time scales for the following three principal interaction features of endophilin N-BAR at 22–27 °C. Assembled dimers remain associated for hours to days, bind the membrane in tens of milliseconds, and following higher-order oligomerization, remain membrane-bound for tens of minutes.
Implications of Dimerization Affinity and Kinetics
An influence of monomeric species on the mechanism of endophilin membrane association has been suggested previously. Particularly, the dimerization dissociation constant for N-BAR was measured at 4 °C as 10 μm (16), a far lower affinity than that determined here at 27 °C.
The higher affinity for endophilin dimerization found here relative to previously reported results is, in fact, closer to a value that may be predicted by consideration of dimer-interface surface area. Notably, endophilin exhibits high dimer interface area, as do BAR domains in general (27, 66, 67), calculated using PISA (68) as 2743 Å2 for the human N-BAR structure (24). This value is over two times larger than the maximum of a recent tabulation of homodimers classified as weak affinity, which have measured KD values down to 12 nm (69). Using the affinity curation of Nooren and Thornton (70) this interface area predicts a subnanomolar KD, consistent with our experimentally determined value at 27 °C.
Our finding of subnanomolar affinity rationalizes previous studies that are consistent with the predominance of N-BAR dimers over monomers at sub/low micromolar concentrations: distance measurements via EPR methods in solution at low micromolar concentrations (17), biochemical analysis of extracts from rat brain (71) and from cultured cells exogenously expressing endophilin A2 (45), and fluorescence microscopy analysis of endophilin A2 and A1 fluorescent protein chimeras in cultured cells (35). An analogous notion has been advanced for the BAR protein sorting nexin 9 (72). We note in passing that kassn for endophilin dimerization may be calculated from our measured dissociation equilibrium and rate constants (Fig. 2), as kassn = kdissn/KD = (1.4 × 10−4 s−1)/(0.4 × 10−9 m) = 3.5 × 105 m−1 s−1 at 27 °C. We conclude that our results do not support a role for dimerization in the kinetic mechanism of membrane binding.
Deoligomerization Kinetics
Our value for k2− suggests far slower dissociation of membrane-bound oligomers for N-BAR relative to a peptide examined recently (44), likely reflecting intersubunit interactions of higher complexity for N-BAR. We note that disassembly of membrane lattice components in live cells is commonly not spontaneous, but depends on additional agents, as has been illuminated in recent in vitro studies (73–75).
Electrostatic Influence on Membrane Binding
Equilibria of electrostatic membrane binding are well predicted by Gouy-Chapman-Stern theory (76–78). However, predictions for electrostatic influences on kinetics (79–81) of membrane binding are less well developed. Trends in kinetics for varying vesicle acidic lipid content observed here resemble those previously reported for a C2 domain (54), a C1 domain (61), and the HIV-1 Nef protein (58). The increase in association rates with DOPG content for N-BAR (Fig. 8A) indicates that a stage in the N-BAR binding mechanism in which PG hydrogen-bonding networks are disassembled (82) is not rate-limiting.
Physiological Relevance of the Separation of Time Scales of Endophilin Interactions
Live-cell imaging reports slower apparent membrane recruitment times (5–15 s) (7, 9) relative to the present stopped-flow measurements (Figs. 5B and 6D). Numerous features not present in our in vitro experiments likely contribute to this difference, with dynamin GTPase activity (83) and actin scaffolding (5) representing factors with documented influences on endophilin membrane recruitment in vivo.
It has been suggested that BAR domains oligomerize at clathrin-coated pits (84). Based on the marked extension of membrane residence time observed here and attributed to oligomerization for endophilin N-BAR, this process may underlie the detection of endophilin in internalized vesicle preparations (85, 86). However, recycling of endophilin between membranes and the cytoplasm would likely require mechanisms for the dissociation of endophilin oligomers to thereby regulate the membrane residence time of the protein. Indeed, a regulatory role for membrane-mediated oligomerization of synaptotagmin has been proposed (87). Recent work has shown the effects of phosphorylation within endophilin H1I on membrane binding and tubulation, and in turn, functional recycling of endophilin between membrane and cytoplasm. A direction for future studies is thus to pursue whether phosphorylation within H0 (88) may provide alternative regulation by impacting endophilin oligomerization on the membrane (89).
Acknowledgments
We thank Dr. E. M. Ostap for generously providing access to the stopped-flow instrument. We thank Ken Chang for assisting with fluorescence measurements and for reading the manuscript. We thank Drs. J. Saven, P. Janmey, and T. Svitkina for additional access to instrumentation, Dr. T. Troxler for guidance with time-correlated single photon counting/lifetime measurements, and Drs. N. Mochizuki, P. De Camilli, and R. Langen for supplying plasmids. Time-correlated single photon counting measurements were collected at the Ultrafast Optical Processes Laboratory at the University of Pennsylvania supported through National Institutes of Health/NIGMS Grant 9P41GM104605.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM097552 (to T. B.).
A, protein conjugated to acceptor fluorophore (AF-488); D, protein conjugated to donor fluorophore (PB).
The parameter definitions used are: kassn and kdissn are protein dimer association and dissociation rates; KD is a protein dimerization equilibrium constant; k1 and k1− are net rate constants for membrane binding and unbinding of non-oligomerized N-BAR; and k2 and k2− are oligomerization and disassembly rates for protein subunits on the membrane surface.
- N-BAR
- Bin/amphiphysin/Rvs domain with N-terminal amphipathic helix
- AF-488
- Alexa Fluor® 488
- PB
- Pacific BlueTM
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPG
- 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- DPH-PC
- 2-(3-(diphenylhexatrienyl)propanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine
- FCS
- fluorescence correlation spectroscopy
- GUV
- giant unilamellar vesicle
- PI(4)P
- l-α-phosphatidylinositol (4)-phosphate
- PI(4,5)P2
- l-α-phosphatidylinositol (4,5)-bis-phosphate
- DMSO
- dimethyl sulfoxide
- SEC
- size exclusion chromatography.
REFERENCES
- 1. McMahon H. T., Boucrot E. (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12, 517–533 [DOI] [PubMed] [Google Scholar]
- 2. Ringstad N., Gad H., Löw P., Di Paolo G., Brodin L., Shupliakov O., De Camilli P. (1999) Endophilin/SH3p4 is required for the transition from early to late stages in clathrin-mediated synaptic vesicle endocytosis. Neuron 24, 143–154 [DOI] [PubMed] [Google Scholar]
- 3. Llobet A., Gallop J. L., Burden J. J. E., Camdere G., Chandra P., Vallis Y., Hopkins C. R., Lagnado L., McMahon H. T. (2011) Endophilin drives the fast mode of vesicle retrieval in a ribbon synapse. J. Neurosci. 31, 8512–8519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sundborger A., Soderblom C., Vorontsova O., Evergren E., Hinshaw J. E., Shupliakov O. (2011) An endophilin-dynamin complex promotes budding of clathrin-coated vesicles during synaptic vesicle recycling. J. Cell Sci. 124, 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferguson S. M., Raimondi A., Paradise S., Shen H., Mesaki K., Ferguson A., Destaing O., Ko G., Takasaki J., Cremona O., O' Toole E., De Camilli P. (2009) Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev. Cell 17, 811–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai J., Hu Z., Dittman J. S., Pym E. C., Kaplan J. M. (2010) Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell 143, 430–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perera R. M., Zoncu R., Lucast L., De Camilli P., Toomre D. (2006) Two synaptojanin 1 isoforms are recruited to clathrin-coated pits at different stages. Proc. Natl. Acad. Sci. U.S.A. 103, 19332–19337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mettlen M., Stoeber M., Loerke D., Antonescu C. N., Danuser G., Schmid S. L. (2009) Endocytic accessory proteins are functionally distinguished by their differential effects on the maturation of clathrin-coated pits. Mol. Biol. Cell 20, 3251–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taylor M. J., Perrais D., Merrifield C. J. (2011) A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 9, e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farsad K., Ringstad N., Takei K., Floyd S. R., Rose K., De Camilli P. (2001) Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J. Cell Biol. 155, 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frost A., Unger V. M., De Camilli P. (2009) The BAR domain superfamily: Membrane-molding macromolecules. Cell 137, 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ringstad N., Nemoto Y., De Camilli P. (1997) The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc. Natl. Acad. Sci. U.S.A. 94, 8569–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang-Ileto B., Frere S. G., Chan R. B., Voronov S. V., Roux A., Di Paolo G. (2011) Synaptojanin 1-mediated PI(4,5)P2 hydrolysis is modulated by membrane curvature and facilitates membrane fission. Dev. Cell 20, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simpson F., Hussain N. K., Qualmann B., Kelly R. B., Kay B. K., McPherson P. S., Schmid S. L. (1999) SH3-domain-containing proteins function at distinct steps in clathrin-coated vesicle formation. Nat. Cell Biol. 1, 119–124 [DOI] [PubMed] [Google Scholar]
- 15. de Heuvel E., Bell A. W., Ramjaun A. R., Wong K., Sossin W. S., McPherson P. S. (1997) Identification of the major synaptojanin-binding proteins in brain. J. Biol. Chem. 272, 8710–8716 [DOI] [PubMed] [Google Scholar]
- 16. Gallop J. L., Jao C. C., Kent H. M., Butler P. J., Evans P. R., Langen R., McMahon H. T. (2006) Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 25, 2898–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jao C. C., Hegde B. G., Gallop J. L., Hegde P. B., McMahon H. T., Haworth I. S., Langen R. (2010) Roles of amphipathic helices and the Bin/Amphiphysin/Rvs (BAR) domain of endophilin in membrane curvature generation. J. Biol. Chem. 285, 20164–20170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhatia V. K., Madsen K. L., Bolinger P. Y., Kunding A., Hedegård P., Gether U., Stamou D. (2009) Amphipathic motifs in BAR domains are essential for membrane curvature sensing. EMBO J. 28, 3303–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayton G. S., Lyman E., Krishna V., Swenson R. D., Mim C., Unger V. M., Voth G. A. (2009) New insights into BAR domain-induced membrane remodeling. Biophys. J. 97, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizuno N., Jao C. C., Langen R., Steven A. C. (2010) Multiple modes of endophilin-mediated conversion of lipid vesicles into coated tubes: implications for synaptic endocytosis. J. Biol. Chem. 285, 23351–23358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mim C., Cui H., Gawronski-Salerno J. A., Frost A., Lyman E., Voth G. A., Unger V. M. (2012) Structural basis of membrane bending by the N-BAR protein endophilin. Cell 149, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mim C., Unger V. M. (2012) Membrane curvature and its generation by BAR proteins. Trends Biochem. Sci. 37, 526–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cui H., Mim C., Vázquez F. X., Lyman E., Unger V. M., Voth G. A. (2013) Understanding the role of amphipathic helices in N-BAR domain driven membrane remodeling. Biophys. J. 104, 404–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Masuda M., Takeda S., Sone M., Ohki T., Mori H., Kamioka Y., Mochizuki N. (2006) Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 25, 2889–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baumgart T., Capraro B. R., Zhu C., Das S. L. (2011) Thermodynamics and mechanics of membrane curvature generation and sensing by proteins and lipids. Annu. Rev. Phys. Chem. 62, 483–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peter B. J., Kent H. M., Mills I. G., Vallis Y., Butler P. J. G., Evans P. R., McMahon H. T. (2004) BAR domains as sensors of membrane curvature: The amphiphysin BAR structure. Science 303, 495–499 [DOI] [PubMed] [Google Scholar]
- 27. Masuda M., Mochizuki N. (2010) Structural characteristics of BAR domain superfamily to sculpt the membrane. Semin. Cell Dev. Biol. 21, 391–398 [DOI] [PubMed] [Google Scholar]
- 28. Yoon Y., Zhang X., Cho W. (2012) PtdIns(4,5)P2 specifically induces membrane penetration and deformation by Bin/Amphiphysin/Rvs (BAR) domains. J. Biol. Chem. 287, 34078–34090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cui H. S., Lyman E., Voth G. A. (2011) Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophys. J. 100, 1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cui H. S., Ayton G. S., Voth G. A. (2009) Membrane binding by the endophilin N-BAR domain. Biophys. J. 97, 2746–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. MacDonald R. C., MacDonald R. I., Menco B. P., Takeshita K., Subbarao N. K., Hu L. (1991) Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta 1061, 297–303 [DOI] [PubMed] [Google Scholar]
- 32. Kingsley P. B., Feigenson G. W. (1979) The synthesis of a perdeuterated phospholipid: 1,2-dimyristoyl-sn-glycero-3-phosphocholine-d72. Chem. Phys. Lipids 24, 135–147 [Google Scholar]
- 33. Angelova M. I., Dimitrov D. S. (1986) Liposome electroformation. Faraday Discuss. Chem. Soc. 81, 303–311 [Google Scholar]
- 34. Tian A., Baumgart T. (2009) Sorting of lipids and proteins in membrane curvature gradients. Biophys. J. 96, 2676–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ross J. A., Chen Y., Müller J., Barylko B., Wang L., Banks H. B., Albanesi J. P., Jameson D. M. (2011) Dimeric endophilin A2 stimulates assembly and GTPase activity of dynamin 2. Biophys. J. 100, 729–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knight J. D., Falke J. J. (2009) Single-molecule fluorescence studies of a PH domain: New insights into the membrane docking reaction. Biophys. J. 96, 566–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sevcsik E., Trexler A. J., Dunn J. M., Rhoades E. (2011) Allostery in a disordered protein: oxidative modifications to α-synuclein act distally to regulate membrane binding. J. Am. Chem. Soc. 133, 7152–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goldberg J. M., Speight L. C., Fegley M. W., Petersson E. J. (2012) Minimalist probes for studying protein dynamics: thioamide quenching of selectively excitable fluorescent amino acids. J. Am. Chem. Soc. 134, 6088–6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wendt H., Berger C., Baici A., Thomas R. M., Bosshard H. R. (1995) Kinetics of folding of leucine zipper domains. Biochemistry 34, 4097–4107 [DOI] [PubMed] [Google Scholar]
- 40. Jonsson T., Waldburger C. D., Sauer R. T. (1996) Nonlinear free energy relationships in Arc repressor unfolding imply the existence of unstable, native-like folding intermediates. Biochemistry 35, 4795–4802 [DOI] [PubMed] [Google Scholar]
- 41. Jia H., Satumba W. J., Bidwell G. L., 3rd, Mossing M. C. (2005) Slow assembly and disassembly of λ-Cro repressor dimers. J. Mol. Biol. 350, 919–929 [DOI] [PubMed] [Google Scholar]
- 42. Merzlyakov M., Chen L., Hristova K. (2007) Studies of receptor tyrosine kinase transmembrane domain interactions: the EmEx-FRET method. J. Membr. Biol. 215, 93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang N., Lin T., Ostap E. M. (2002) Dynamics of myo1c (myosin-Iβ) lipid binding and dissociation. J. Biol. Chem. 277, 42763–42768 [DOI] [PubMed] [Google Scholar]
- 44. Kreutzberger A. J., Pokorny A. (2012) On the origin of multiphasic kinetics in peptide binding to phospholipid vesicles. J. Phys. Chem. B 116, 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gortat A., San-Roman M. J., Vannier C., Schmidt A. A. (2012) Single point mutation in the Bin/Amphiphysin/Rvs (BAR) sequence of endophilin impairs dimerization, membrane shaping, and Src homology 3 domain-mediated partnership. J. Biol. Chem. 287, 4232–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suresh S., Edwardson J. M. (2010) The endophilin N-BAR domain perturbs the structure of lipid bilayers. Biochemistry 49, 5766–5771 [DOI] [PubMed] [Google Scholar]
- 47. García De La Torre J., Huertas M. L., Carrasco B. (2000) Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Q., Kaan H. Y., Hooda R. N., Goh S. L., Sondermann H. (2008) Structure and plasticity of endophilin and sorting Nexin 9. Structure 16, 1574–1587 [DOI] [PubMed] [Google Scholar]
- 49. Laurent T. C., Killander J. (1964) Theory of gel filtration and its experimental verification. J. Chromatogr. 14, 317–321 [Google Scholar]
- 50. Zhu C., Das S. L., Baumgart T. (2012) Nonlinear sorting, curvature generation, and crowding of endophilin N-BAR on tubular membranes. Biophys. J. 102, 1837–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwarz G. (1987) Basic kinetics of binding and incorporation with supramolecular aggregates. Biophys. Chem. 26, 163–169 [DOI] [PubMed] [Google Scholar]
- 52. Tang J., Yin H., Qiu J., Tucker M. J., DeGrado W. F., Gai F. (2009) Using two fluorescent probes to dissect the binding, insertion, and dimerization kinetics of a model membrane peptide. J. Am. Chem. Soc. 131, 3816–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tang J., Signarvic R. S., DeGrado W. F., Gai F. (2007) Role of helix nucleation in the kinetics of binding of mastoparan X to phospholipid bilayers. Biochemistry 46, 13856–13863 [DOI] [PubMed] [Google Scholar]
- 54. Nalefski E. A., Newton A. C. (2001) Membrane binding kinetics of protein kinase C βII mediated by the C2 domain. Biochemistry 40, 13216–13229 [DOI] [PubMed] [Google Scholar]
- 55. McKenna J. M., Ostap E. M. (2009) Kinetics of the interaction of myo1c with phosphoinositides. J. Biol. Chem. 284, 28650–28659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Corbin J. A., Dirkx R. A., Falke J. J. (2004) GRP1 pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid. Biochemistry 43, 16161–16173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Krishnaswamy S., Jones K. C., Mann K. G. (1988) Prothrombinase complex assembly: kinetic mechanism of enzyme assembly on phospholipid vesicles. J. Biol. Chem. 263, 3823–3834 [PubMed] [Google Scholar]
- 58. Gerlach H., Laumann V., Martens S., Becker C. F., Goody R. S., Geyer M. (2010) HIV-1 Nef membrane association depends on charge, curvature, composition, and sequence. Nat. Chem. Biol. 6, 46–53 [DOI] [PubMed] [Google Scholar]
- 59. Arbuzova A., Wang J., Murray D., Jacob J., Cafiso D. S., McLaughlin S. (1997) Kinetics of interaction of the myristoylated alanine-rich C kinase substrate, membranes, and calmodulin. J. Biol. Chem. 272, 27167–27177 [DOI] [PubMed] [Google Scholar]
- 60. Hui E., Bai J., Wang P., Sugimori M., Llinas R. R., Chapman E. R. (2005) Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc. Natl. Acad. Sci. U.S.A. 102, 5210–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dries D. R., Newton A. C. (2008) Kinetic analysis of the interaction of the C1 domain of protein kinase C with lipid membranes by stopped-flow spectroscopy. J. Biol. Chem. 283, 7885–7893 [DOI] [PubMed] [Google Scholar]
- 62. Feeser E. A., Ignacio C. M., Krendel M., Ostap E. M. (2010) Myo1e binds anionic phospholipids with high affinity. Biochemistry. 49, 9353–9360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gregory S. M., Cavenaugh A., Journigan V., Pokorny A., Almeida P. F. (2008) A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys. J. 94, 1667–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu J., Sun Y., Drubin D. G., Oster G. F. (2009) The mechanochemistry of endocytosis. PLoS. Biol. 7, e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Milosevic I., Giovedi S., Lou X., Raimondi A., Collesi C., Shen H., Paradise S., O'Toole E., Ferguson S., Cremona O., De Camilli P. (2011) Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72, 587–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gallop J. L., McMahon H. T. (2005) BAR domains and membrane curvature: bringing your curves to the BAR. Biochem. Soc Symp. 223–231 [DOI] [PubMed] [Google Scholar]
- 67. Frost A., Unger V. M., De Camilli P. (2009) Boomerangs, bananas, and blimps: Structure and function of F-BAR domains in the context of the BAR domain superfamily in The Pombe Cdc15 Homology Proteins (Aspenström P., ed) pp. 1–10, Landes Bioscience, Austin, TX [Google Scholar]
- 68. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 69. Dey S., Pal A., Chakrabarti P., Janin J. (2010) The subunit interfaces of weakly associated homodimeric proteins. J. Mol. Biol. 398, 146–160 [DOI] [PubMed] [Google Scholar]
- 70. Nooren I. M., Thornton J. M. (2003) Structural characterisation and functional significance of transient protein-protein interactions. J. Mol. Biol. 325, 991–1018 [DOI] [PubMed] [Google Scholar]
- 71. Ringstad N., Nemoto Y., De Camilli P. (2001) Differential expression of endophilin 1 and 2 dimers at central nervous system synapses. J. Biol. Chem. 276, 40424–40430 [DOI] [PubMed] [Google Scholar]
- 72. Lundmark R., Carlsson S. R. (2009) SNX9 - a prelude to vesicle release. J. Cell Sci. 122, 5–11 [DOI] [PubMed] [Google Scholar]
- 73. Böcking T., Aguet F., Harrison S. C., Kirchhausen T. (2011) Single-molecule analysis of a molecular disassemblase reveals the mechanism of Hsc70-driven clathrin uncoating. Nat. Struct. Mol. Biol. 18, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mima J., Hickey C. M., Xu H., Jun Y., Wickner W. (2008) Reconstituted membrane fusion requires regulatory lipids, SNAREs, and synergistic SNARE chaperones. EMBO J. 27, 2031–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lata S., Schoehn G., Jain A., Pires R., Piehler J., Gottlinger H. G., Weissenhorn W. (2008) Helical structures of ESCRT-III are disassembled by VPS4. Science 321, 1354–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kim J., Mosior M., Chung L. A., Wu H., McLaughlin S. (1991) Binding of peptides with basic residues to membranes containing acidic phospholipids. Biophys. J. 60, 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mosior M., McLaughlin S. (1991) Peptides that mimic the pseudosubstrate region of protein kinase C bind to acidic lipids in membranes. Biophys. J. 60, 149–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McLaughlin S. (1989) The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem. 18, 113–136 [DOI] [PubMed] [Google Scholar]
- 79. Schreiber G. (2002) Kinetic studies of protein-protein interactions. Curr. Opin. Struct. Biol. 12, 41–47 [DOI] [PubMed] [Google Scholar]
- 80. Berg O. G., Blomberg C. (1978) Association kinetics with coupled diffusion III. Ionic-strength dependence of the lac repressor-operator association. Biophys. Chem. 8, 271–280 [DOI] [PubMed] [Google Scholar]
- 81. Lohman T. M., DeHaseth P. L., Record M. T., Jr. (1978) Analysis of ion concentration effects of the kinetics of protein-nucleic acid interactions. Application to lac repressor-operator interactions. Biophys. Chem. 8, 281–294 [DOI] [PubMed] [Google Scholar]
- 82. Subramanian M., Jutila A., Kinnunen P. K. (1998) Binding and dissociation of cytochrome c to and from membranes containing acidic phospholipids. Biochemistry 37, 1394–1402 [DOI] [PubMed] [Google Scholar]
- 83. Taylor M. J., Lampe M., Merrifield C. J. (2012) A feedback loop between dynamin and actin recruitment during clathrin-mediated endocytosis. PLoS Biol. 10, e1001302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Youn J. Y., Friesen H., Kishimoto T., Henne W. M., Kurat C. F., Ye W., Ceccarelli D. F., Sicheri F., Kohlwein S. D., McMahon H. T., Andrews B. J. (2010) Dissecting BAR domain function in the yeast amphiphysins Rvs161 and Rvs167 during endocytosis. Mol. Biol. Cell 21, 3054–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Takamori S., Holt M., Stenius K., Lemke E. A., Grønborg M., Riedel D., Urlaub H., Schenck S., Brügger B., Ringler P., Müller S. A., Rammner B., Gräter F., Hub J. S., De Groot B. L., Mieskes G., Moriyama Y., Klingauf J., Grubmüller H., Heuser J., Wieland F., Jahn R. (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846 [DOI] [PubMed] [Google Scholar]
- 86. Borner G. H., Harbour M., Hester S., Lilley K. S., Robinson M. S. (2006) Comparative proteomics of clathrin-coated vesicles. J. Cell Biol. 175, 571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wu Y., He Y., Bai J., Ji S. R., Tucker W. C., Chapman E. R., Sui S. F. (2003) Visualization of synaptotagmin I oligomers assembled onto lipid monolayers. Proc. Natl. Acad. Sci. U.S.A. 100, 2082–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kaneko T., Maeda A., Takefuji M., Aoyama H., Nakayama M., Kawabata S., Kawano Y., Iwamatsu A., Amano M., Kaibuchi K. (2005) Rho mediates endocytosis of epidermal growth factor receptor through phosphorylation of endophilin A1 by Rho-kinase. Genes Cells 10, 973–987 [DOI] [PubMed] [Google Scholar]
- 89. Matta S., Van Kolen K., da Cunha R., van den Bogaart G., Mandemakers W., Miskiewicz K., De Bock P. J., Morais V. A., Vilain S., Haddad D., Delbroek L., Swerts J., Chávez-Gutiérrez L., Esposito G., Daneels G., Karran E., Holt M., Gevaert K., Moechars D. W., De Strooper B., Verstreken P. (2012) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75, 1008–1021 [DOI] [PubMed] [Google Scholar]