

Background: The transcription factor IRF3 is critical for type I interferon induction and innate antiviral immune response.
Results: FoxO1 mediates IRF3 ubiquitination and degradation.
Conclusion: FoxO1 is a negative regulator of cellular antiviral response.
Significance: Our study reveals a new mechanism for control of type I interferon induction and cellular antiviral response.
Keywords: Innate Immunity, Interferon, Signal Transduction, Ubiquitination, Virus
Abstract
Viral infection causes activation of the transcription factor IRF3, which is critical for production of type I interferons (IFNs) and innate antiviral immune response. How virus-induced type I IFN signaling is controlled is not fully understood. Here we identified the transcription factor FoxO1 as a negative regulator for virus-triggered IFN-β induction. Overexpression of FoxO1 inhibited virus-triggered ISRE activation, IFN-β induction as well as cellular antiviral response, whereas knockdown of FoxO1 had opposite effects. FoxO1 interacted with IRF3 in a viral infection-dependent manner and promoted K48-linked polyubiquitination and degradation of IRF3 in the cytosol. Furthermore, FoxO1-mediated degradation of IRF3 was independent of the known E3 ubiquitin ligases for IRF3, including RBCK1 and RAUL. Our findings thus suggest that FoxO1 negatively regulates cellular antiviral response by promoting IRF3 ubiquitination and degradation, providing a previously unknown mechanism for control of type I IFN induction and cellular antiviral response.
Introduction
Viral infection triggers a series of cellular signaling events that consequently lead to the production of type I interferons (IFNs).2 Type I IFNs further activate expression of many downstream genes, the products of which are involved in the inhibition of viral replication and clearance of infected cells (1, 2).
During the viral infection and replication process, viral molecular features called pathogen-associated molecular patterns (PAMPs) such as single-stranded (ss) RNA, double-stranded (ds) RNA, and dsDNA are generated. Host pattern recognition receptors (PRRs) detect these PAMPs and initiate signaling cascades that induce expression of type I IFNs. The membrane-associated receptor Toll-like receptor (TLR) 3 recognizes dsRNA and triggers TRIF-mediated signaling pathways that lead to the activation of type I IFNs (3–5). Cytosolic receptors such as RIG-I and MDA5 also act as viral RNA sensors. Upon the recognition of viral RNA, RIG-I and MDA5 undergo conformational changes and interact with downstream mitochondrial-localized adapter protein VISA (also known as MAVS, Cardif, and IPS-1) via their N-terminal CARD domains (6–9). Another mitochondrial- and endoplasmic reticulum (ER)-localized adapter protein MITA (also known as STING) interacts with VISA and recruits the downstream kinase TBK1 and transcription factor IRF3 to the VISA-associated complex, thereby promoting activation of IRF3 and expression of type I IFNs (10, 11). Recently, we have found that GSK3β is required for IRF3-mediated type I IFNs induction via activation of TBK1 (12).
IRF3 is a member of the transcription factor family called interferon-regulatory factor (IRF). After activation, IRF3 translocates from cytosol to the nucleus, binds to conserved enhancer motifs called ISREs or IRF-Es in the promoters and initiates transcription of a number of genes such as IFNB1 and IFNA (13). The activation of IRF3 is tightly controlled by post-transcriptional modifications. The upstream kinases TBK1 and IKKϵ phosphorylate IRF3, which is required for its dimerization and activation. Many molecules, such as WDR5, TRIM56, MIB1/2, and TRIM32 are involved in this process by various mechanisms (14–17). IRF3 is sumolylated or desumolylated by viral or host components, which interferes with its activity and stability (18, 19). While Herc5-mediated ISG15 modification of IRF3 positively regulates its activation (20), RBCK1 and RAUL are E3 ubiquitin ligases that induce K48-linked ubiquitination and degradation of IRF3, thereby turning down expression of type I IFNs (21, 22). Whether and how other molecules are involved in regulating this process is of great interest.
FoxO1 (Forkhead box protein O1) belongs to the Fork-head transcription factor family that has a conserved DNA-binding domain termed as “forkhead box.” It has been reported that FoxO transcription factors are involved in many signaling pathways and regulate numerous cellular progresses, including tumor development, nervous system, metabolism, longevity, autophagy, and insulin action (23–26). Recently, FoxO3 was reported to inhibit IRF7 transcription and negatively regulate innate immune response (27). In this study, we identified FoxO1 as a negative regulator of virus-induced type I IFNs signaling pathways. FoxO1 was associated with IRF3 in a viral infection-dependent manner and promoted K48-linked polyubiquitination and degradation of IRF3, thereby inhibiting excessive expression of IFN-β and cellular antiviral response. Our findings thus uncovered a negative feedback regulatory mechanism of cellular antiviral response by FoxO1.
EXPERIMENTAL PROCEDURES
Constructs
ISRE, NF-κB, IFN-β, and IRF1 promoter luciferase reporter plasmids, mammalian expression plasmids for HA- or Flag-tagged IRF3 were previously described (11, 12, 18). Mammalian cDNA expression plasmids were purchased from Origene company. Flag- and HA-tagged FoxO1 and its mutants were constructed by standard molecular biology techniques. Flag-tagged VISA, MITA, TBK1, and IKKϵ were previously described (11, 12).
Reagents and Antibodies
Recombinant human IFN-β (Peprotech), mouse monoclonal antibodies against Flag, HA, and β-actin (Sigma), mouse polyclonal antibody against GFP and rabbit polyclonal antibody against IRF3 (Santa Cruz Biotechnology), rabbit polyclonal antibodies against FoxO1 (Cell Signaling Technology) were purchased from the indicated manufacturers. SeV, VSV, NDV-GFP, and VSV-GFP were previously described (17, 28).
Transfection and Reporter Gene Assays
293 cells (∼1 × 105) were seeded on 24-well plates and transfected on the following day by standard calcium phosphate precipitation method. In the same experiment, empty control plasmid was added to ensure that each transfection receives the same amount of total DNA. To normalize for transfection efficiency, 0.01 μg of pRL-TK Renilla luciferase reporter plasmid was added to each transfection. Luciferase assays were performed using a dual-specific luciferse assay kit (Promega), and the firefly luciferase activities were normalized based on Renilla luciferase activities.
Coimmunoprecipitation, Ubiquitination, and Western Blot Analysis
Real-time PCR
Total RNA was isolated from cells using Trizol reagent (TAKARA, Japan) and subjected to real-time PCR analysis to measure expression of mRNA. The mRNA levels of specific genes were normalized to GAPDH mRNA. Gene-specific primer sequences were as following: IFNB1: TTGTTGAGAACCTCCTGGCT (forward), TGACTATGGTCCAGGCACAG (reverse); CCL5: GGCAGCCCTCGCTGTCATCC (forward), GCAGCAGGGTGTGGTGTCCG (reverse); ISG56: TCATCAGGTCAAGGATAGTC (forward), CCACACTGTATTTGGTGTCTAGG (reverse); ISG15: AGGACAGGGTCCCCCTTGCC (forward), CCTCCAGCCCGCTCACTTGC (reverse); GAPDH: GAGTCAACGGATTTGGTCGT (forward), GACAAGCTTCCCGTTCTCAG (reverse).
VSV Plaque Assays
Cells were grown in 24-well plate and transfected with the indicated plasmids. Twenty-four hours after transfection, cells were transfected by Lipofactamine with poly(I:C) (1 μg) or control buffer for another 24 h prior to VSV infection. At 1 h post-infection, cells were washed with warm PBS and then fresh medium was added. The supernatant was harvested 24 h later and used to infect confluent cultured Vero cells. Plaque assays were then performed as described (11, 12).
Virus Manipulation
Cells were grown in 24-well plates and transfected with the indicated plasmids prior to virus infection. The culture medium was replaced by serum-free DMEM containing NDV-GFP or VSV-GFP viruses (MOI, 0.1). One hour later, the cells were washed with PBS and then fed with DMEM containing 10% FBS. NDV-GFP or VSV-GFP replication was visualized by monitoring the GFP expression level in fluorescence microscopy or by Western immunoblot analysis.
Size-exclusion Chromatography
Cells (1 × 107) were lysed in 1.2 ml of lysis buffer. The lysate was centrifuged for 0.5 h at 15,000 rpm. The supernatant was recovered and loaded onto a Superdex 200 gel filtration chromatography column pre-equilibrated with lysis buffer. The samples were eluted from the column in lysis buffer at a flow rate of 0.5 ml/min and collected in fractions of 0.5 ml. The fractions were precipitated with 20% trichloroacetic acid and analyzed by Western blots with antibodies against IRF3 and FoxO1, respectively.
RNAi
Double-strand oligonucleotides corresponding to the target sequences were cloned into the pSuper.Retro RNAi plasmid (Oligoengine Inc.). The following sequences were targeted for human FoxO1 and RAUL cDNA, respectively: FoxO1-RNAi: 1: CCTCATGGATGGAGATACA; 2: CTCAAATGCTAGTACTATT; 3: CTGCATCCATGGACAACAA; 4: CCATGGACAACAACAGTAA; RAUL-RNAi: GCAAGAAGTTCAAGGAGAT.
The nonspecific-RNAi and pGCSIL-FoxO1-RNAi were provided by Dr. Wei-Guo Zhu (Peking University) and previously described (23). The pSuper-RBCK1-RNAi was previously described (21).
RESULTS
Overexpression of FoxO1 Inhibits Virus-triggered IFN-β Induction
To identify candidate molecules involved in virus-triggered IFN-β induction, we screened ∼15,000 independent human and mouse cDNA expression plasmids by IFN-β promoter luciferase reporter assays and found that human FoxO1 (clone 45A1) inhibited Sendai virus (SeV)-induced activation of the IFN-β promoter (supplemental Fig. S1A). To further confirm this result, we made Flag-tagged expression plasmid encoding human FoxO1 independently of the clone 45A1 and performed additional luciferase reporter assays. As shown in Fig. 1A, overexpression of Flag-FoxO1 strongly inhibited SeV-induced activation of ISRE, NF-κB and the IFN-β promoter in a dose-dependent manner. Results from real-time PCR experiments indicated that overexpression of FoxO1 inhibited SeV-induced expression of downstream genes, such as IFNB1 and CCL5 (Fig. 1B). Poly(I:C), a synthetic analog of viral dsRNA, is recognized by TLR3 on the membrane or RLR in the cytoplasm, respectively. We found that overexpression of FoxO1 potently inhibited poly(I:C)-triggered both RLR (Fig. 1C) and TLR3 (Fig. 1D) -mediated activation of the IFN-β promoter and ISRE. This effect is not cell-type specific because overexpression of FoxO1 also inhibited SeV-induced activation of the IFN-β promoter in HCT116 and HeLa cells (Fig. 1E). In similar reporter assays, overexpression of FoxO1 did not affect IFN-γ-triggered activation of IRF1 promoter and SeV-triggered activation of AP-1 (supplemental Fig. S1, B and C). Furthermore, overexpression of FoxO1 did not affect IFN-β-induced expression of RIG-I or MDA5 (supplemental Fig. S1D). These data suggest that FoxO1 specifically inhibits virus-triggered induction of downstream genes.
FIGURE 1.

Overexpression of FoxO1 inhibits virus-triggered induction of downstream genes. A, FoxO1 inhibits SeV-induced activation of ISRE, NF-κB, and IFN-β promoter in a dose-dependent manner. The 293 cells (1 × 105) were transfected with the IFN-β promoter, ISRE and NF-κB luciferase reporter (0.1 μg) and the indicated amounts of Flag-FoxO1 plasmids. Twenty hours after transfection, cells were infected with SeV or left uninfected for 10 h before reporter assays were performed. B, FoxO1 inhibits SeV-induced expression of IFNB1 and CCL5 genes. The 293 cells (4 × 105) were transfected with control or FoxO1 expression plasmids (2 μg each) for 20 h. Cells were then infected with SeV or left uninfected for 10 h before real-time PCR analysis was performed. C, FoxO1 inhibits cytosolic poly(I:C)-triggered activation of the IFN-β promoter and ISRE. The 293 cells (1 × 105) were transfected with control or FoxO1 expression plasmids (0.25 μg) and the indicated reporter plasmids (0.1 μg). Twenty hours after transfection, cells were mock-transfected or transfected with poly(I:C) (1 μg) for 16 h before luciferase assays were performed. D, FoxO1 inhibits TLR3-mediated activation of the IFN-β promoter and ISRE. The 293-TLR3 cells (1 × 105) were transfected with FoxO1 expression (0.5 μg) and the indicated reporter (0.1 μg each) plasmids. Twenty hours after transfection, cells were treated with poly(I:C) (50 μg/ml) or left untreated for 12 h before luciferase assays were performed. E, FoxO1 inhibits SeV-induced activation of the IFN-β promoter in HCT116 or HeLa cells. The experiments were performed as in A except that HeLa cells and HCT116 cells were used.
Knockdown of FoxO1 Potentiates SeV-induced Expression of IFNB1
We next determined whether endogenous FoxO1 was involved in virus-triggered IFN-β induction. We constructed four RNAi plasmids (pSuper-FoxO1-RNAi 1–4) and obtained a previously reported RNAi plasmid (pGCSIL-FoxO1-RNAi) (23) for FoxO1, all of which could inhibit the expression of transfected and endogenous FoxO1 (Fig. 2A). In reporter assays, knockdown of FoxO1 potentiated SeV-induced activation of IFN-β promoter, ISRE and NF-κB in 293 cells (Fig. 2, B and C). Consistently, results from real-time PCR experiments confirmed that SeV-induced expression of IFNB1 was increased by knockdown of FoxO1 (Fig. 2D). In addition, knockdown of FoxO1 potentiated cytosolic poly(I:C)-induced activation of IFN-β promoter, but did not affect IFN-γ-induced activation of the IRF1 promoter (supplemental Fig. S2, A and B). These results suggest that endogenous FoxO1 inhibits virus-triggered IFN-β expression.
FIGURE 2.

Knockdown of FoxO1 potentiates SeV-induced expression of IFN-β. A, effects of FoxO1-RNAi plasmids on the expression of transfected FoxO1. Left panels: the 293 cells (4 × 105) were transfected with expression plasmids for Flag-FoxO1 and HA-β-actin (0.1 μg each), and the indicated RNAi plasmids (2 μg). Twenty-four hours after transfection, cell lysates were analyzed by immunoblot with anti-HA and anti-Flag. Right panels: the 293 cells (4 × 105) were transfected with control or the indicated FoxO1-RNAi plasmids (2 μg each). Thirty-six hours later, cells were lysed and the cell lysates were analyzed by immunoblot with the indicated antibodies. NS-RNAi, Nonspecific RNAi; pFoxO1-RNAi, pGCSIL-FoxO1-RNAi. B, effects of FoxO1-RNAi plasmids on SeV-induced activation of the IFN-β promoter. The 293 cells (1 × 105) were transfected with the indicated FoxO1-RNAi (0.5 μg) and IFN-β reporter (0.1 μg) plasmids. Thirty-six hours after transfection, cells were left uninfected or infected with SeV for 10 h before luciferase assays were performed. C, knockdown of FoxO1 potentiates SeV-induced ISRE and NF-κB activation. The experiments were performed as in B except that the ISRE and NF-κB reporter was used. D, effects of FoxO1-RNAi plasmids on SeV-induced expression of IFNB1gene. The 293 cells (4 × 105) were transfected with control or FoxO1-RNAi plasmids (3 μg). Thirty-six hours after transfection, cells were left uninfected or infected with SeV for 10 h before real-time PCR analysis was performed. E, effects of FoxO1-RNAi on VSV replication. The 293 cells (1 × 105) were transfected with the pFoxO1- RNAi plasmids (0.5 μg each). Thirty-six hours later, cells were mock-transfected or transfected with poly(I:C) (1 μg) for 16 h and then infected with VSV (MOI, 0.1). The supernatants were harvested 24 h after infection for standard plaque assays. F, effects of FoxO1-RNAi on VSV-GFP replication. The 293 (1 × 105) cells were transfected with pFoxO1-RNAi plasmids (1 μg). Thirty-six hours later, cells were infected with VSV-GFP (MOI = 0.1) for 24 h and imaged by microscopy or analyzed by immunoblots with the indicated antibodies. The amounts of VSV-GFP were quantitated using the Bio-Rad Quantity One Program and were normalized to that of β-actin.
We further found that knockdown of FoxO1 could potentiate SeV- but not IFN-β-triggered expression of RIG-I and MDA5 (supplemental Fig. S2, C and D), indicating that FoxO1 functions in virus-triggered type I IFNs signaling rather than downstream of type I IFNs. Because FoxO1 negatively regulated virus-triggered IFN-β expression, we next determined whether FoxO1 was involved in the regulation of cellular antiviral response. In plaque assays with vesicular stomatitis virus (VSV), we found that knockdown of FoxO1 significantly inhibited VSV replication and further promoted cytoplasmic poly(I:C)-mediated inhibition of VSV replication (Fig. 2E). Similarly, the replication of GFP-tagged VSV and Newcastle disease virus (NDV) was inhibited in FoxO1-knockdown cells compared with control cells as monitored by GFP expression (Fig. 2F and supplemental Fig. S2E). To examine whether FoxO1 regulates viral replication directly by interfering the replication machinery or indirectly by regulating type I IFNs, we determined the effects of FoxO1-RNAi on virus replication in VISA-knockdown cells, in which viral infection-induced expression of type I IFNs was impaired. The results showed that viral replication was comparable between cells transfected with VISA-RNAi alone and those with both VISA-RNAi and FoxO1-RNAi (supplemental Fig. S2F), indicating that FoxO1 negatively regulates viral replication indirectly through type I IFNs. Collectively, these results suggest that endogenous FoxO1 plays a key role in the regulation of type I IFN production and cellular antiviral response.
FoxO1 Negatively Regulates Virus-triggered Signaling at IRF3 Level
A series of components have been reported to participate in virus-triggered IFN-β induction, such as RIG-I, VISA, MITA, TBK1, and IRF3. To determine the molecular order of FoxO1 in the regulation of virus-triggered IFN-β induction, we transfected plasmids encoding these molecules together with the IFN-β promoter or ISRE reporters in the presence or absence of FoxO1, and found that FoxO1 could markedly inhibit activation of the IFN-β promoter and ISRE mediated by overexpression of IRF3 and the upstream adaptors or kinases, including VISA, MITA and TBK1 (Fig. 3, A and B). Conversely, knockdown of FoxO1 potentiated ISRE activation mediated by IRF3 and the upstream molecules (Fig. 3C). These results indicate that FoxO1 functions at the level or downstream of IRF3 for regulation of virus-triggered IFN-β expression.
FIGURE 3.
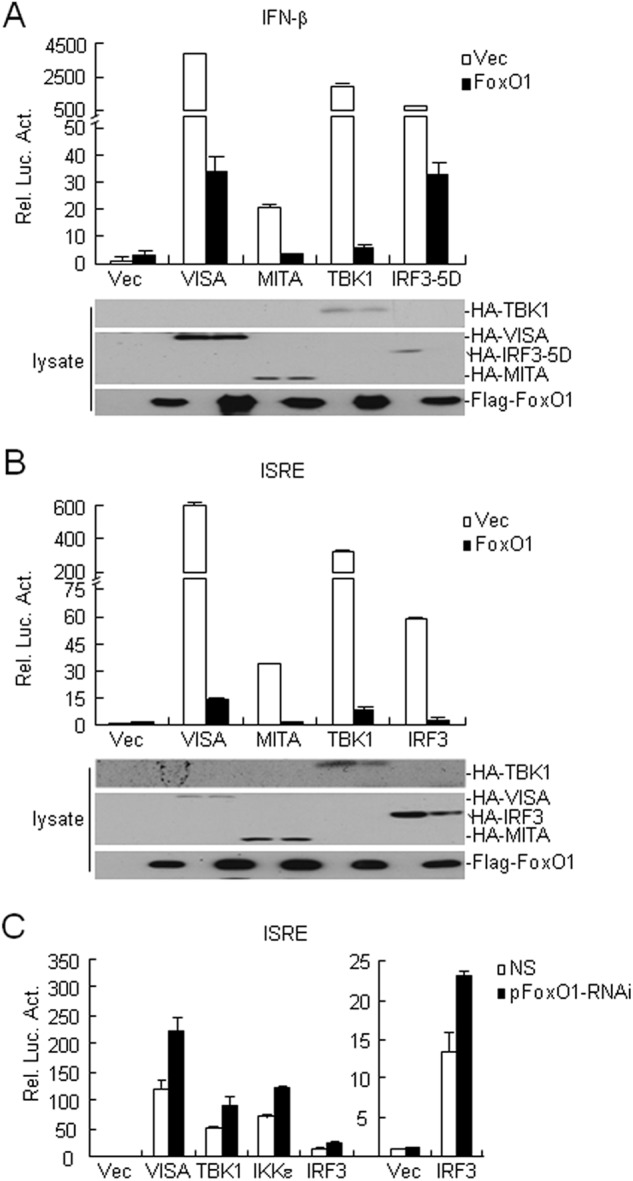
FoxO1 negatively regulates SeV-induced activation of ISRE at IRF3. A, overexpression of FoxO1 inhibits VISA-, MITA-, TBK1-, and IRF3 (5D)-mediated IFN-β promoter activation. The 293 cells (1 × 105) were transfected with control or FoxO1 expression plasmids and the indicated plasmids (0.1 μg each). Reporter assays were performed 24 h after transfection. Expression of transduced proteins were detected by immunoblot with indicated antibodies. B, overexpression of FoxO1 inhibits VISA-, MITA-, TBK1-, and IRF3-mediated ISRE activation. The experiments were performed as in A except that the ISRE reporter was used. C, knockdown of FoxO1 potentiates VISA-, IKKϵ-, TBK1-, and IRF3-mediated ISRE activation. The 293 cells (1 × 105) were transfected with control or FoxO1-RNAi plasmids (0.5 μg). Thirty-six hours later, cells were further transfected with the indicated plasmids (0.1 μg each) and ISRE reporter plasmids. Reporter assays were performed 24 h after transfection.
FoxO1 Is Associated with IRF3 in a Viral Infection-dependent Manner
Because FoxO1 is a transcription factor that functions at the level or downstream of IRF3, we hypothesized that FoxO1 might interact with IRF3 and interfere its transcriptional activity. In transient transfection and co-immunoprecipitation experiments, overexpression of FoxO1 interacted with IRF3 constitutively (Fig. 4A). We further found that GFP-tagged IRF3 colocalized with cherry-FoxO1 by confocal immunofluorescent microscopy analysis (supplemental Fig. S3A). In our gel filtration experiments, we found that FoxO1 and IRF3 were mostly isolated in different fractions (IRF3, lanes 6–8 and lanes 17–20; FoxO1, lanes 9–11 and lanes 12–15) in uninfected cells (Fig. 4B). After viral infection, increased FoxO1 and IRF3 were detected in overlapping fractions (lanes 8–11) (Fig. 4B), suggesting that viral infection may induce FoxO1 and IRF3 complex formation. Consistent with these observations, results from endogenous co-immunoprecipitation experiments indicated that FoxO1 did not interact with IRF3 without stimulation but was associated with IRF3 at 8 h after SeV infection (Fig. 4C). These results suggest that FoxO1 interacts with IRF3 in a viral infection-dependent manner.
FIGURE 4.

FoxO1 interacts with IRF3 in a viral infection-dependent manner. A, FoxO1 interacts with IRF3. The 293 cells (2 × 106) were transfected with the indicated plasmids. Twenty hours after transfection, co-immunoprecipitation was performed with anti-HA or control IgG. The immunoprecipitates were analyzed by immunoblot with anti-Flag (upper panel). The lysates were analyzed by immunoblots with anti-Flag or anti-HA (lower panels). B, analysis of protein complexes containing FoxO1 and IRF3 by size-exclusion chromatography. The 293 cells (1 × 107) were infected with SeV for 8 h or left uninfected before lysis. Cell lysates were analyzed by size-exclusion chromatography on Superdex 200 column. The individual fractions were analyzed by Western blots with anti-FoxO1 and anti-IRF3 antibodies, respectively. C, endogenous FoxO1 interacts with IRF3 after viral infection. The 293 cells (1 × 107) were left uninfected or infected with SeV for the indicated time points. Cells were lysed, and the lysates were immunoprecipitated with anti-IRF3 or control IgG. The immunoprecipitates were analyzed by immunoblot with anti-FoxO1 (upper panel). The expression levels of the endogenous FoxO1, IRF3, and β-actin were analyzed by immunoblot analysis (lower panels). D, immunoblot analysis of the subcellular fractions. The 293 cells were infected with SeV for the indicated time points. Cell fractionations were performed and the fractions were analyzed by immunoblots with the indicated antibodies (upper four panels). The whole cellular levels of FoxO1 and IRF3 upon viral infection were analyzed by immunoblots with indicated antibodies (lower three panels). E, FoxO1 interacts with IRF3 in the cytosol. The 293 cells (1 × 107) were left uninfected or infected with SeV for 8 h. Cellular fractions were prepared as in C. The fractions were immunoprecipitated with anti-IRF3. The immunoprecipitates were analyzed by immunoblot with anti-FoxO1 (upper panel). The expression levels of the endogenous FoxO1 and IRF3 were detected by immunoblot analysis (lower four panels).
Previously, it has been reported that FoxO1 shuttles between cytosol and nucleus after activation (29, 30). We next determined the localization of FoxO1 in cells in response to viral infection. Surprisingly, cherry-tagged FoxO1 was mainly localized in the cytosol (supplemental Fig. S3A). We then performed cellular fractionation and immunoblot analysis and found that endogenous FoxO1 was mainly localized in the cytosol and barely detectable in the nucleus in the presence or absence of SeV infection in 293 cells (Fig. 4D). Previously, it has been reported that FBS could retain FoxO1 and FoxO3 (another member of Fork-head transcription factor) in the cytosol. In our experiments, we found that there was increased FoxO1 in the nucleus in the absence of FBS and viral infection resulted in translocation of FoxO1 from the nucleus to cytosol (supplemental Fig. S3B). Interestingly, SeV-induced activation of IFN-β promoter was inhibited and potentiated by overexpression or knockdown of FoxO1 in the absence of FBS, respectively (supplemental Fig. S3, C and D). We next determined whether FoxO1 interacts with IRF3 in the cytosol or nucleus. As shown in Fig. 4E, FoxO1 interacted with IRF3 in the cytosol after viral infection. These data collectively suggest that FoxO1 interacts with IRF3 in the cytosol in a viral infection-dependent manner.
FoxO1 Targets IRF3 for Degradation
When examining the interaction between FoxO1 and IRF3, we repeatedly observed that overexpression of FoxO1 resulted in a decreased level of IRF3 (Fig. 4A and supplemental S4A). We thus hypothesized that FoxO1 might regulate the stability of IRF3 protein. To test this hypothesis, we co-transfected Flag-tagged FoxO1 together with HA-tagged IKKϵ, VISA, MITA, or IRF3 and performed immunoblot analysis. As shown in Fig. 5A, FoxO1 specifically down-regulated the amount of IRF3 protein. In similar experiments, FoxO1 down-regulated the expression of IRF3–5A and IRF-5D, which are inactive and active mutants of IRF3, respectively (supplemental Fig. S4A). Our experiments indicated that FoxO1 also down-regulated IRF7, which forms dimer with IRF3 and activates IFN expression, but had minimal effects on IRF5 level (supplemental Fig. S4B). We also found that overexpression of FoxO1 promoted degradation of endogenous IRF3 with or without viral infection (Fig. 5B), whereas knockdown of FoxO1 delayed the degradation of IRF3 after SeV infection (Fig. 5C and supplemental Fig. S4C). In addition, knockdown of FoxO1 resulted in increased level of IRF7 after SeV infection (supplemental Fig. S4D). In contrast, unlike FoxO3-mediated transcriptional inhibition of IRF7, FoxO1 did not affect transcription of IRF3 in the presence or absence of SeV infection (Fig. 5D). These data demonstrate that FoxO1 negatively regulates IRF3 at protein level.
FIGURE 5.

FoxO1 promotes K48-linked ubiquitination and degradation of IRF3. A, FoxO1 specifically destabilizes IRF3. The 293 (4 × 105) cells were transfected with the indicated plasmids for 20 h before immunoblot analysis was performed with the indicated antibodies. B, overexpression of FoxO1 destabilizes endogenous IRF3. The 293 cells (4 × 105) were transfected with control or FoxO1 expression plasmids (3 μg). Twenty hours after transfection, cells were left uninfected or infected with SeV for 8 h before immunoblot analysis was performed with the indicated antibodies. The amounts of pIRF3 were quantitated using the Bio-Rad Quantity One Program and were normalized to that of IRF3. C, knockdown of FoxO1 delays the degradation of IRF3 following viral infection. The 293 cells (4 × 105) were transfected with control or pFoxO1-RNAi plasmids (3 μg). Thirty-six hours after transfection, cells were left uninfected or infected with SeV for the indicated time points before immunoblot analysis was performed with the indicated antibodies. D, effect of FoxO1 on IRF3 gene transcription. The 293 cells (4 × 105) were transfected with FoxO1 expression or FoxO1-RNAi plasmids (2 μg). Twenty-four or thirty-six hours after transfection, cells were left uninfected or infected with SeV for 8 h. Cells were then harvested for real-time PCR analysis. E, FoxO1 mediates IRF3 degradation in a proteasome-dependent manner. The 293 cells (4 × 105) were transfected with the indicated plasmids (0.5 μg each) and the indicated amounts of FoxO1 expression plasmid. Twenty hours later, cells were treated with MG132 (10 μm) for 6 h before immunoblot analysis was performed. F, overexpression of FoxO1 promotes ubiquitination of IRF3. The 293 cells (2 × 106) were transfected with the indicated plasmids. Twenty hours after transfection, cell lysates were denatured and immunoprecipitated with anti-Flag and the immunoprecipitates were analyzed by immunoblot with anti-Myc (upper panel). The expression levels of the proteins were examined by immunoblots with the indicated antibodies (lower panels). G, FoxO1 mediated K48-linked ubiquitination of IRF3. The 293 cells (2 × 106) were transfected with the indicated plasmids. Twenty hours after transfection, cell lysates were denatured. The lysates were analyzed by immunoprecipitation with anti-Flag and immunoblots with the anti-HA (upper panel) antibodies. The levels of the transfected proteins were examined by immunoblots with the anti-HA (middle panels) and anti-Flag (lower panels) antibodies, respectively. K48, ubiquitin with all lysines except K48 were mutated; K63, ubiquitin with all lysines except K63 were mutated. H, effect of FoxO1-RNAi on SeV-induced ubiquitination of IRF3. The 293 cells (1 × 107) were transfected with control or pFoxO1-RNAi plasmids. Thirty-six hours after transfection, cells were left uninfected or infected with SeV for 8 h. Cells were lysed and the lysates were immunoprecipitated with anti-IRF3. The immunoprecipitates were analyzed by immunoblot with anti-ubiquitin (upper panel). The expression of the related proteins was examined by immunoblots with the anti-FoxO1 (middle panels) and anti-β-actin (lower panels) antibodies, respectively.
To further confirm this conclusion, we treated cells with actinomycin-D and CHX, which are inhibitors of transcription or protein synthesis, respectively, and examined the half-life of IRF3 protein in the presence or absence of FoxO1. As shown in supplemental Fig. S4, E and F, overexpression of FoxO1 significantly accelerated degradation of IRF3 in cells treated with actinomycin-D or CHX. FoxO1-mediated degradation of IRF3 could be restored by treatment with the proteasome inhibitor MG132 but not with the autophagy inhibitor 3-MA (Fig. 5E and supplemental Fig. S4G). These data indicate that FoxO1 controls half-life of IRF3 protein in a proteasome-dependent manner.
FoxO1 Promotes K48-linked Ubiquitination and Degradation of IRF3
Having established that FoxO1 negatively regulates virus-triggered IFN-β activation by promoting IRF3 degradation in a proteasome-dependent pathway, we next examined the role of FoxO1 in regulating ubiquitination of IRF3. As shown in Fig. 5F, overexpression of FoxO1 potentiated ubiquitination and degradation of IRF3. Because K48-linked polyubiquitin modification leads to the target proteins for proteasome recognition and degradation, we determined whether FoxO1 promoted K48- or K63-linked ubiquitination of IRF3. As shown in Fig. 5G, overexpression of FoxO1 significantly increased K48-linked but not K63-linked ubiquitination of IRF3. We further examined whether endogenous FoxO1 played a role in regulating virus-triggered IRF3 ubiquitination. As shown in Fig. 5H, ubiquitination of IRF3 was strongly increased after SeV infection, which was diminished in FoxO1-knockdown 293 cells. Previous studies have identified several E3 ubiquitin ligases that catalyze K48-linked ubiquitination of IRF3, including RBCK1 and RAUL (21, 22). However, knockdown of neither RBCK1 nor RAUL had any effect on FoxO1-mediated inhibition of SeV-triggered activation of ISRE or degradation of IRF3 (supplemental Fig. S5, A and B). Conversely, RBCK1- or RAUL-mediated inhibition of SeV-triggered ISRE activation or degradation of IRF3 did not require FoxO1 (supplemental Fig. S5, C and D), indicating that FoxO1 mediates degradation of IRF3 via an unknown E3 or a different mechanism that involves the ubiquitin-proteasome system. Taken together, these results suggest that FoxO1 promotes K48-linked polyubiquitination and proteasome-dependent degradation of IRF3.
DISCUSSION
Viral infection leads to production of type I IFNs that are responsible for inhibition of virus replication, clearance of virus-infected cells, and facilitation of adaptive immune response. However, uncontrolled and excessive immune response causes pathological immunity to the host. To prevent harmful effects during acute infection, host cells have developed distinct strategies to control excessive innate immune response. In the present study, we identified the transcription factor FoxO1 as a negative feedback regulator of innate cellular antiviral response.
Our results demonstrated that FoxO1 specifically inhibited virus-triggered expression of type I IFNs and the cellular antiviral response. Overexpression of FoxO1 inhibited virus-triggered activation of the IFN-β promoter and ISRE in various types of cells, whereas knockdown of FoxO1 potentiated virus-triggered IFN-β induction and ISRE activation and inhibited viral replication. Interestingly, neither overexpression nor knockdown of FoxO1 had effect on IFN-β-induced expression of RIG-I or MDA5. Taken together these data suggest that FoxO1 physiologically restricts virus-triggered type I IFN signaling and cellular antiviral response.
To explore the mechanisms by which FoxO1 regulates virus-triggered type I IFN induction, we first examined the molecular order of the involvement of FoxO1 in this process and found that FoxO1 regulated ISRE activation at the level or downstream of IRF3. Considering FoxO1 is a transcription factor, we hypothesized that FoxO1 might interact with IRF3 and interfere with its transcriptional activity. However, our study demonstrated that FoxO1 interacted with IRF3 in the cytosol at 8 h after virus infection, excluding the possibility that FoxO1 interferes with the transcriptional activity of IRF3 in the nucleus. Our study also showed that the protein level of FoxO1 was increased at 8–12 h after viral infection, while the mRNA level of FoxO1 remained unaltered after SeV infection for indicated times (supplemental Fig. S6A), indicating that viral infection triggers a post-transcriptional regulation of FoxO1. In this context, it is possible that viral infection-induced up-regulation of FoxO1 in the cytosol accounts for FoxO1-IRF3 association after viral infection. How FoxO1 protein was increased by viral infection requires further investigations.
Interestingly, we routinely observed that overexpression of FoxO1 specifically resulted in decreased protein level of IRF3 but had no effect on the mRNA level of IRF3, indicating that FoxO1 regulates the stability of IRF3 protein. Furthermore, overexpression of FoxO1 did not affect the ratio of phosphorylated IRF3 to the total IRF3 (pIRF3:IRF3) after SeV infection, though the levels of phosphorylated IRF3 and total IRF3 were dramatically decreased, indicating that FoxO1 does not regulates the IRF3 phosphorylation. It should be noted that the basal level of IRF3 was higher in 293 cells transfected with FoxO1-RNAi than controls. There are several reasons behind this phenomenon. First, FoxO1 may interact with IRF3 weakly and induce degradation of IRF3 under steady condition, which is beyond our detection limit. In this context, we found that a small part of IRF3 and FoxO1 were co-isolated from the same fractions in our gel filtration experiments. Second, FoxO1 may have additional targets which could destabilize IRF3 without virus infection. Nonetheless, our data clearly demonstrates that FoxO1 interacts with and promotes degradation of IRF3 after viral infection.
Protein stability is regulated by proteasome-mediated degradation or autophagy-mediated protein hydrolysis. In our experiments, we found that FoxO1-mediated degradation of IRF3 was inhibited by the proteasome inhibitor MG132 but not by the autophagy inhibitor 3-MA. In addition, FoxO1 promoted K48-linked ubiquitination of IRF3 and knockdown of FoxO1 strongly inhibited virus-induced ubiquitination and degradation of IRF3. Previously, it has been reported that several E3 ubiquitin ligases such as RBCK1 and RAUL induce ubiquitination and degradation of IRF3 (21, 22). However, neither of them was involved in FoxO1-mediated degradation of IRF3, indicating that FoxO1 and these E3s function independently and/or redundantly for regulation of IRF3. Future studies are required to identify the potential E3 ubiquitin ligase that is responsible for FoxO1-mediated IRF3 degradation and to fully elucidate the mechanisms by which FoxO1 restricts virus-triggered type I IFN induction and cellular antiviral response. Nonetheless, our results clearly demonstrate that FoxO1 regulates IRF3 stability through ubiquitin-proteasome system after viral infection.
FoxO1 has recently been reported to regulate regulatory T (Treg) cell development in the thymus and deficiency of FoxO1 in Treg cells renders mice severe autoimmune diseases (31). In this study, we found that FoxO1 is important for control of excessive immune response by regulating turnover of IRF3. Thus, our study not only provides new insight into the molecular mechanisms for termination of excessive immune responses, but also provides a potential target for drug development against viral infection-related and autoimmune diseases.
Acknowledgments
We thank Dr. Yan-Yi Wang (Wuhan Institute of Virology), Yu Liu, and Shu Li (Wuhan University) for discussions. We also thank Dr. Wei-Guo Zhu for reagents.
This work was supported by grants from the Chinese Ministry of Science and Technology (2012CB910201, 2010DFA31100), the National Science Foundation of China (91029302, 31130020, and 31221061), the Chinese 111 project (B06018), and the academic award for excellent Ph.D. candidates funded by Ministry of Education of China.

This article contains supplemental Figs. S1–S6.
- IFN
- type I interferon
- PAMP
- pathogen-associated molecular pattern
- PRR
- pattern recognition receptor
- TLR
- Toll-like receptor
- IRF
- interferon-regulatory factor
- Fox
- Forkhead box protein
- VSV
- vesicular stomatitis virus.
REFERENCES
- 1. Akira S., Uematsu S., Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 2. Hiscott J. (2007) Convergence of the NF-κB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 18, 483–490 [DOI] [PubMed] [Google Scholar]
- 3. Alexopoulou L., Holt A. C., Medzhitov R., Flavell R. A. (2001) Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 [DOI] [PubMed] [Google Scholar]
- 4. Han K. J., Su X., Xu L. G., Bin L. H., Zhang J., Shu H. B. (2004) Mechanisms of the TRIF-induced interferon-stimulated response element and NF-κB activation and apoptosis pathways. J. Biol. Chem. 279, 15652–15661 [DOI] [PubMed] [Google Scholar]
- 5. Oshiumi H., Matsumoto M., Funami K., Akazawa T., Seya T. (2003) TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-β induction. Nat. Immunol. 4, 161–167 [DOI] [PubMed] [Google Scholar]
- 6. Xu L. G., Wang Y. Y., Han K. J., Li L. Y., Zhai Z., Shu H. B. (2005) VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 19, 727–740 [DOI] [PubMed] [Google Scholar]
- 7. Seth R. B., Sun L., Ea C. K., Chen Z. J. (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 [DOI] [PubMed] [Google Scholar]
- 8. Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 [DOI] [PubMed] [Google Scholar]
- 9. Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K. J., Takeuchi O., Akira S. (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 [DOI] [PubMed] [Google Scholar]
- 10. Ishikawa H., Barber G. N. (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhong B., Yang Y., Li S., Wang Y. Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P., Shu H. B. (2008) The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 [DOI] [PubMed] [Google Scholar]
- 12. Lei C. Q., Zhong B., Zhang Y., Zhang J., Wang S., Shu H. B. (2010) Glycogen synthase kinase 3β regulates IRF3 transcription factor-mediated antiviral response via activation of the kinase TBK1. Immunity 33, 878–889 [DOI] [PubMed] [Google Scholar]
- 13. Au W. C., Moore P. A., Lowther W., Juang Y. T., Pitha P. M. (1995) Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. U.S.A. 92, 11657–11661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y. Y., Liu L. J., Zhong B., Liu T. T., Li Y., Yang Y., Ran Y., Li S., Tien P., Shu H. B. (2010) WDR5 is essential for assembly of the VISA-associated signaling complex and virus-triggered IRF3 and NF-κB activation. Proc. Natl. Acad. Sci. U.S.A. 107, 815–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuchida T., Zou J., Saitoh T., Kumar H., Abe T., Matsuura Y., Kawai T., Akira S. (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 [DOI] [PubMed] [Google Scholar]
- 16. Li S., Wang L., Berman M., Kong Y. Y., Dorf M. E. (2011) Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang J., Hu M. M., Wang Y. Y., Shu H. B. (2012) TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ran Y., Liu T. T., Zhou Q., Li S., Mao A. P., Li Y., Liu L. J., Cheng J. K., Shu H. B. (2011) SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J. Mol. Cell Biol. 3, 283–292 [DOI] [PubMed] [Google Scholar]
- 19. Kubota T., Matsuoka M., Chang T. H., Tailor P., Sasaki T., Tashiro M., Kato A., Ozato K. (2008) Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 283, 25660–25670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi H. X., Yang K., Liu X., Liu X. Y., Wei B., Shan Y. F., Zhu L. H., Wang C. (2010) Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell Biol. 30, 2424–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang M., Tian Y., Wang R. P., Gao D., Zhang Y., Diao F. C., Chen D. Y., Zhai Z. H., Shu H. B. (2008) Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Research 18, 1096–1104 [DOI] [PubMed] [Google Scholar]
- 22. Yu Y., Hayward G. S. (2010) The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33, 863–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y., Yang J., Liao W., Liu X., Zhang H., Wang S., Wang D., Feng J., Yu L., Zhu W. G. (2010) Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675 [DOI] [PubMed] [Google Scholar]
- 24. Greer E. L., Brunet A. (2005) FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24, 7410–7425 [DOI] [PubMed] [Google Scholar]
- 25. van der Horst A., Burgering B. M. (2007) Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 8, 440–450 [DOI] [PubMed] [Google Scholar]
- 26. Barthel A., Schmoll D., Unterman T. G. (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16, 183–189 [DOI] [PubMed] [Google Scholar]
- 27. Litvak V., Ratushny A. V., Lampano A. E., Schmitz F., Huang A. C., Raman A., Rust A. G., Bergthaler A., Aitchison J. D., Aderem A. (2012) A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 490, 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Y., Chen R., Zhou Q., Xu Z., Li C., Wang S., Mao A., Zhang X., He W., Shu H. B. (2012) LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc. Natl. Acad. Sci. U.S.A. 109, 11770–11775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rena G., Woods Y. L., Prescott A. R., Peggie M., Unterman T. G., Williams M. R., Cohen P. (2002) Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 21, 2263–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang X., Gan L., Pan H., Guo S., He X., Olson S. T., Mesecar A., Adam S., Unterman T. G. (2002) Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 277, 45276–45284 [DOI] [PubMed] [Google Scholar]
- 31. Ouyang W., Liao W., Luo C. T., Yin N., Huse M., Kim M. V., Peng M., Chan P., Ma Q., Mo Y., Meijer D., Zhao K., Rudensky A. Y., Atwal G., Zhang M. Q., Li M. O. (2012) Nature 491, 554–559 [DOI] [PMC free article] [PubMed] [Google Scholar]