

Background: Dysregulation of cell proliferation, usually controlled by cyclin-dependent kinases (CDKs), is a hallmark of cancer.
Results: Ubiquitin C-terminal hydrolase L1 (UCH-L1) potentiates CDK activities to enhance cell proliferation.
Conclusion: Potentiating CDK activities are a newly identified function of UCH-L1 and are involved in cancer growth.
Significance: These findings should lead to a better understanding of, and novel therapies against, cancer.
Keywords: Cancer, Cyclin-dependent Kinase (CDK), Cell Growth, Cell Proliferation, Protein Complexes, Protein-Protein Interactions, Tumor Marker, UCH-L1
Abstract
Dysregulation of cell proliferation and the cell cycle are associated with various diseases, such as cancer. Cyclin-dependent kinases (CDKs) play central roles in cell proliferation and the cell cycle. Ubiquitin C-terminal hydrolase L1 (UCH-L1) is expressed in a restricted range of tissues, including the brain and numerous types of cancer. However, the molecular functions of UCH-L1 remain elusive. In this study, we found that UCH-L1 physically interacts with CDK1, CDK4, and CDK5, enhancing their kinase activity. Using several mutants of UCH-L1, we showed that this enhancement is dependent upon interaction levels between UCH-L1 and CDKs but is independent of the known ubiquitin-related functions of UCH-L1. Gain- and loss-of-function studies revealed that UCH-L1 enhances proliferation of multiple cell types, including human cancer cells. Inhibition of the interaction between UCH-L1 and cell cycle-associated CDK resulted in the abolishment of UCH-L1-induced enhancement of cell proliferation. RNA interference of UCH-L1 reduced the growth of human xenograft tumors in mice. We concluded that UCH-L1 is a novel regulator of the kinase activities of CDKs. We believe that our findings from this study will significantly contribute to our understanding of cell cycle-associated diseases.
Introduction
The cell cycle is one of the most fundamental of biological processes. Cyclin-dependent kinases (CDKs)3 are central regulators of the mammalian cell cycle and cell proliferation (1). The CDKs are activated by interacting with cyclin regulatory subunits. Human CDK4 shares 90% sequence homology with CDK6. CDK4/6 are activated by D-type cyclins and function during early to middle G1 phase. CDK2-cyclin E and CDK2-cyclin B complexes function in late G1 phase and S phase, respectively. CDK1-cyclin B and CDK1-cyclinA complexes promote G2/M transition (1). In contrast to these cell cycle-associated CDKs, CDK5 is not a part of the core cell cycle machinery. CDK5 is activated by the neuron-specific p35 regulatory subunit and, therefore, functions in postmitotic neurons (2, 3).
It is well established that dysregulation of cell cycle/proliferation is one of the hallmarks of cancer cells (4). Dysregulation of the cell cycle or CDK5 is also associated with neurodegenerative diseases (5, 6). Therefore, elucidating the mechanisms underlying dysregulation of the cell cycle machinery in disease states is of critical importance.
Ubiquitin C-terminal hydrolase L1 (UCH-L1/PGP9.5) is normally expressed in restricted types of cells or organs, such as neurons, testes, and ovaries (7–9). The expression of UCH-L1 has also been observed in various human cancers, such as non-small-cell lung cancer, pancreatic cancer, invasive colorectal cancer, esophageal squamous cell carcinoma, myeloma, invasive breast cancer, parathyroid carcinoma, glioma cell lines, the acute lymphoblastic leukemia cell line, the chronic lymphocytic leukemia cell line, and Burkitt's lymphoma cell lines (10–19). This suggests that UCH-L1 is involved in cancer progression. Indeed, a recent study showed that UCH-L1 transgenic mice are prone to malignancy, primarily lymphomas and lung tumors (20).
The I93M mutation in UCH-L1 has been observed in a family with dominant inherited Parkinson's disease (21). We have shown that UCH-L1I93M transgenic mice exhibit a partial loss of dopaminergic neurons (22), indicating that the I93M mutation participates in the cause of familial Parkinson's disease. The E7A mutation in UCH-L1 has been reported recently as a causative mutation in a family with early-onset progressive neurodegeneration (23).
UCH-L1 has been reported to cleave small adducts, such as amino acids, from the C terminus of ubiquitin in a cell-free system. However, it is not able to hydrolase the Lys-48 isopeptide bond in vitro (24). Under in vitro conditions, UCH-L1 can also cleave ubiquitin gene products, α-linked ubiquitin oligomers (ubiquitin B, ubiquitin C), or α-linked ubiquitin fused to ribosomal protein S27a (ubiquitin A), albeit very slowly (24). UCH-L1 was also reported to act as a ubiquitin ligase in a cell-free system (25). Other than these enzymatic activities, we have shown that UCH-L1 stabilizes monoubiquitin through interaction with monoubiquitin (26). However, the molecular functions of UCH-L1 involving cancer progression or cell cycle control remain largely unknown.
In addition to monoubiquitin, we have shown that UCH-L1 physically interacts with multiple proteins, including α/β-tubulin, LAMP-2A, Hsc70, and Hsp90 (27–29). The levels of interactions of these proteins with UCH-L1C90S, which lacks hydrolase activity but maintains binding affinity for monoubiquitin, were not altered compared with that of UCH-L1WT (27, 28). From these observations, we hypothesized that UCH-L1 can function independently of a ubiquitin system. Another deubiquitinating enzyme, UBP43/USP18, is known to negatively regulate interferon signaling independently of its isopeptidase activity (30). In this study, we further screened for UCH-L1-interacting proteins to clarify the molecular functions of UCH-L1.
EXPERIMENTAL PROCEDURES
Plasmids
The pCI-neo-hUCH-L1 plasmid containing human WT UCH-L1 and UCH-L1 variants, with or without a FLAG tag, was prepared as described previously (27, 28) or generated using a QuikChange site-directed mutagenesis kit (Stratagene). The plasmids for expressing human CDK1, CDK2, CDK4, CDK5, cyclin A1, cyclin D1, p35, Rb, p27 (Kip1), and p21 (Cip1), with or without a FLAG tag, were constructed in a pCI-neo vector (Promega). The cDNA encoding these genes were purchased from Open Biosystems. The plasmids for expressing deletion mutants of UCH-L1 with a FLAG tag were constructed in pCI-neo. The plasmids for expressing the deletion mutants of UCH-L1 and CDK4 with a GFP tag were constructed in pEGFP-C1 (Clontech). The pET-CDK4 bacterial expression plasmids for expressing CDK4 with a His tag at the C terminus and an S tag at the N terminus were constructed by ligating the cDNAs encoding CDK4 into the pET-29b(+) vector (Novagen). The pCI-neo-CDK5 plasmid containing human D144N CDK5 was generated using a QuikChange site-directed mutagenesis kit.
Cell Culture and Transfection
Neuro2a, COS-7, and HeLa cells were cultured as described previously (31, 32). H727 and MCF7 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. NIH-3T3 cells stably expressing human UCH-L1 with a FLAG-HA double tag at the N terminus were cultured as described previously (27). Transient transfection of COS-7 cells with each vector was performed using the FuGENE 6 transfection reagent (Roche) as described previously (33). HeLa cells were transfected with plasmids using Lipofectamine LTX (Invitrogen) as described previously (34).
Antibody Array
An antibody array assay was performed using an antibody array (Hypromatrix) according to the protocols of the manufacturer. Briefly, lysate was prepared from NIH-3T3 cells stably expressing human UCH-L1 with a FLAG-HA double tag. The array containing 400 antibodies was incubated with lysate, and binding of UCH-L1 was detected using an anti-HA antibody conjugated to horseradish peroxidase (Sigma).
Immunoblotting
SDS-PAGE was performed under reducing conditions, and immunoblotting was performed according to standard procedures as described previously (35, 36). Anti-CDK4, anti-CDK5, anti-CDK6, anti-histone H1, anti-p35, and anti-cyclin D1 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-CDK1, anti-Rb, and anti-phospho-Rb (Ser-795) antibodies were from Cell Signaling Technology, Inc. Anti-CDK2 and anti-p27 antibodies were purchased from BD Biosciences. Anti-UCH-L1, anti-phospho-histone H1, anti-phospho-p27 (Ser-10), and anti-β-actin were from UltraClone, Upstate, Zymed Laboratories Inc., Invitrogen, and Sigma, respectively. An anti-DYKDDDDK (FLAG) antibody was from Wako.
Immunoprecipitation
Immunoprecipitation was performed as described previously (27).
Preparation of Recombinant Proteins and Pull-down Assay
Recombinant GST and GST-tagged UCH-L1 were prepared as described previously (27). Recombinant CDK4 with a His tag at the C terminus and an S tag at the N terminus was prepared as described previously (32).
A pull-down assay was performed according to the methods described previously (37, 38). Glutathione-Sepharose beads (GE Healthcare) were blocked with 3% BSA for 15 h to prevent nonspecific binding of proteins to the beads and washed three times with PBS containing 0.05% Triton X-100. Twenty microliters of glutathione beads blocked with BSA, 150 pmol of recombinant CDK4, and 100 pmol of recombinant GST-UCH-L1 or GST were mixed and incubated for 2 h in PBS containing 0.05% Triton X-100. After the beads were washed three times with PBS containing 0.05% Triton X-100, proteins were eluted with SDS sample buffer.
Assessment of Cell Proliferation
One day before transfection, cells were seeded at 1 × 104, 2 × 104 (HeLa and Neuro2a), or 4 × 104 (H727 and MCF7) cells/well in 24-well plates. After transfection with plasmids or siRNA, cells were incubated for the indicated time periods. Viable and dead cell numbers were counted using a Vi-CELL XR cell viability analyzer (Beckman Coulter). The relative number of viable cells was measured using a CellTiter-Glo luminescent cell viability assay (Promega). In some experiments, CDK4 inhibitor (219476, Merck Millipore) was added to cells.
The BrdU incorporation assay was performed using a cell proliferation ELISA BrdU kit (Roche). Briefly, HeLa cells transfected with UCH-L1 or an empty vector were incubated with BrdU for 3 h and then fixed. Cells were then immunostained with anti-UCH-L1 and anti-BrdU antibodies. After BrdU-positive cells were counted, the percentage of BrdU-positive cells among total cells transfected with empty vector or UCH-L1 was calculated.
In Vitro Kinase Assay
In vitro kinase assays of CDKs were performed according to the procedure of Kawauchi et al. (39). Purified CDK1-cyclin B1, CDK4-cyclin D1, CDK5-p35, histone H1, Rb, p27, and UCH-L1 were purchased from Upstate, Biaffin GmbH, Upstate, New England BioLabs, QED Bioscience, Santa Cruz Biotechnology, Inc., and Boston Biochem, respectively. Purified proteins were incubated in 50 μl of kinase buffer (50 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 200 μm ATP, 1 μm DTT, 0.1% BSA) at 30 °C for the indicated time and analyzed by immunoblotting.
CDK4 activity in cultured cells was measured according to the method described by Smith et al. (40). Briefly, CDK4 was immunoprecipitated from the cell lysate using anti-CDK4 antibody (Santa Cruz Biotechnology, Inc.) and protein G-Sepharose beads (GE Healthcare). Beads were then mixed with purified Rb in kinase buffer and incubated at 30 °C for 30 min. Phosphorylation levels of Rb were analyzed by immunoblotting.
siRNA Preparation and Transfection
Double-stranded siRNAs targeting UCH-L1, EGFP, and CDK4 were purchased from RNAi Co., Ltd. (Tokyo, Japan). Sequences targeted by siRNA were selected using siDirect (RNAi Co., Ltd) and were as follows: mouse UCH-L1 siRNA-A, 5′-GUU AGC CCU AAA GUU UAC UUC-3′ (sense) and 5′-AGU AAA CUU UAG GGC UAA CUU-3′ (antisense); mouse UCH-L1 siRNA-B, 5′-CUG AAG CCG AUG GAG AUU AAC-3′ (sense) and 5′-UAA UCU CCA UCG GCU UCA GCU-3′ (antisense); EGFP siRNA for mouse cells, 5′-GCC ACA ACG UCU AUA UCA UGG-3′ (sense) and 5′-AUG AUA UAG ACG UUG UGG CUG-3′ (antisense); human UCH-L1 siRNA-A, 5′-GAC AAG AAG UUA GUC CUA AAG-3′ (sense) and 5′-UUA GGA CUA ACU UCU UGU CCC-3′ (antisense); human UCH-L1 siRNA-B, 5′-GCA CAA UCG GAC UUA UUC ACG-3′ (sense) and 5′-UGA AUA AGU CCG AUU GUG CCA-3′ (antisense); EGFP siRNA for human cells, 5′-CAG CAC GAC UUC UUC AAG UCC-3′ (sense) and 5′-ACU UGA AGA AGU CGU GCU GCU-3′ (antisense); and human CDK4 siRNA, 5′-GUG CCA CAU CCC GAA CUG ACC-3′ (sense) and 5′-UCA GUU CGG GAU GUG GCA CAG-3′ (antisense). EGFP siRNAs were used as a control. Neuro2a cells were transfected with siRNA using X-treamGENE (Roche). H727 and MCF7 cells were transfected with siRNAs using Lipofectamine RNAiMAX (Invitrogen).
Xenograft Cancer Models
Male 5-week-old BALB/c nu/nu mice were purchased from CLEA Japan (Tokyo, Japan). These athymic nude mice were anesthetized by intraperitoneal injection of Somnopentyl (50 mg/kg) and inoculated with 3 × 107 MCF7 cells containing 50% Matrigel (BD Biosciences) into the left flank on day 0. After confirmation of tumor growth at 50 mm3 or more, the siRNAs were injected intratumorally into mice on days 14, 21, and 28. To achieve effective delivery, the siRNAs were mixed with atelocollagen (AteloGene Local Use, Koken, Tokyo, Japan). The siRNA-atelocollagen complexes were prepared according to the guidelines of the manufacturer (1.0 mg/kg body weight, ∼20 μg/200 μl/injection). Tumor size was measured from above the skin, and tumor volume was estimated by measuring in two directions and calculated by the following formula: tumor volume (mm3) = (length) × (width)2 × 0.5. Forty days after inoculation of the cells, tumors were dissected and weighed. All animal experiments were approved by the animal experimentation committee of the National Center of Neurology and Psychiatry.
Statistical Analysis
For comparisons between two groups, statistical difference was determined by Student's t test. For comparison of more than two groups, Dunnett's multiple comparison test or Fisher's protected least significant difference test was used. * and ** indicate p < 0.05 and p < 0.01, respectively.
RESULTS
UCH-L1 Interacts with CDK1, CDK4, CDK5, and CDK6
To clarify the molecular functions of UCH-L1, we screened for UCH-L1-interacting proteins using an array containing 400 antibodies (supplemental Table S1). The array was incubated with cell lysate containing HA-tagged UCH-L1, and binding of UCH-L1 was detected using an anti-HA antibody (Fig. 1A). Signal intensities of the spots with antibodies against CDK1, CDK2, CDK4, and CDK6 were higher than that of the average intensity of 400 spots, suggesting interactions of CDK family proteins with UCH-L1 (Fig. 1, A and B). To confirm the interactions between UCH-L1 and CDKs, coimmunoprecipitation assays were performed using COS-7 cells transiently expressed with FLAG-tagged UCH-L1. Endogenous CDK1, CDK4, CDK5, and CDK6 were coprecipitated with FLAG-tagged UCH-L1, whereas endogenous CDK2 was not (Fig. 1, C and D). We also detected the interaction of endogenous UCH-L1 with endogenous CDK1 or CDK4 using Neuro2a cells, which endogenously express UCH-L1 (Fig. 1E).
FIGURE 1.

UCH-L1 interacts with CDK1, CDK4, CDK5, and CDK6. A, an antibody array containing 400 antibodies was incubated with cell lysates expressing HA-tagged UCH-L1. UCH-L1 was detected using an anti-HA antibody. B, graphical representation of signal intensities detected in A. The plotted values represent the relative intensities of the spots with antibodies against CDKs (average intensity of 400 spots = 1). C and D, COS-7 cells were transfected with FLAG-tagged UCH-L1 (WT, I93M, or C90S) or an empty vector (control). Proteins were immunoprecipitated (IP) and analyzed by immunoblotting. E, interaction of endogenous UCH-L1 with CDK1 and CDK4. Cell lysates of Neuro2a cells were immunoprecipitated using the indicated antibodies and analyzed by immunoblotting.
We found that the levels of CDK1, CDK4, and CDK5 interacting with UCH-L1I93M are higher than the amount interacting with UCH-L1WT (2.3-fold, 3.1-fold, and 2.7-fold increases, respectively) (Fig. 1C). The interactions of CDK with UCH-L1C90S were not changed notably compared with those of UCH-L1WT (Fig. 1C). UCH-L1C90S, in which the active center cysteine residue is substituted with serine, exhibits no hydrolase activity and, presumably, possesses no ligase activity (25, 41). The hydrolase and ligase activities of UCH-L1I93M are reduced compared with those of UCH-L1WT (25, 41). Overexpression of UCH-L1C90S or UCH-L1I93M stabilizes monoubiquitin at levels comparable with that of UCH-L1WT (26, 27). Thus, the levels of interactions between UCH-L1 and CDKs are independent of the known ubiquitin-related functions of UCH-L1.
In contrast to endogenous CDK2, overexpressed CDK2 interacted with FLAG-tagged UCH-L1 (supplemental Fig. S1). We hypothesized that the endogenous interaction between UCH-L1 and CDK2 is below the limit of detection. Given that endogenous CDK2 did not interact with UCH-L1, we did not conduct any further analyses on CDK2.
UCH-L1 Enhances the Kinase Activity of CDK
We next tested whether UCH-L1 affects the kinase activities of CDKs. To examine the direct effects of UCH-L1 on kinase activities of CDKs, we first utilized a cell-free kinase assay. We used histone H1 as an in vitro substrate of CDK1-cyclin B, Rb for CDK4-cyclin D, and p27 for CDK5-p35 (39). Intriguingly, a kinase assay in which purified CDK1-cyclin B, histone H1, and UCH-L1 were mixed in vitro revealed that UCH-L1 enhanced the phosphorylation of histone H1 by CDK1-cyclin B (1.6-fold increase at 30 min) (Fig. 2A). Similarly, UCH-L1 enhanced the phosphorylation of Rb and p27 by CDK4-cyclin D and CDK5-P35, respectively, in a cell-free kinase assay (1.8-fold and 1.8-fold increases at 30 min, respectively) (Fig. 2, B and C). These results indicate that UCH-L1 enhances the kinase activity of CDK1, CDK4, and CDK5.
FIGURE 2.

UCH-L1 enhances kinase activity of CDK1, CDK4, and CDK5. A–C, in vitro kinase assay with purified proteins. Proteins were mixed and incubated as indicated, and the phosphorylation of substrates was analyzed by immunoblotting. D–I, cell-based kinase assay. COS-7 cells were cotransfected with the indicated plasmids. Phosphorylation of Rb (D and H) or p27 (E) was analyzed by immunoblot analysis. Relative levels of phospho-Rb and phospho-p27 were quantified and normalized against Rb and p27 levels, respectively (F, G, and I). Data are mean ± S.E. (F, n = 4; G, n = 3; I, n = 3). *, p < 0.05; **, p < 0.01; n.s., not significant.
We also assessed the effects of UCH-L1 on the kinase activity of CDK4 and CDK5 using a cell-based kinase assay (39). UCH-L1, CDK4, cyclin D, and Rb were coexpressed in COS-7 cells, and the phosphorylation levels of Rb were measured using immunoblotting. Consistent with the in vitro assay, UCH-L1 increased the phosphorylation levels of Rb (Fig. 2, D and F). In assays expressing UCH-L1, CDK5, p35, and p27, UCH-L1 was observed to also enhance the phosphorylation levels of p27 (Fig. 2, E and G). In the absence of overexpressed cyclin D (Fig. 2D, lanes 7–9) and p35 (E, lanes 4-6), UCH-L1 had no effect on the phosphorylation of Rb and p27, respectively, indicating that UCH-L1 increased the phosphorylation levels of Rb and p27 through CDK4-cyclin D or CDK5-p35. As a negative control for overexpression of UCH-L1, we also examined the effects of other proteins interacting with CDKs on kinase activity. We found that neurodegenerative disease-linked mutant SOD1 also interacts with CDK4 but that mutant SOD1 did not enhance CDK4 activity (supplemental Fig. S2, A and B).
To investigate the mechanism of how UCH-L1 enhances the kinase activity of CDK-cyclin, we used UCH-L1I93M and UCH-L1C90S in cell-based kinase assays. Compared with UCH-L1WT, UCH-L1I93M further elevated the phosphorylation of Rb (lanes 2 and 3 in Fig. 2, D and F) and p27 (lanes 2 and 3 in Fig. 2, E and G). UCH-L1C90S enhanced the phosphorylation of Rb at similar levels to UCH-L1WT (Fig. 2, H and I). Taken together with the results showing that UCH-L1I93M displays increased levels of interaction with CDKs (Fig. 1C), these results indicate that the enhancement of CDK activities by UCH-L1 is independent of the known ubiquitin-related functions of UCH-L1 but correlates with the interaction levels between UCH-L1 and CDKs.
Because CDKs are activated by interacting with cyclin regulatory subunits, we examined whether UCH-L1 augments the interaction between CDKs and cyclins. Coimmunoprecipitation assays showed that UCH-L1 does not affect interaction levels between CDK4 and cyclin D, CDK5 and p35, and CDK1 and cyclin B (supplemental Fig. S3, A–C). We also tested whether UCH-L1 competes with the CDK inhibitors p27 (Kip1) or p21 (Cip1) for binding to CDKs. UCH-L1 did not affect the level of interaction between CDK1 and p21, CDK1 and p27, CDK4 and p21, and CDK4 and p27 (supplemental Fig. S3, D and E), indicating that UCH-L1 does not compete with p27 or p21 for the binding to CDKs. Our results support the idea that UCH-L1 directly enhances the kinase activity of CDKs.
Interaction Regions between UCH-L1 and CDK4
We next assessed the interaction regions between UCH-L1 and CDKs to investigate the mechanism(s) of interaction of UCH-L1 with CDKs. Because CDKs share high sequence homology (CDK4 shares 73, 74, and 90% sequence homology with CDK1, CDK5, and CDK6, respectively) (supplemental Fig. S4) and UCH-L1 had similar effects on the enhancement of kinase activity of CDK1, CDK4, and CDK5 (Fig. 2), we focused on CDK4 and mainly used CDK4 in our experiments.
We investigated whether UCH-L1 directly interacts with CDK4 using a pull-down assay. Recombinant CDK4 was pulled down with recombinant UCH-L1 (Fig. 3A). This result indicates that UCH-L1 directly interacts with CDK4. To determine the region of UCH-L1 that interacts with CDK4, we performed coimmunoprecipitation assays using deletion mutants of UCH-L1 incorporating FLAG tags (Fig. 3B). CDK4 was found to interact with amino acid (aa) residues 60–223 or 100–223 in UCH-L1 but not with aa 1–150 (Fig. 3C), indicating that CDK4 interacted with a region within aa 151–223 of UCH-L1. We generated several deletion mutants of UCH-L1 with GFP and FLAG tags (Fig. 3D) and carried out coimmunoprecipitation assays. CDK4 interacted with aa 148–190, 160–190, 148–223, and 160–223 but not with aa 188–223 of UCH-L1 (Fig. 3E). These results indicate that CDK4 mainly interacts with aa 160–190 of UCH-L1. Aa 160–190 are distinct from Asp-30, which is required for monoubiquitin binding (Fig. 3F). This is consistent with results showing that UCH-L1 CDK potentiation activity is independent of the known ubiquitin-related functions of UCH-L1.
FIGURE 3.

CDK4 interacts with aa 160–190 of UCH-L1, which are distinct from the ubiquitin-binding region. A, direct interaction between UCH-L1 and CDK4. Recombinant GST or GST-tagged UCH-L1 and His-tagged recombinant CDK4 were mixed and pulled down using glutathione beads. Proteins were analyzed by immunoblotting with an anti-CDK4 antibody. B and D, schematic representation of deletion mutants of UCH-L1. C and E, COS-7 cells transfected with the indicated constructs or empty vector (control in C). Proteins were immunoprecipitated and analyzed by immunoblotting. F, structural model for human UCH-L1. Cys-90 is shown in blue, Asp-30 in magenta, and the CDK-4-interacting-region (aa 160–190) in yellow. The structural model of the NCBI (accession no. 38174) was used.
Coimmunoprecipitation assays using a series of alanine substitutions of UCH-L1 (39 mutants) revealed that the R63A, E174A, D176A, and H185A mutations notably increase interactions with CDK4 (supplemental Fig. S5A). Glu-174, Asp-176, and His-185 are within aa 148–190, with Arg-63 located adjacent to this region (supplemental Fig. S5B), supporting the notion that CDK4 interacts with aa 160–190 of UCH-L1.
To determine the region of CDK4 that interacts with UCH-L1, we performed coimmunoprecipitation assays using several deletion mutants of CDK4 (Fig. 4A). UCH-L1 interacted with aa 151–218 of CDK4 but not with aa 2–80 and 219–294 of CDK4 (Fig. 4B). UCH-L1 interacted only slightly with aa 81–150 (Fig. 4B). The interacting region, aa 151–218, of CDK4 contains a T-loop (Fig. 4A) that conformationally regulates CDKs kinase activity (42), suggesting that UCH-L1 may affect the conformation of CDKs in enhancing kinase activity. This region of CDK4 shares a particularly high homology: 82, 84, and 97% sequence homology with the corresponding regions of CDK1, CDK5, and CDK6, respectively (supplemental Fig. S4).
FIGURE 4.
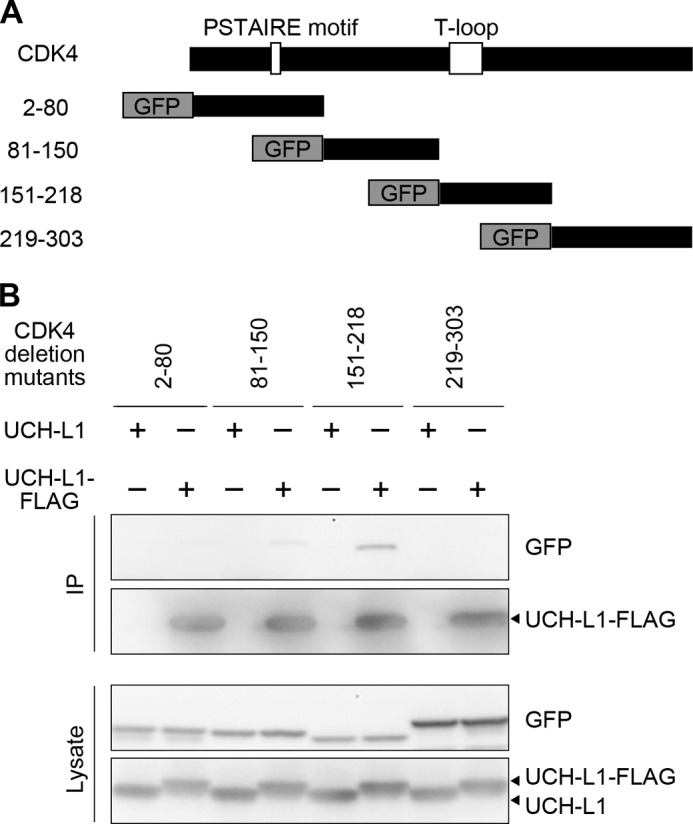
UCH-L1 interacts with aa 151–218 of CDK4, which contains a T-loop. A, schematic representation of deletion mutants of CDK4. B, COS-7 cells were cotransfected with the indicated plasmids. Proteins were immunoprecipitated (IP) and analyzed by immunoblotting.
Overexpression of UCH-L1 Enhances Cell Proliferation
Given that UCH-L1 augments the kinase activity of CDK1 and CDK4, we hypothesized that UCH-L1 enhances cell proliferation. We assessed the effects of UCH-L1 overexpression on the proliferation of HeLa cells that do not express detectable levels of UCH-L1 (43). Compared with transfection of the empty vector, overexpression of UCH-L1 significantly increased viable cell numbers without affecting cell death (Fig. 5A), indicating that overexpression of UCH-L1 enhances proliferation in these cells. When relative viable cell numbers were measured by ATP-based cell viability assays, overexpression of UCH-L1 in HeLa cells also increased the viable cell numbers compared with control transfections (Fig. 5B). Therefore, we used this assay for the measurement of viable cell numbers in several experiments. We examined the effect of UCH-L1 on the proliferation of COS-7 cells, which also do not express detectable levels of UCH-L1 (25). In these cells, overexpression of UCH-L1 enhanced cell proliferation (supplemental Fig. S6, A and B).
FIGURE 5.

UCH-L1 enhances cell proliferation. A, B, and D, HeLa cells were transfected with UCH-L1 or an empty vector (control). At 48 h post-transfection, viable and dead cell numbers were counted (A), and relative numbers of viable cells were measured using an ATP-based cell viability assay (B). At 22 h post-transfection, cells were incubated with BrdU for 3 h, and BrdU-positive cells were counted (D). BrdU-positive cell numbers area expressed as the percentage of total cells counted. Data are mean ± S.E. (n = 4). n.s., not significant. C, HeLa cells were transfected with UCH-L1 or empty vector. Zero hours (day 1), 24 h (day 2), and 48 h (day 3) after transfection, relative numbers of viable cells were measured. Data are mean ± S.E. (n = 4). E, HeLa cells were transfected with UCH-L1 (WT, I93M, or C90S) or control vector, and relative numbers of viable cells were measured 48 h after transfection. Data are mean ± S.E. (n = 4). F, HeLa cells were incubated for 48 h with conditioned medium of HeLa cells transfected with UCH-L1 or empty vector, and relative numbers of viable cells were measured 48 h after transfection. Data are mean ± S.E. (n = 4). G, HeLa cells were cotransfected with the indicated plasmids. At 48 h post-transfection, proteins were immunoprecipitated (IP) and analyzed by immunoblotting. H and I, ectopic expression of CDK5 inhibits UCH-L1-induced enhancement of cell proliferation. HeLa cells were cotransfected with the indicated plasmids. At 48 h post-transfection, viable cell numbers were measured. Data are mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01; n.s., not significant.
For further confirmation of the effect of UCH-L1 on cell proliferation, we examined proliferation over time and BrdU incorporation in HeLa cells. In the time course proliferation assay, UCH-L1 enhanced proliferation of HeLa cells (p < 0.01, two-way analysis of variance) (Fig. 5C). Consistent with the enhancement of proliferation, overexpression of UCH-L1 increased the incorporation of BrdU (Fig. 5D).
To investigate whether enhancement of the kinase activity of CDKs is involved in the enhancement of cell proliferation by UCH-L1, we used UCH-L1I93M and UCH-L1C90S in proliferation assays. Both enhanced proliferation (Fig. 5E). UCH-L1I93M had a tendency to enhance proliferation compared with UCH-L1WT (p < 0.05, Student's t test), whereas UCH-L1C90S did not significantly enhance proliferation compared with UCH-L1WT (p > 0.05, Student's t test). Together with the result showing that UCH-L1I93M further enhanced kinase activities of CDKs compared with UCH-L1WT (Fig. 2), enhancement of proliferation by UCH-L1 is independent of the known ubiquitin-related functions of UCH-L1 and correlates with the levels of enhancement of CDK4 by UCH-L1. These results suggest that enhancement of cell proliferation by UCH-L1 is mediated by CDKs. We investigated this further by observing the effects of UCH-L1 overexpression on the proliferation of cells treated with CDK4 inhibitors and in cells where CDK4 expression was knocked down (supplemental Fig. S7). We observed that UCH-L1 did not significantly affect proliferation (supplemental Fig. S7).
To exclude the possibility that UCH-L1 enhances cell proliferation through humoral factors, we tested the effect of conditioned media from UCH-L1-overexpressed cells on proliferation. Treatment of cells with conditioned medium from cells overexpressing UCH-L1 had no effect on proliferation compared with the control conditioned medium (Fig. 5F). This indicates that enhancement of cell proliferation by UCH-L1 is not mediated by alternating humoral factors such as increasing growth factors and decreasing growth inhibition factors.
Ectopic Expression of CDK5 Attenuates the Interaction between UCH-L1 and CDK4 and Inhibits the Enhancement of Proliferation by UCH-L1
Because of high sequence homology among the CDKs, we assumed that ectopic expression of CDK5 may result in the inhibition of interactions of UCH-L1 with cell cycle-associated CDKs. It may also inhibit the enhancement of proliferation by UCH-L1. We observed that overexpression of CDK5 attenuated the interaction between overexpressed CDK4 and UCH-L1 (1.5-fold decrease) (Fig. 5G, lanes 2 and 4). In HeLa cells overexpressing CDK5, overexpression of UCH-L1 did not enhance cell proliferation (Fig. 5H). To exclude the possibility of detrimental effects from the overexpression of CDK5, we used a kinase-dead CDK5 (D144N) (44). We confirmed that CDK5D144N interacted with UCH-L1 at a level comparable with that with CDK5WT (supplemental Fig. S8). In cells overexpressing CDK5D144N, overexpression of UCH-L1 did not enhance proliferation (Fig. 5I). These results strongly suggest that UCH-L1 enhances cell proliferation through interaction with cell cycle-associated CDKs.
Knockdown of UCH-L1 Reduces Cell Proliferation
To assess the role of endogenous UCH-L1 on CDK activity and cell proliferation, we examined the loss of functional effects of UCH-L1 using siRNA in the mouse neuroblastoma cell line Neuro2a, which endogenously expresses UCH-L1. We used two different siRNA sequences for knockdown of mouse UCH-L1. Both siRNAs decreased protein levels of UCH-L1 to ∼25% (Fig. 6A). Knockdown of UCH-L1 by each siRNA significantly decreased the levels of cell proliferation in Neuro2a cells without increasing cell death (Fig. 6B). As a control, we tested the effects of transfecting UCH-L1 siRNAs on proliferation of HeLa cells, as these do not express detectable levels of UCH-L1. Proliferation rates were not reduced in these HeLa cells (supplemental Fig. S9). We tested the effect of UCH-L1 knockdown on the kinase activity of CDK4. CDK4 was immunoprecipitated from Neuro2a cells in which UCH-L1 was knocked down and subjected to in vitro kinase assays. Knockdown of UCH-L1 by each siRNA reduced kinase activity (Fig. 6, C and D). We confirmed that the total levels of cyclin D were unaltered in cells where UCH-L1 expression was knocked down (Fig. 6A). These results indicate that reduced CDK4 activity in vitro is not a consequence of reduced proliferation and that endogenous UCH-L1 also enhances the kinase activity of CDK4 and cell proliferation.
FIGURE 6.

Knockdown of UCH-L1 reduces cell proliferation and kinase activity of CDK4. A and B, Neuro2a cells were transfected with the indicated siRNA, incubated for 72 h, and analyzed by immunoblot analysis (A). Viable and dead cell numbers were counted (B). Data are mean ± S.E. (n = 4). C and D, CDK4 was immunoprecipitated from Neuro2a cells transfected with the indicated siRNA and subjected to kinase assays (C). Relative levels of phospho-Rb were quantified and normalized against Rb levels. Data are mean ± S.E. (n = 3). E–H, human cancer cell lines H727 and MCF7 were transfected with the indicated siRNA. After cells were incubated for 4 days (H727) or 6 days (MCF7), lysates were analyzed by immunoblotting (E and G), and viable and dead cell numbers were counted (F and H). Data are mean ± S.E. (F, n = 10; H, n = 4). *, p < 0.05; **, p < 0.01.
We also examined the role of endogenous UCH-L1 on the proliferation of human cancer cells that endogenously express UCH-L1. We prepared two different siRNA sequences for knockdown of human UCH-L1. In a human lung carcinoid cell line, H727, hUCH-L1 siRNA-A and siRNA-B decreased protein levels of UCH-L1 to 46 and 58%, respectively (Fig. 6E). Knockdown of UCH-L1 by each siRNA reduced the viable cell number without increasing cell death, indicating that knockdown of UCH-L1 reduces the proliferation of H727 cells (Fig. 6F). Because both inhibitory effects on proliferation and knockdown efficiency of siRNA-B were high compared with those of siRNA-A, we used siRNA-B for the following experiments. In the human breast adenocarcinoma cell line MCF7, knockdown of UCH-L1 also reduced viable cell numbers without increasing cell death, indicating that knockdown of UCH-L1 reduces proliferation of MCF7 cells (Fig. 6, G and H). We confirmed that siRNA-B did not reduce proliferation levels of HeLa cells (supplemental Fig. S9). Thus, endogenous UCH-L1 enhances proliferation of certain types of human cancer cells.
Local Administration of UCH-L1 siRNA into a Subcutaneous Xenograft Tumor Reduced Tumor Growth
To examine whether endogenous UCH-L1 also enhances proliferation of tumor cells in vivo, we performed subcutaneous xenograft studies. MCF7 cells (3 × 107) were inoculated into nude mice. After confirmation of tumor formation, siRNAs were delivered into tumors once a week over 3 weeks (Fig. 7A). Measurement of the width of tumors and estimation of tumor volume over time showed that administration of UCH-L1 siRNA significantly reduced tumor growth compared with that of control siRNA (p < 0.05, two-way analysis of variance) (Fig. 7, B and C). Measurement of the wet weight of tumors also demonstrated that administration of UCH-L1 siRNA significantly reduced tumor growth (Fig. 7, D and E).
FIGURE 7.

RNAi of UCH-L1 reduced growth of xenograft tumors. A, experimental paradigm for xenograft studies. MCF7 cells (3 × 107) were inoculated into nude mice on day 0. After confirmation of tumor formation, siRNAs were delivered into tumors on days 14, 21, and 28. B, tumor volume estimated from width of tumors. Tumor size was measured from above the skin on days 14, 21, 28, 35, and 40. Data are mean ± S.E. (n = 5). C, representative appearance of mice with xenograft tumors (day 40). D, morphology of excised tumors (day 40). E, wet weight of xenograft tumors (day 40). Data are mean ± S.E. (n = 5) *, p < 0.05.
To exclude the possibility that UCH-L1 siRNA reduced tumor growth via alterations in the immune system, splenocytes were isolated from siRNA-treated nude mice, and T cell-mediated cytotoxicity was measured. We observed that UCH-L1 siRNA did not affect T cell-mediated toxicity (supplemental Fig. S10). UCH-L1 siRNA did not affect body weight or numbers of red blood cells and white cells in mice with xenograft tumors compared with control siRNA (supplemental Fig. S10).
DISCUSSION
Enhancement of CDK Activity by UCH-L1
In this study, we have shown that UCH-L1 physically interacts with CDK1, CDK4, and CDK5 and enhances CDKs activity. Activation of CDKs requires cyclin binding and phosphorylation of a conserved threonine residue by the CDK-activating kinase (45). In contrast, phosphorylation of a conserved threonine-tyrosine pair in CDKs or binding of CDK inhibitors is known to block kinase activity (45). UCH-L1 enhances kinase activity of CDKs in a cell-free system containing only buffer and purified proteins (Fig. 2, A–C). UCH-L1 did not compete with the CDK inhibitors p27 (Kip1) or p21 (Cip1) for binding to CDKs (supplemental Fig. S3). UCH-L1 is not a kinase nor a phosphatase, indicating that UCH-L1 enhances kinase activity independently of phosphorylation of CDKs and CDK inhibitors. In the cell-based kinase assay, UCH-L1 enhanced CDK activity only in the presence of overexpressed cyclin or p35 (Fig. 2, D and E). UCH-L1 does not modify the interaction levels between CDKs and cyclin (supplemental Fig. S3). Thus, UCH-L1 does not activate CDKs but potentiates CDK/cyclin activity. To our knowledge, this is the first report of a potentiator of CDKs/cyclins.
Using UCH-L1 mutants, we have shown that the enhancement of CDK activities by UCH-L1 is independent of the known ubiquitin-related functions of UCH-L1 but correlates with interaction levels between UCH-L1 and CDKs. Compared with UCH-L1WT, UCH-L1I93M further enhanced CDK4 activity and interacted with CDKs at higher levels (Figs. 1C and 2, D–G). UCH-L1C90S interacted with CDKs and enhanced CDK4 activity at levels comparable with UCH-L1WT (Figs. 1C and 2, H and I). Similarly, another deubiquitinating enzyme, UBP43/USP18, is known to negatively regulate interferon signaling independently of its isopeptidase activity (30).
The precise mechanism(s) of how UCH-L1 enhances CDKs activities is currently unknown. Because UCH-L1 interacts with a region of CDK4 containing a T-loop (Fig. 4, A and B), which conformationally regulates kinase activity of CDKs (42), UCH-L1 possibly affects the conformation of CDKs in enhancing kinase activity. However, further structural studies are needed to elucidate this point.
Enhancement of Proliferation by UCH-L1
We have shown that overexpression of UCH-L1 enhances proliferation of two cell lines, HeLa and COS-7, both of which do not express UCH-L1 (Fig. 5 and supplemental Fig. S6). Knockdown of UCH-L1 reduced proliferation in three cell lines, Neuro-2a, H727, and MCF7, all of which endogenously express UCH-L1 (Fig. 6). These results indicate that UCH-L1 enhances proliferation in certain types of cells. UCH-L1 is expressed in restricted types of cells or organs, including neurons, testes, and ovaries. Previous studies have shown that UCH-L1 is expressed in spermatogonial cells and that the levels of proliferation of spermatogonial cells in UCH-L1-deficient mice (gad mice) are decreased compared with that in wild-type mice (8, 46). This indicates that UCH-L1 is required for the normal proliferation of these cells in vivo, and our results are consistent with these findings.
The following observations indicate that enhancement of proliferation by UCH-L1 is at least partly mediated by enhancement of CDK1 and CDK4 activities by UCH-L1 (supplemental Fig. S11). First, UCH-L1 enhances CDK1 and CDK4 activities (Fig. 2), which are known to be essential for proliferation and to enhance proliferation, respectively. CDK1 is the only essential CDK for the mammalian cell cycle (47). CDK4-deficient mice are small, and mouse embryonic fibroblasts derived from these mice exhibit a delayed S phase re-entry (48). Second, in cells overexpressing CDK5, which attenuates the interaction of UCH-L1 with CDK4 and, presumably, with CDK1, overexpression of UCH-L1 did not alter proliferation (Fig. 5H). Third, enhancement of proliferation by UCH-L1 is independent of the known ubiquitin-related functions of UCH-L1 but correlates with the interaction levels between UCH-L1 and CDKs and with the enhanced levels of CDK4 by UCH-L1 (Figs. 1C; 2, D–I; and 5E). Fourth, in cells treated with CDK4 inhibitors or where CDK4 expression was knocked down, UCH-L1 did not enhance proliferation (supplemental Fig. S7). Fifth, the enhancement of cell proliferation by UCH-L1 is not mediated by alterations in humoral factors (Fig. 5F).
However, this does not necessarily rule out the possibility that enhancement of proliferation via UCH-L1 is partially mediated by other mechanisms. Other than CDKs, UCH-L1 has been reported to interact with JAB1, a protein involved in cell proliferation (49).
Roles of UCH-L1 in Human Diseases
Dysregulation of the cell cycle and proliferation is a hallmark of cancer cells. Although UCH-L1 is expressed in restricted types of cells or organs, UCH-L1 has been reported to be expressed in various human cancers. In vitro and in vivo studies have shown that UCH-L1 enhances the invasive potential of tumor cells (50). Another study showed that UCH-L1 transgenic mice are prone to malignancy, primarily lymphomas and lung tumors (20). UCH-L1 enhances proliferation in premalignant tissues of UCH-L1-c-myc double transgenic mice (20). Thus, in certain types of tumors, UCH-L1 functions as an oncogenic protein rather than as a tumor suppresser. Consistent with this notion, we have shown that endogenous UCH-L1 enhances proliferation of tumor cells both in vitro and in vivo. This is the first report to show that endogenous UCH-L1 in cancer cells enhances proliferation in vivo.
In contrast to the expression of UCH-L1 in various cancers, the UCHL1 gene is silenced in human colorectal and ovarian cancers and renal cell carcinoma via promoter methylation (51, 52). Transfection of UCH-L1 in renal cancer cell lines resulted in growth inhibition, suggesting that UCHL1 functions as a tumor suppressor gene in these cells (52). Taken together, these observations indicate that UCH-L1 possesses a dual role as an oncogenic protein and a tumor suppressor. The molecular mechanisms to describe how UCH-L1 functions in opposite ways are currently unknown and require further investigation.
Dysregulation of the cell cycle is also associated with neurodegenerative diseases. A number of laboratories have reported a correlation between neuronal loss and the appearance of cell cycle-related proteins, including CDK4, in the brains of Alzheimer's disease patients (5, 53–57). Mitosis-associated proteins have also been reported to be aberrantly expressed in dopaminergic neurons in patients with Parkinson's disease (6). In a mouse model of Parkinson's disease, CDK5 has been shown to be a mediator of dopaminergic neuronal loss (40). We found that UCH-L1I93M further elevates the kinase activity of CDK4 and CDK5 compared with UCH-L1WT (Fig. 2, D–G). Whether enhancement of CDK activities by UCH-L1 is involved in the pathogenesis of neurodegenerative diseases remains an interesting issue to be resolved. We believe that our findings in this study will significantly contribute to our understanding of cell cycle-associated diseases.
This work was supported by grants-in-aid for scientific research on nervous and mental disorders from the Ministry of Health, Labour, and Welfare of Japan (to T. K.); by grants-in-aid for young scientists (to T. K.) and grants-in-aid for scientific research (to K. W.) of the Japan Society for the Promotion of Science; by a Research grant in priority area research of the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to K. W.); by grants-in-aid for scientific research of the Ministry of Health, Labour, and Welfare, Japan (to K. W.) and the by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO), Japan (to K. W.).

This article contains supplemental Figs. S1–S11 and Table S1.
- CDK
- cyclin-dependent kinase
- UCH-L1
- ubiquitin C-terminal hydrolase L1
- EGFP
- enhanced GFP
- aa
- amino acid(s).
REFERENCES
- 1. Morgan D. O. (1997) Cyclin-dependent kinases. Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291 [DOI] [PubMed] [Google Scholar]
- 2. Lew J., Huang Q. Q., Qi Z., Winkfein R. J., Aebersold R., Hunt T., Wang J. H. (1994) A brain-specific activator of cyclin-dependent kinase 5. Nature 371, 423–426 [DOI] [PubMed] [Google Scholar]
- 3. Nikolic M., Dudek H., Kwon Y. T., Ramos Y. F., Tsai L. H. (1996) The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 10, 816–825 [DOI] [PubMed] [Google Scholar]
- 4. Hartwell L. H., Kastan M. B. (1994) Cell cycle control and cancer. Science 266, 1821–1828 [DOI] [PubMed] [Google Scholar]
- 5. Yang Y., Mufson E. J., Herrup K. (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J. Neurosci. 23, 2557–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Höglinger G. U., Breunig J. J., Depboylu C., Rouaux C., Michel P. P., Alvarez-Fischer D., Boutillier A. L., Degregori J., Oertel W. H., Rakic P., Hirsch E. C., Hunot S. (2007) The pRb/E2F cell-cycle pathway mediates cell death in Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 104, 3585–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilkinson K. D., Lee K. M., Deshpande S., Duerksen-Hughes P., Boss J. M., Pohl J. (1989) The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 246, 670–673 [DOI] [PubMed] [Google Scholar]
- 8. Kwon J., Wang Y. L., Setsuie R., Sekiguchi S., Sakurai M., Sato Y., Lee W. W., Ishii Y., Kyuwa S., Noda M., Wada K., Yoshikawa Y. (2004) Developmental regulation of ubiquitin C-terminal hydrolase isozyme expression during spermatogenesis in mice. Biol. Reprod. 71, 515–521 [DOI] [PubMed] [Google Scholar]
- 9. Sekiguchi S., Kwon J., Yoshida E., Hamasaki H., Ichinose S., Hideshima M., Kuraoka M., Takahashi A., Ishii Y., Kyuwa S., Wada K., Yoshikawa Y. (2006) Localization of ubiquitin C-terminal hydrolase L1 in mouse ova and its function in the plasma membrane to block polyspermy. Am. J. Pathol. 169, 1722–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howell V. M., Gill A., Clarkson A., Nelson A. E., Dunne R., Delbridge L. W., Robinson B. G., Teh B. T., Gimm O., Marsh D. J. (2009) Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. J. Clin. Endocrinol. Metab. 94, 434–441 [DOI] [PubMed] [Google Scholar]
- 11. Tezel E., Hibi K., Nagasaka T., Nakao A. (2000) PGP9.5 as a prognostic factor in pancreatic cancer. Clin. Cancer Res. 6, 4764–4767 [PubMed] [Google Scholar]
- 12. Yamazaki T., Hibi K., Takase T., Tezel E., Nakayama H., Kasai Y., Ito K., Akiyama S., Nagasaka T., Nakao A. (2002) PGP9.5 as a marker for invasive colorectal cancer. Clin. Cancer Res. 8, 192–195 [PubMed] [Google Scholar]
- 13. Hibi K., Westra W. H., Borges M., Goodman S., Sidransky D., Jen J. (1999) PGP9.5 as a candidate tumor marker for non-small-cell lung cancer. Am. J. Pathol. 155, 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyoshi Y., Nakayama S., Torikoshi Y., Tanaka S., Ishihara H., Taguchi T., Tamaki Y., Noguchi S. (2006) High expression of ubiquitin carboxy-terminal hydrolase-L1 and -L3 mRNA predicts early recurrence in patients with invasive breast cancer. Cancer Sci. 97, 523–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takase T., Hibi K., Yamazaki T., Nakayama H., Taguchi M., Kasai Y., Ito K., Akiyama S., Nagasaka T., Nakao A. (2003) PGP9.5 overexpression in esophageal squamous cell carcinoma. Hepatogastroenterology 50, 1278–1280 [PubMed] [Google Scholar]
- 16. Otsuki T., Yata K., Takata-Tomokuni A., Hyodoh F., Miura Y., Sakaguchi H., Hatayama T., Hatada S., Tsujioka T., Sato Y., Murakami H., Sadahira Y., Sugihara T. (2004) Expression of protein gene product 9.5 (PGP9.5)/ubiquitin-C-terminal hydrolase 1 (UCHL-1) in human myeloma cells. Br. J. Haematol. 127, 292–298 [DOI] [PubMed] [Google Scholar]
- 17. Giambanco I., Bianchi R., Ceccarelli P., Pula G., Sorci G., Antonioli S., Bocchini V., Donato R. (1991) “Neuron-specific” protein gene product 9.5 (PGP 9.5) is also expressed in glioma cell lines and its expression depends on cellular growth state. FEBS Lett. 290, 131–134 [DOI] [PubMed] [Google Scholar]
- 18. Ovaa H., Kessler B. M., Rolén U., Galardy P. J., Ploegh H. L., Masucci M. G. (2004) Activity-based ubiquitin-specific protease (USP) profiling of virus-infected and malignant human cells. Proc. Natl. Acad. Sci. U.S.A. 101, 2253–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al-Katib A. M., Mohammad R. M., Maki A., Smith M. R. (1995) Induced expression of a ubiquitin COOH-terminal hydrolase in acute lymphoblastic leukemia. Cell Growth & Differ. 6, 211–217 [PubMed] [Google Scholar]
- 20. Hussain S., Foreman O., Perkins S. L., Witzig T. E., Miles R. R., van Deursen J., Galardy P. J. (2010) The de-ubiquitinase UCH-L1 is an oncogene that drives the development of lymphoma in vivo by deregulating PHLPP1 and Akt signaling. Leukemia 24, 1641–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leroy E., Boyer R., Auburger G., Leube B., Ulm G., Mezey E., Harta G., Brownstein M. J., Jonnalagada S., Chernova T., Dehejia A., Lavedan C., Gasser T., Steinbach P. J., Wilkinson K. D., Polymeropoulos M. H. (1998) The ubiquitin pathway in Parkinson's disease. Nature 395, 451–452 [DOI] [PubMed] [Google Scholar]
- 22. Setsuie R., Wang Y. L., Mochizuki H., Osaka H., Hayakawa H., Ichihara N., Li H., Furuta A., Sano Y., Sun Y. J., Kwon J., Kabuta T., Yoshimi K., Aoki S., Mizuno Y., Noda M., Wada K. (2007) Dopaminergic neuronal loss in transgenic mice expressing the Parkinson's disease-associated UCH-L1 I93M mutant. Neurochem. Int. 50, 119–129 [DOI] [PubMed] [Google Scholar]
- 23. Bilguvar K., Tyagi N. K., Ozkara C., Tuysuz B., Bakircioglu M., Choi M., Delil S., Caglayan A. O., Baranoski J. F., Erturk O., Yalcinkaya C., Karacorlu M., Dincer A., Johnson M. H., Mane S., Chandra S. S., Louvi A., Boggon T. J., Lifton R. P., Horwich A. L., Gunel M. (2013) Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 3489–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Larsen C. N., Krantz B. A., Wilkinson K. D. (1998) Substrate specificity of deubiquitinating enzymes. Ubiquitin C-terminal hydrolases. Biochemistry 37, 3358–3368 [DOI] [PubMed] [Google Scholar]
- 25. Liu Y., Fallon L., Lashuel H. A., Liu Z., Lansbury P. T., Jr. (2002) The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell 111, 209–218 [DOI] [PubMed] [Google Scholar]
- 26. Osaka H., Wang Y. L., Takada K., Takizawa S., Setsuie R., Li H., Sato Y., Nishikawa K., Sun Y. J., Sakurai M., Harada T., Hara Y., Kimura I., Chiba S., Namikawa K., Kiyama H., Noda M., Aoki S., Wada K. (2003) Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum. Mol. Genet. 12, 1945–1958 [DOI] [PubMed] [Google Scholar]
- 27. Kabuta T., Setsuie R., Mitsui T., Kinugawa A., Sakurai M., Aoki S., Uchida K., Wada K. (2008) Aberrant molecular properties shared by familial Parkinson's disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum. Mol. Genet. 17, 1482–1496 [DOI] [PubMed] [Google Scholar]
- 28. Kabuta T., Furuta A., Aoki S., Furuta K., Wada K. (2008) Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 283, 23731–23738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kabuta T., Wada K. (2008) Insights into links between familial and sporadic Parkinson's disease. Physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 4, 827–829 [DOI] [PubMed] [Google Scholar]
- 30. Malakhova O. A., Kim K. I., Luo J. K., Zou W., Kumar K. G., Fuchs S. Y., Shuai K., Zhang D. E. (2006) UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 25, 2358–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kabuta T., Suzuki Y., Wada K. (2006) Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J. Biol. Chem. 281, 30524–30533 [DOI] [PubMed] [Google Scholar]
- 32. Kabuta T., Kinugawa A., Tsuchiya Y., Kabuta C., Setsuie R., Tateno M., Araki T., Wada K. (2009) Familial amyotrophic lateral sclerosis-linked mutant SOD1 aberrantly interacts with tubulin. Biochem. Biophys. Res. Commun. 387, 121–126 [DOI] [PubMed] [Google Scholar]
- 33. Higashi S., Tsuchiya Y., Araki T., Wada K., Kabuta T. (2010) TDP-43 physically interacts with amyotrophic lateral sclerosis-linked mutant CuZn superoxide dismutase. Neurochem. Int. 57, 906–913 [DOI] [PubMed] [Google Scholar]
- 34. Higashi S., Iseki E., Minegishi M., Togo T., Kabuta T., Wada K. (2010) GIGYF2 is present in endosomal compartments in the mammalian brains and enhances IGF-1-induced ERK1/2 activation. J. Neurochem. 115, 423–437 [DOI] [PubMed] [Google Scholar]
- 35. Kabuta T., Hakuno F., Asano T., Takahashi S. (2002) Insulin receptor substrate-3 functions as transcriptional activator in the nucleus. J. Biol. Chem. 277, 6846–6851 [DOI] [PubMed] [Google Scholar]
- 36. Fujiwara Y., Furuta A., Kikuchi H., Aizawa S., Hatanaka Y., Konya C., Uchida K., Yoshimura A., Tamai Y., Wada K., Kabuta T. (2013) Discovery of a novel type of autophagy targeting RNA. Autophagy 9, 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kabuta T., Take K., Kabuta C., Hakuno F., Takahashi S. (2008) Differential subcellular localization of insulin receptor substrates depends on C-terminal regions and importin beta. Biochem. Biophys. Res. Commun. 377, 741–746 [DOI] [PubMed] [Google Scholar]
- 38. Kabuta T., Hakuno F., Cho Y., Yamanaka D., Chida K., Asano T., Wada K., Takahashi S. (2010) Insulin receptor substrate-3, interacting with Bcl-3, enhances p50 NF-κB activity. Biochem. Biophys. Res. Commun. 394, 697–702 [DOI] [PubMed] [Google Scholar]
- 39. Kawauchi T., Chihama K., Nabeshima Y., Hoshino M. (2006) Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat. Cell Biol. 8, 17–26 [DOI] [PubMed] [Google Scholar]
- 40. Smith P. D., Crocker S. J., Jackson-Lewis V., Jordan-Sciutto K. L., Hayley S., Mount M. P., O'Hare M. J., Callaghan S., Slack R. S., Przedborski S., Anisman H., Park D. S. (2003) Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 13650–13655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishikawa K., Li H., Kawamura R., Osaka H., Wang Y. L., Hara Y., Hirokawa T., Manago Y., Amano T., Noda M., Aoki S., Wada K. (2003) Alterations of structure and hydrolase activity of parkinsonism-associated human ubiquitin carboxyl-terminal hydrolase L1 variants. Biochem. Biophys. Res. Commun. 304, 176–183 [DOI] [PubMed] [Google Scholar]
- 42. Jeffrey P. D., Russo A. A., Polyak K., Gibbs E., Hurwitz J., Massagué J., Pavletich N. P. (1995) Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320 [DOI] [PubMed] [Google Scholar]
- 43. Mann D. A., Trowern A. R., Lavender F. L., Whittaker P. A., Thompson R. J. (1996) Identification of evolutionary conserved regulatory sequences in the 5′ untranscribed region of the neural-specific ubiquitin C-terminal hydrolase (PGP9.5) gene. J. Neurochem. 66, 35–46 [DOI] [PubMed] [Google Scholar]
- 44. Kaminosono S., Saito T., Oyama F., Ohshima T., Asada A., Nagai Y., Nukina N., Hisanaga S. (2008) Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J. Neurosci. 28, 8747–8755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morgan D. O. (1995) Principles of CDK regulation. Nature 374, 131–134 [DOI] [PubMed] [Google Scholar]
- 46. Kwon J., Kikuchi T., Setsuie R., Ishii Y., Kyuwa S., Yoshikawa Y. (2003) Characterization of the testis in congenitally ubiquitin carboxy-terminal hydrolase-1 (Uch-L1) defective (gad) mice. Exp. Anim. 52, 1–9 [DOI] [PubMed] [Google Scholar]
- 47. Santamaría D., Barrière C., Cerqueira A., Hunt S., Tardy C., Newton K., Cáceres J. F., Dubus P., Malumbres M., Barbacid M. (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815 [DOI] [PubMed] [Google Scholar]
- 48. Rane S. G., Dubus P., Mettus R. V., Galbreath E. J., Boden G., Reddy E. P., Barbacid M. (1999) Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nat. Genet. 22, 44–52 [DOI] [PubMed] [Google Scholar]
- 49. Caballero O. L., Resto V., Patturajan M., Meerzaman D., Guo M. Z., Engles J., Yochem R., Ratovitski E., Sidransky D., Jen J. (2002) Interaction and colocalization of PGP9.5 with JAB1 and p27(Kip1). Oncogene 21, 3003–3010 [DOI] [PubMed] [Google Scholar]
- 50. Kim H. J., Kim Y. M., Lim S., Nam Y. K., Jeong J., Kim H. J., Lee K. J. (2009) Ubiquitin C-terminal hydrolase-L1 is a key regulator of tumor cell invasion and metastasis. Oncogene 28, 117–127 [DOI] [PubMed] [Google Scholar]
- 51. Okochi-Takada E., Nakazawa K., Wakabayashi M., Mori A., Ichimura S., Yasugi T., Ushijima T. (2006) Silencing of the UCHL1 gene in human colorectal and ovarian cancers. Int. J. Cancer 119, 1338–1344 [DOI] [PubMed] [Google Scholar]
- 52. Kagara I., Enokida H., Kawakami K., Matsuda R., Toki K., Nishimura H., Chiyomaru T., Tatarano S., Itesako T., Kawamoto K., Nishiyama K., Seki N., Nakagawa M. (2008) CpG hypermethylation of the UCHL1 gene promoter is associated with pathogenesis and poor prognosis in renal cell carcinoma. J. Urol. 180, 343–351 [DOI] [PubMed] [Google Scholar]
- 53. Hoozemans J. J., Brückner M. K., Rozemuller A. J., Veerhuis R., Eikelenboom P., Arendt T. (2002) Cyclin D1 and cyclin E are co-localized with cyclo-oxygenase 2 (COX-2) in pyramidal neurons in Alzheimer disease temporal cortex. J. Neuropathol. Exp. Neurol. 61, 678–688 [DOI] [PubMed] [Google Scholar]
- 54. McShea A., Harris P. L., Webster K. R., Wahl A. F., Smith M. A. (1997) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am. J. Pathol. 150, 1933–1939 [PMC free article] [PubMed] [Google Scholar]
- 55. Busser J., Geldmacher D. S., Herrup K. (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J. Neurosci. 18, 2801–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagy Z., Esiri M. M., Smith A. D. (1997) Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol. 93, 294–300 [DOI] [PubMed] [Google Scholar]
- 57. Arendt T., Rödel L., Gärtner U., Holzer M. (1996) Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. Neuroreport. 7, 3047–3049 [DOI] [PubMed] [Google Scholar]