

Background: Prolactin, but not its best studied mediator STAT5a, is associated with breast cancer progression.
Results: In stiff but not compliant collagen matrices, prolactin promotes tumorigenic processes via an enhanced ERK1/2 cascade.
Conclusion: Extracellular matrix stiffness powerfully modulates the spectrum of prolactin signals and actions.
Significance: Prolactin and stiff matrices interact in a feed-forward loop in breast cancer, suggesting new therapeutic approaches.
Keywords: Breast Cancer, Collagen, Extracellular Matrix, Focal Adhesion Kinase, Prolactin, Density, Desmoplasia
Abstract
Clinically, circulating prolactin levels and density of the extracellular matrix (ECM) are individual risk factors for breast cancer. As tumors develop, the surrounding stroma responds with increased deposition and cross-linking of the collagen matrix (desmoplasia). In mouse models, prolactin promotes mammary carcinomas that resemble luminal breast cancers in women, and increased collagen density promotes tumor metastasis and progression. Although the contributions of the ECM to the physiologic actions of prolactin are increasingly understood, little is known about the functional relationship between the ECM and prolactin signaling in breast cancer. Here, we examined consequences of increased ECM stiffness on prolactin signals to luminal breast cancer cells in three-dimensional collagen I matrices in vitro. We showed that matrix stiffness potently regulates a switch in prolactin signals from physiologic to protumorigenic outcomes. Compliant matrices promoted physiological prolactin actions and activation of STAT5, whereas stiff matrices promoted protumorigenic outcomes, including increased matrix metalloproteinase-dependent invasion and collagen scaffold realignment. In stiff matrices, prolactin increased SRC family kinase-dependent phosphorylation of focal adhesion kinase (FAK) at tyrosine 925, FAK association with the mitogen-activated protein kinase mediator GRB2, and pERK1/2. Stiff matrices also increased co-localization of prolactin receptors and integrin-activated FAK, implicating altered spatial relationships. Together, these results demonstrate that ECM stiffness is a powerful regulator of the spectrum of prolactin signals and that stiff matrices and prolactin interact in a feed-forward loop in breast cancer progression. Our study is the first reported evidence of altered ECM-prolactin interactions in breast cancer, suggesting the potential for new therapeutic approaches.
Introduction
Prolactin (PRL)2 is a protein hormone produced by the pituitary gland in mammals as well as in peripheral tissues, such as the mammary gland in women (1). It is a critical regulator of mammary physiology, driving alveolar proliferation and lactation (2). Epidemiologic and experimental studies have also pointed to roles in breast cancer. A large prospective study nested within the Women's Health Study correlated elevated serum PRL with the risk of estrogen receptor α-positive breast cancer independent of circulating estrogen (3), and smaller studies also associated high activity of PRL in established tumors with treatment failure, earlier recurrence after recession, and worse overall survival (for reviews, see Refs. 3–6). However, the mechanism(s) whereby PRL contributes to these outcomes is poorly understood.
PRL binding to its receptor (PRLR), a class I cytokine receptor, initiates signals through many different pathways, including JAK2-STAT5 and MEK1/2-ERK1/2 kinases (7); however, these pathways have quite distinct effects on cell behavior. Mouse models have demonstrated that PRL signaling through JAK2-STAT5 accounts for the vast majority of its physiological actions in the mammary gland (8, 9). However, activation of STAT5 in clinical breast cancers predicts sensitivity to antiestrogen therapies and more favorable outcomes (10–12) modeled by experimental murine PRL-induced mammary carcinomas (13). PRL also initiates strong phosphorylation of ERK1/2 in breast cancer cells in vitro (7) and in the murine mammary gland in vivo (14). Moreover, it cooperates with growth factors to prolong phosphorylated ERK1/2 (pERK1/2) (15) associated with increased matrix metalloproteinase (MMP) expression and invasion (16). We have shown previously that PRL signals to STAT5 and mitogen-activated protein kinase-stimulated activating protein 1 are inversely related in vitro and in vivo (13, 17). Consistently, PRL-induced carcinomas that exhibit lower levels of phosphorylated STAT5 (pSTAT5) express higher levels of MMPs (13), some of which are driven by mitogen-activated protein kinase-activated activating protein 1 enhancers (18). Furthermore, reduction and/or inhibition of STAT5 in breast cancer cells in vitro increases PRL-stimulated invasiveness (17, 19). Together, these studies indicate that variations in the relative strength of PRL-activated pathways can have profoundly different outcomes in breast cancer. However, the factors that regulate the balance of PRL-initiated signals are not understood.
Epidemiological studies also link breast density and the risk and progression of breast cancer (20–22). Collagen I is a major component of the extracellular matrix (ECM) in the developing and adult mammary gland and of the increased fibrillar collagens that elevate mammographic density (23, 24). As cancers progress, the stiffness of the ECM around the tumor increases (desmoplasia) as a result of altered collagen deposition, cross-linking, and remodeling (25). This increased ECM density increases breast cancer invasiveness in vitro and metastasis in vivo (for reviews, see Refs. 26 and 27). Cells sense the stiffness of the ECM through Rho-mediated contraction (26, 27). In compliant matrices, the ECM can be contracted with minimal mechanical tension to the cells. Conversely, an ECM that is too stiff for cell-induced contraction results in mechanically based signal transduction through focal adhesions. This mechanical tension in high density matrices increases basal levels of pERK1/2 and initiates ERK1/2-dependent increases in proliferation and changes in morphology and in the transcriptome (28). Culture in high density collagen I gels also increases the association of upstream modulators of ERK1/2, such as SRC family kinases (SFKs), with focal adhesion kinase (FAK) (28). These studies establish the FAK-SFK-ERK1/2 signaling cascade as a key regulator of the switch between normal and disease-like actions of cells in different collagen densities. PRL also has been shown to activate these kinases (29–32), suggesting that ECM stiffness and PRL may cross-talk through this signaling pathway.
To study the effect of matrix stiffness on PRL actions in breast cancer cells, we examined PRL-induced signaling and cell behavior in two well characterized, luminal breast cancer cell lines cultured in compliant and stiff three-dimensional collagen I matrices in vitro. We report that matrix stiffness is a potent modulator of the spectrum of PRL signals. A stiff collagen matrix favored protumorigenic PRL actions, increasing co-localization of PRLR and FAK, PRL activation of the FAK-SFK-GRB2-ERK1/2 cascade, invasive behavior, and proinvasive collagen realignment. In contrast, a compliant collagen matrix favored more physiologically normal PRL responses associated with increased pSTAT5. Our studies demonstrate that increased stiffness of the ECM is sufficient to switch the outcome of PRL signals in breast cancer cells from differentiation toward enhanced tumorigenic processes. This work provides one explanation for the apparent disparity between PRL activity and poor outcomes in breast cancer and the positive therapeutic response and favorable outcome predicted by STAT5 activation. Our findings have direct implications for PRL-directed therapies.
EXPERIMENTAL PROCEDURES
Reagents
Antibodies used for these studies were purchased as follows: pFAK-Tyr397 (catalog number 44624G), PRLR extracellular domain (catalog number 35-9200), and pSTAT5-Tyr694 (catalog number 71-6900) from Invitrogen; ERK1/2 (catalog number 9102), pERK1/2-Thr202/204 (catalog number 9101), pFAK-Tyr925 (catalog number 3284S), and FAK (catalog number 3285) from Cell Signaling Technology (Danvers, MA); FAK clone 4.47 (catalog number 05-537) from EMD Millipore (Billerica, MA); and STAT5 (catalog number sc-835x) from Santa Cruz Biotechnology (Santa Cruz, CA). An antibody to the PRLR (LFA-102) was a gift from Novartis Pharma AG (Basel, Switzerland). β1 integrin-blocking antibody (clone mAb13, catalog number 552828) and rat IgG2a,k isotype antibody (catalog number 555841) were purchased from BD Biosciences. Inhibitors used for these studies were purchased as follows: SFK inhibitor PP1 (catalog number EI275) from Biomol International (Plymouth Meeting, PA) and SFK inhibitor SU6656 (catalog number 572635) from Millipore (Billerica, MA). Phalloidin-FITC (catalog number P5282) and 1,10-phenanthroline (catalog number 131377) were purchased from Sigma-Aldrich. Recombinant hPRL (Lot AFP795) was obtained from Dr. A. F. Parlow (National Hormone and Pituitary Program, NIDDK, National Institutes of Health, Torrance, CA). Type I rat tail collagen (catalog number CB354249) was obtained from Fisher Scientific. All other reagents were obtained from Fischer Scientific or Sigma.
Cell Culture
Estrogen receptor α-positive, PRLR+ T47D (17), and MCF-7 breast cancer cells (33) were maintained as described prior to culture in collagen gels. The cells were plated at 6 × 105 or 1.5 × 106 (immunoprecipitation experiments) cells/ml in low (LD) (1.2 mg/ml) or high (HD) (2.8 mg/ml) density type I rat tail collagen at concentrations optimized for compliance and stiffness as described previously (34). After 24 h in collagen, the gels were released from the edges of the dishes, floating the gels and permitting cells to contract the LD matrix, and cells were serum-starved for 24 h prior to hormone treatment. For some experiments, β1 integrin signals were blocked by pretreating T47D cells with isotype control or β1-blocking antibody clone mAb13 (500 ng/ml) for 15 min prior to plating in LD or HD collagen. After 24 h, the gels were released as above and serum-starved in the presence of isotype control or mAb13 (500 ng/ml) for 24 h prior to hormone treatment. For other experiments, cells were pretreated for 1 h with small molecule inhibitors or a volume equivalent of vehicle (DMSO) prior to treatment with PRL. For longer term studies, 6 × 105 T47D cells in LD or HD collagen were cultured in 5% horse serum in place of serum-free medium. Cells were treated every 48 h with or without PRL (4 nm), and a half-medium exchange was performed every 4 days.
Immunoblotting
Immunoblotting was performed as described previously (35). Signals were visualized using enhanced chemiluminescence (Thermo Fisher) and quantified by scanning densitometry (VisionWorksLS, v7.1, UVP, Upland, CA). Immunoprecipitation was performed as described previously utilizing 125 μg of cellular protein and 0.6 μg of the rabbit FAK antibody (28).
Quantitative Real Time PCR
Quantitative real time PCR was performed as described previously (35) and analyzed via the ΔΔC(t) method using 18 S ribosomal RNA. The following primer sequences were utilized: 18 S F, 5′-CGC CGC TAG AGG TGA AAT TCT; 18 S R, 5′-CGA ACC TCC GAC TTT CGT TCT; MMP2 F, 5′-CTG CAA CCT GTT TGT GCT GAA; MMP2 R, 5′-GGC TTG CGA GGG AAG AAG T; MMP9 F, 5′-CGG AGT GAG TTG AAC CAG; and MMP9 R, 5′-GTC CCA GTG GGG ATT TAC.
Invasion Assays
Invasion assays were performed as described (36). Briefly, T47D cells (3 × 105/well) were mixed with low (1.2 mg/ml) or high (2.8 mg/ml) density type I collagen in the presence or absence of 200 ng/ml (8 nm) PRL, plated in Transwell permeable supports with polycarbonate membranes containing 12-μm pores (Corning, Inc., Tewksbury, MA), and allowed to polymerize for 20 min at room temperature. RPMI 1640 medium containing 10% horse serum was placed in the lower chamber, and the system was incubated at 37 °C for 24 h. Traversed cells were counted after staining with Giemsa stain. For some experiments, cells were pretreated with vehicle or the MMP inhibitor 1,10-phenanthroline (1 mm) in dimethylformamide for 15 min prior to collagen plating and PRL treatment. This concentration of MMP inhibitor did not affect numbers of viable cells or metabolic activity as determined by the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega Corp., Madison, WI) (data not shown).
Quantitative Zymography
Quantitative zymography of MMP-2 was performed as described (37). Briefly, conditioned medium was collected after 24 h of hormone treatment, separated by non-denaturing SDS-PAGE with 2 mg/ml gelatin, and then incubated for 18 h in enzyme renaturing buffer (50 mm Tris, pH 7.5, 200 mm NaCl, 5 mm CaCl2, and 0.02% Nonidet P-40). Digested gelatin was visualized by staining in 0.02% Coomassie R-250, 30% methanol, and 10% acetic acid and quantified by scanning densitometry. Pro-MMP-2 was identified at 72 kDa, whereas active MMP-2 was identified at 60 kDa.
Immunofluorescence
Immunofluorescence was performed as described (34) except that cells were permeabilized in 0.1% Triton X-100. Primary antibodies for PRLR (Novartis) and FAK-Tyr(P)397 were added to the gels for 45 min at room temperature followed by extensive washing in PBS with 0.1% Tween 20 (PBS-T) and subsequent secondary antibody addition (anti-rabbit TRITC and anti-human FITC), DAPI, or phalloidin-FITC. Confocal images were obtained on a Nikon C1-LU3D-Eclipse confocal microscope at 100× magnification with EZ-3.8 imaging software for data collection. Quantification of co-localization was performed as described (38). Fluorescent images were obtained on an E600 Eclipse fluorescence microscope with an RGB camera and Nikon NIS-Elements imaging software. Images were analyzed utilizing NIH ImageJ software.
Multiphoton Microscopy and Second Harmonic Generation
Multiphoton microscopy (39) and second harmonic generation (40) were performed at the University of Wisconsin Laboratory for Optical and Computational Instrumentation as described (41). Z-stacks of 5 μm that spanned the thickness of each gel were obtained utilizing the Laboratory for Optical and Computational Instrumentation-developed WiscScan software package. Collagen images were obtained at 890 nm, and NADH images were obtained at 780 nm utilizing a Plan Fluor 20× objective lens and a 445 ± 0.5-nm narrow bandpass filter to detect the second harmonic generation signal. Data were analyzed utilizing NIH ImageJ software.
RESULTS
High Collagen Density Shifts PRL Signals toward the ERK1/2 Cascade without Altering PRLR Expression
PRL can initiate multiple signaling cascades, including the JAK2-STAT5 and RAS-MEK-ERK1/2 pathways. To investigate the effect of matrix stiffness on the relative strengths of the PRL signaling repertoire, we cultured breast cancer cells in LD or HD matrices. In T47D cells cultured in LD collagen gels, PRL initiated robust phosphorylation of STAT5 (Fig. 1A). However, this was reduced 2-fold in cells cultured in HD conditions (p < 0.01). In contrast, T47D cells in HD culture responded to PRL with a strong phosphorylation of ERK1/2 compared with LD conditions (p < 0.01) (Fig. 1B). These differences in downstream phosphorylation events were not influenced by changes in PRLR expression; levels of neither the long PRLR isoform nor the ΔS-1 isoform were changed by collagen density (Fig. 1C). Similar effects on PRL-initiated signals were observed in another luminal breast cancer cell line, MCF-7 cells (data not shown).
FIGURE 1.

Stiff matrices increase PRL signaling to ERK1/2 and reduce that to STAT5 without altering PRLR levels. A and B, serum-starved T47D cells in LD or HD collagen I gels were treated with or without PRL (4 nm) for 20 min. Cell lysates were immunoblotted with the indicated antibodies. Top panels, representative immunoblots. Bottom panels, quantification of immunoblots by densitometry. Means ± S.D. are shown. n = 3 (A); n = 4 (B). The asterisks denote significant differences between treatments as determined by two-way ANOVA followed by paired t tests. **, p < 0.01; ***, p < 0.001. C, T47D cells in LD or HD collagen I gels were harvested after 24 h of serum starvation, and cell lysates were immunoblotted for PRLR or ERK1/2. Top panel, representative immunoblot. Bottom panel, quantification of the long PRLR isoform (lPRLR) compared with total ERK1/2 by densitometry. Means ± S.D. are shown. n = 3. D and E, serum-starved T47D cells in LD or HD collagen I gels were treated with isotype control antibody (−) or β1 integrin-blocking antibody mAb13 (+) during plating and subsequent treatments as in A and B. Cell lysates were immunoblotted with the indicated antibodies. Top panels, representative immunoblots. Bottom panels, quantification of immunoblots by densitometry. Means ± S.D. are shown. n = 3 (D); n = 4 (E). The asterisks denote significant differences between treatments as determined by two-way ANOVA followed by paired t tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars represent S.D.
Integrin heterodimers containing the β1 integrin subunit link the ECM to downstream signaling cascades and play critical roles in mammary differentiation, including activation of the JAK2-STAT5 pathway by PRL, as well as breast cancer progression via linkage to associated adaptors, such as FAK and downstream signals (for reviews, see Refs. 42 and 43). Matrix stiffness did not affect β1 integrin expression in T47D cells (data not shown) as we reported previously (28). To evaluate the importance of this integrin in the changes in the relative strength of PRL signals observed, we used a blocking antibody that prevents β1 integrin from binding to the ECM (44). Blocking β1 integrin signals reduced the ability of PRL to phosphorylate STAT5 compared with isotype-matched controls in both LD (p < 0.05) and HD (p < 0.01) matrices (Fig. 1D) as well as decreased PRL-induced ERK1/2 phosphorylation in HD matrices (p < 0.001) (Fig. 1E). These data demonstrate that β1 integrin is a critical link between the ECM and PRL-initiated signals to STAT5 in our system as reported for normal mammary epithelial cells (45, 46) independent of matrix stiffness as well as in the strengthened PRL signals to ERK1/2 in stiff matrices.
PRL Increases Formation of Well Differentiated Colonies in Low Density Collagen but Increases Disorganized Structures in High Density Collagen
To examine the long term effect of this change in PRL signals on cell behavior, we examined effects on morphology. T47D cells cultured in LD floating collagen gels form well differentiated structures, whereas HD culture results in a non-differentiated morphology (34). In light of the change in the balance of PRL-induced signals with matrix stiffness, we hypothesized that PRL enhances morphological changes. To test this, we cultured T47D cells in LD or HD collagen with 5% horse serum in the presence or absence of PRL for 7 days. As described previously (34), culture in compliant matrices significantly increased the ratio of well differentiated colonies with strong apical lateral localization of cortical F-actin to disorganized structures compared with HD culture (p < 0.001) (Fig. 2A). Interestingly, PRL significantly further increased the proportion of well differentiated colonies in compliant cultures (p < 0.001) but increased that of irregular cell clusters with disorganized F-actin staining in stiff matrices (p < 0.001) (Fig. 2).
FIGURE 2.

PRL increases formation of well differentiated colonies in compliant matrices but disorganized structures in stiff matrices. T47D cells were cultured in low or high density collagen gels for 7 days in 5% horse serum with or without PRL (4 nm) and then stained for DAPI and phalloidin-FITC as described under “Experimental Procedures.” A, well differentiated colonies and disorganized colonies were counted in individual 10× fields, and the ratio was determined. Means ± S.E. (error bars) are shown. n = 16. The asterisks denote significant differences between treatments as determined by two-way ANOVA followed by unpaired t test. ***, p < 0.001. B, representative organized and disorganized colonies stained with DAPI (left column) and F-actin (right column). Scale bar, 50 μm.
PRL Increases MMP-2 Expression and Activity in High but Not Low Density Matrices
We hypothesized that the observed shift in PRL signals toward ERK1/2 in HD culture would result in protumorigenic outcomes, such as increased MMP-2 expression and activity (47). To test this hypothesis, we treated T47D and MCF-7 cells in LD or HD collagen culture with or without PRL for 24 h and examined MMP2 transcript levels. In compliant matrices, PRL did not alter MMP2 mRNA (Fig. 3, A and B). Stiff matrices modestly increased unstimulated levels of MMP2 mRNA but permitted PRL to significantly increase MMP2 mRNA levels up to 3.5-fold in MCF-7 cells (p < 0.05). MMP9 transcript levels followed a similar pattern. Stiff matrices increased unstimulated levels of MMP9 mRNA 1.54-fold over unstimulated levels in MCF-7 cells in compliant matrices (p < 0.05). Under these conditions, PRL was able to further increase MMP9 mRNA levels 2.15-fold (p < 0.05). Similar results were observed in T47D cells. To confirm that the increased MMP2 mRNA levels resulted in increased protein expression and enzyme activity, we examined conditioned media from MCF-7 cells cultured in LD or HD collagen in the presence or absence of PRL for 24 h by gelatin zymography. Consistent with the mRNA levels, PRL did not affect either total (pro-MMP-2 plus active MMP-2) or active MMP-2 in LD culture. However, in stiff matrices, PRL significantly increased both total (p < 0.05) and active (p < 0.05) MMP-2 (Fig. 3C).
FIGURE 3.
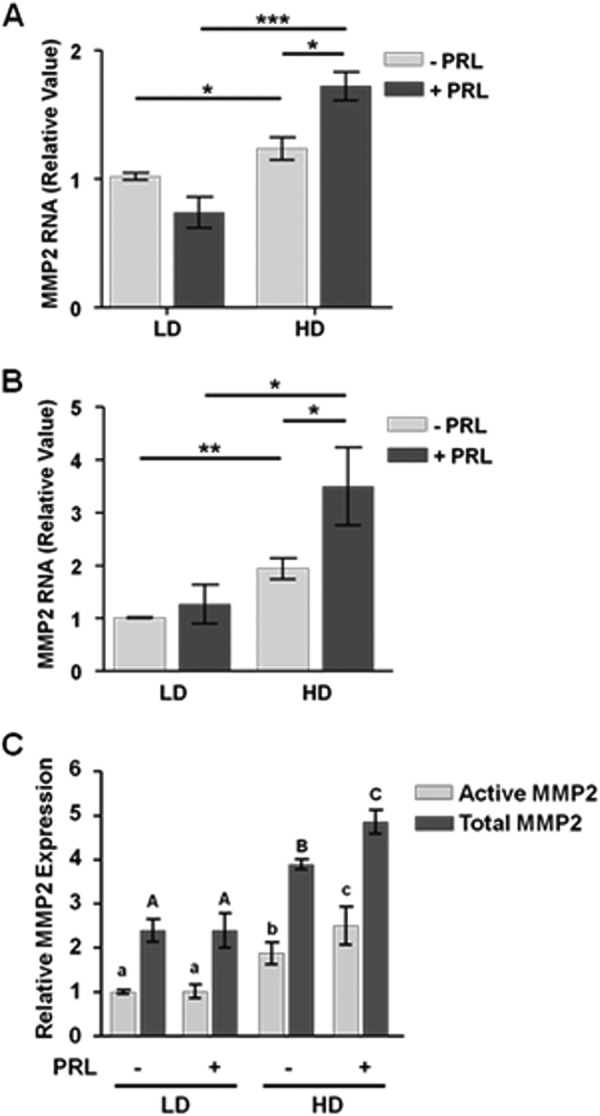
PRL increases MMP2 mRNA and activity in stiff but not compliant matrices. Serum-starved T47D (A) or MCF-7 (B and C) cells in LD or HD collagen gels were treated with or without PRL (4 nm) for 24 h. A and B, MMP2 and 18 S ribosomal RNA transcripts were analyzed by quantitative real time PCR as described under “Experimental Procedures,” and results were normalized to LD, no-PRL samples. Means ± S.D. are shown. n = 3. The asterisks denote significant differences between treatments as determined by two-way ANOVA followed by paired t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, conditioned medium from MCF-7 cells treated as above was assayed for gelatinase activity as described under “Experimental Procedures.” Latent and active MMP-2 was quantified by densitometry and expressed as a percentage of active MMP-2 in LD collagen without PRL treatment. Means ± S.D. are shown. n = 3. Different lowercase letters denote significant differences in active MMP-2 between groups (p < 0.05), and different capital letters denote significant differences in total MMP-2 between groups (p < 0.05). Error bars represent S.D.
PRL Increases Cell Invasiveness in a Density- and MMP-dependent Manner
The ability to invade through a collagen matrix is a hallmark of metastatic cells and is a key step in disease progression (48). To determine whether the PRL-induced increases in MMP-2 expression led to increased invasiveness, we performed Transwell assays with T47D cells in different collagen densities. Consistent with our measured MMP-2 activity, PRL did not stimulate invasion in compliant matrices. Stiff matrices significantly increased cell invasiveness and permitted PRL to further augment invasion 5-fold above LD culture (p < 0.001) (Fig. 4A). To confirm that the increase in PRL-induced invasiveness was mediated by MMP activity, we utilized the pan-MMP inhibitor 1,10-phenanthroline. This compound blocked virtually all cell invasion in HD culture, consistent with the pattern of MMP-2 activity (p < 0.001) (Fig. 4B).
FIGURE 4.

PRL increases invasiveness in stiff matrices. A, serum-starved T47D cells were plated with or without PRL (8 nm) in LD or HD collagen gels onto Transwell permeable supports and permitted to invade toward 10% horse serum for 24 h. Traversed cells were counted as described under “Experimental Procedures.” Results are presented as percentage of LD, no-PRL treatment. Means ± S.D. are shown. n = 3. B, serum-starved T47D cells cultured as in A were pretreated with vehicle or 1,10-phenanthroline for 15 min prior to plating in high density collagen with or without PRL (8 nm) treatment. Results are presented as percentage of HD vehicle-treated samples. Means ± S.D. are shown. n = 3. The asterisks denote significant differences between treatments as determined by two-way ANOVA followed by paired t test. **, p < 0.01; ***, p < 0.001. Error bars represent S.D. C, serum-starved T47D cells were cultured in LD or HD collagen and treated with or without PRL for 72 h, and the collagen fibers were imaged by second harmonic generation as described under “Experimental Procedures.” Representative images are shown. White, collagen; green, NADH. Arrows denote collagen fibers perpendicular to the cell membrane, whereas arrowheads denote collagen fibers parallel to the cell membrane. Scale bar, 50 μm.
PRL Induces Collagen Matrix Reorganization in HD, but not LD, Collagen Culture
Collagen alignment around tumor boundaries is a prognostic marker for human breast cancer survival (49). Collagen fibers that align perpendicularly to the tumor or cell boundary facilitate invasion, whereas parallel collagen alignment reduces migration and is a signature for a less aggressive outcome (41, 49, 50). To visualize the effect of PRL on collagen alignment in three-dimensional collagen culture, we utilized multiphoton microscopy combined with second harmonic generation. Consistent with our biochemical and morphological data, cells in LD collagen presented a non-aggressive collagen signature. Collagen was poorly organized throughout the matrix and paralleled cell membranes regardless of PRL treatment (Fig. 4, C and D). Tubule-like structures were also observed in LD collagen, consistent with our previous observations (Fig. 4D) (34). In contrast, HD collagen exhibited more organized fibers with some perpendicular to the cells (Fig. 4E). Strikingly, PRL treatment in HD culture stimulated radial collagen alignment, evident as long, straight collagen fibers that displayed a starburst pattern emanating from foci on the cell periphery (Fig. 4F, white arrow).
PRL-induced Increase in pERK1/2 Is Mediated through SFK Activation of FAK
High density collagen culture increases basal levels of pERK1/2 through the SFK-FAK signaling cascade (28). We hypothesized that this pathway would be enhanced by PRL in HD culture. As shown in Fig. 5A, PRL induced SFK-dependent FAK phosphorylation at Tyr925 (51) in both LD and HD culture. However, in HD culture, this signal was more robust so that after 15 min levels of FAK Tyr(P)925 were 2-fold higher than in LD culture (p < 0.05). Although FAK-Tyr(P)397 was readily detected in unstimulated cells, PRL did not acutely increase it regardless of matrix stiffness (data not shown). The small molecule inhibitor of SFK activity, PP1, blocked PRL signals to pERK1/2 (Fig. 5B); similar results were observed with SU6656 (data not shown) (52), establishing the role of SFKs in this pathway. pFAK-Tyr925 binds the adaptor GRB2, linking it to the MEK1/2-ERK1/2 cascade (53). As predicted, after 5 min of treatment in HD culture, PRL increased GRB2/FAK association compared with LD conditions (p < 0.01) (Fig. 5C, left). However, after 10 min of PRL treatment, GRB2 had dissociated from FAK, consistent with PRL-stimulated internalization, disassembly, and recycling of focal adhesions (54–56) (Fig. 5C, right).
FIGURE 5.

Culture in stiff matrices enhances PRL-activated, SFK-mediated phosphorylation of FAK and ERK1/2. A, serum-starved T47D cells in LD or HD collagen gels were treated with or without PRL (4 nm) for the indicated times. Cell lysates were immunoblotted with the indicated antibodies and quantified by densitometry. Means ± S.D. (error bars) are shown. n = 3. The asterisks denote significant differences between treatments as determined by two-way ANOVA followed by paired t test. *, p < 0.05. B, serum-starved T47D cells in low or high density collagen gels were pretreated with vehicle or PP1 (20 μm) for 1 h prior to treatment with or without PRL (4 nm) for 20 min. Cell lysates were immunoblotted with the indicated antibodies, and representative experiments are shown. C and D, 1.5 × 106 serum-starved T47D cells in low or high density collagen gels were treated with or without PRL (4 nm) for 5 (left) or 10 min (right). Cell lysates were immunoprecipitated (IP) for FAK and immunoblotted with the indicated antibodies. Representative blots are shown.
HD Culture Increases PRLR Co-localization with FAK
HD culture results in increased formation of focal adhesions as indicated by increased FAK localization to distinct puncta (57). We hypothesized that HD culture would increase PRLR co-localization with FAK, facilitating signaling through this complex. We performed co-immunofluorescence studies on T47D cells in LD and HD culture for PRLR and FAK-Tyr(P)397, the integrin-activated form of FAK present at focal adhesions (58, 59). In LD culture, FAK and PRLR presented diffuse, membrane-specific staining with little apparent co-localization (Fig. 6A). In HD culture, PRLR and FAK exhibited increased co-localization in distinct puncta along with an apparent increase in non-membrane staining consistent with increased focal adhesion turnover observed in metastatic cancer cells (60) (Fig. 6B). Quantification of co-localized PRLR and FAK-Tyr(P)397 by threshold analysis showed a significant increase in PRLR/FAK-Tyr(P)397 co-localization in HD compared with LD conditions (p < 0.001). Based on the analysis parameters of Costes et al. (38), PRLR/FAK-Tyr(P)397 co-localization increased 3-fold between LD and HD culture (Fig. 6C).
FIGURE 6.

HD culture increases co-localization of PRLR and pFAK-Tyr397. Serum-starved T47D cells in LD or HD collagen gels were fixed, stained for PRLR and pFAK-Tyr397, and visualized by confocal microscopy as described under “Experimental Procedures.” Green, PRLR; red, FAK-Tyr(P)397. A, representative image of cells in LD. B, representative image of cells in HD. C, quantification of co-localization utilizing the threshold method of Costes et al. (38). Means ± S.D. (error bars) are shown. n = 3 experiments with at least 20 cells per density in each experiment. The asterisks denote statistically significant differences between treatments determined by two-way ANOVA followed by unpaired t test. ***, p < 0.001. Scale bar, 25 μm.
DISCUSSION
Despite the association of PRL exposure with development and poor outcome of luminal breast cancers clinically (3), expression and activation of the best characterized PRL signaling mediator, STAT5, are associated with more differentiated tumors and better therapeutic responsiveness (10–12). Although PRL activates other signaling cascades that promote tumor progression, such as mitogen-activated protein kinases and PI3K-AKT, the factors that regulate the PRL-induced signaling repertoire are poorly understood. Here, we demonstrated that ECM density is a powerful modulator of the balance of PRL-induced signals with dramatic consequences for the behavior of luminal breast cancer cells.
In compliant matrices, PRL preferentially induced pSTAT5, resulting in prodifferentiation outcomes, including increased formation of well differentiated colonies (Fig. 7, left). Under these conditions, PRL did not stimulate MMP-2 expression or activity or cell invasion. Conversely, in stiff collagen matrices, PRL strongly induced pERK1/2, stimulated MMP-2 expression and activity, and increased invasive behavior (Fig. 7, right). Expression of other metalloproteinases implicated in cancer progression may also be altered (for reviews, see Refs. 61 and 62), although we did not evaluate their role here. Moreover, PRL increased formation of disorganized, non-polar structures and altered collagen alignment as described for aggressive breast cancers (49). In stiff matrices, PRLR and activated FAK co-localized to distinct puncta in three-dimensional matrix adhesions that we have characterized previously (28), and the SFK-FAK-GRB2-mitogen-activated protein kinase cascade mediated the enhanced PRL signals to pERK1/2. Together, our results indicate that PRL and a stiff ECM strongly interact to convert well differentiated, non-invasive T47D cells, which require genetic modification and/or a protumorigenic stimulus to become invasive (63, 64), to a more malignant phenotype.
FIGURE 7.

Interplay between matrix stiffness and prolactin signals in breast cancer. Our data support the following model. In compliant collagen matrices (left), the ECM resembles a normal, non-malignant mammary ECM in vivo. PRLR is not enriched at sites containing FAK, and PRL signals are mediated predominantly by the JAK2-STAT5 signaling cascade (heavy arrows), which drives formation of highly organized, well differentiated colonies. Conversely, increased matrix stiffness increases focal adhesions, clustering integrins, FAK, and other signaling components, including the PRLR (right). This permits PRL to preferentially activate the SFK-FAK-ERK1/2 signaling cascade (heavy arrows), promoting formation of poorly organized colonies and MMP-driven invasion, mimicking aggressive cancers in vivo.
Focal adhesions are critical links between the ECM and intracellular signaling cascades in mammary epithelia (65, 66). They contain FAK, which clusters to integrin binding sites and recruits other components to the complex (59), regulating focal adhesion dynamics and cell migration (for reviews, see Refs. 54–56). In cancer, focal adhesion turnover allows tumor cells to break free of their attachments to the ECM, facilitating invasion and tumor progression (67). In mouse models, mammary tissue-specific ablation of FAK retards tumorigenesis and metastasis and reduces epithelial progenitor populations (35, 68, 69). Additionally, many other protumorigenic growth factor signaling complexes localize to focal adhesions in tumor cells (70, 71). The localization of key extracellularly regulated signaling molecules, such as β1 integrin, SFK, and FAK, is modified by ECM density (26, 72). Our observed increase in co-localization of PRLR and FAK in stiff ECM would facilitate PRL signals through the SFK-FAK-ERK1/2 pathway by bringing PRLR and downstream signaling components into closer proximity. Moreover, acute PRL stimulation under these conditions also increased focal adhesion dissociation, allowing for cellular motility.
The desmoplasia that accompanies tumor progression is well characterized (25). Recent studies demonstrate that collagen alignment as well as density can be altered in invasive tumors, and straight, perpendicularly aligned fibers create avenues for cell movement and invasion (41, 49, 50). Tumor-associated fibroblasts are key producers of excess collagen in and around the primary tumor. Interestingly, PRL augments transcripts for matrix components in normal murine mammary glands (73),3 consistent with epidemiological data associating PRL with increased mammographic density (74, 75). Moreover, a recent report suggests that PRL may act directly on tumor-associated fibroblasts (76) in addition to enhancing collagen realignment in stiff matrices as reported herein. Tyrosine kinase inhibitors, which block activation of ERK1/2 and AKT, inhibit both proliferation and collagen synthesis in tumor-associated fibroblasts (77). In light of the ability of PRL to potently cooperate with growth factors to activate these signaling pathways (15, 16), these interactions deserve further investigation. Together, the altered PRL-initiated signals and consequences for cell behavior described in the current study in the context of the literature begin to define a feed-forward loop in which PRL actions in breast cancer not only are altered by matrix stiffness but where PRL itself directly contributes to the density and organization of the matrix to promote the progression of breast cancer.
The responses of breast cancer cells to PRL in high density matrices described here are quite distinct from those of primary pregnant mammary epithelia in a “normal” ECM. Under physiological conditions in vivo and in three-dimensional laminin culture in vitro, integrin input is independent of FAK but instead activates integrin-like kinase and RAC1 to enhance PRL-induced pSTAT5 and prompt mammary epithelia to undergo differentiation and produce milk proteins (45, 46). When these normal cells are cultured in noncompliant matrices of either laminin or collagen I, PRLR, pSTAT5, and milk protein expression is reduced (78). In combination with our studies, these reports underscore the importance of stromal stiffness in determining the outcome of PRL exposure regardless of the target epithelium. In addition, the characteristics of breast cancer epithelium, including stabilized PRLR (79), likely strengthen the protumorigenic responses to PRL in stiff matrices that we observed here.
We have shown that ECM density can regulate the spectrum of PRL signals, dramatically shifting the outcome of PRL actions in breast cancer cells from prodifferentiation in a low density environment to protumor progression in high density matrices. Our data provide mechanistic insight into how desmoplasia enables PRL to drive cancer progression in marked contrast to its physiologic actions, providing experimental support for the epidemiologic data. Furthermore, they show how PRL itself can contribute to this component of the microenvironment, creating a feed-forward loop in cancer progression. These studies provide novel insights into the complex actions of PRL in mammary biology and point to potential new therapeutic targets in breast cancer.
Acknowledgments
We thank Dr. Jason Damiano and Novartis Pharma AG for the gift of the anti-prolactin receptor antibody. We are grateful to Maria Gracia Garcia, Esteban Carrillo, and Debra Rugowski for technical assistance with the floating collagen gel culture system and members of the Laboratory for Optical and Computational Instrumentation for technical assistance with the multiphoton microscopy and second harmonic generation imaging systems. We also appreciate insightful discussions with Dr. Chad Vezina.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA157675 (to L. A. S.), R01CA142833 and R01CA114462 (to P. J. K.), and R01CA136590 (to the Laboratory for Optical and Computational Instrumentation). This work was also supported by a Regenerative Biology Scholar's Award and Biological Sciences Scholar's Award (to C. E. B.) and by Comparative Biosciences, University of Wisconsin School of Veterinary Medicine.
D. E. Rugowski and L. A. Schuler, unpublished data.
- PRL
- prolactin
- ECM
- extracellular matrix
- FAK
- focal adhesion kinase
- HD
- high density
- LD
- low density
- MMP
- matrix metalloproteinase
- PRLR
- prolactin receptor
- SFK
- SRC family kinase
- pSTAT5
- phosphorylated STAT5
- pERK1/2
- phosphorylated ERK1/2
- F
- forward
- R
- reverse
- TRITC
- tetramethylrhodamine isothiocyanate
- ANOVA
- analysis of variance.
REFERENCES
- 1. Freeman M. E., Kanyicska B., Lerant A., Nagy G. (2000) Prolactin: structure, function, and regulation of secretion. Physiol. Rev. 80, 1523–1631 [DOI] [PubMed] [Google Scholar]
- 2. Oakes S. R., Rogers R. L., Naylor M. J., Ormandy C. J. (2008) Prolactin regulation of mammary gland development. J. Mammary Gland Biol. Neoplasia 13, 13–28 [DOI] [PubMed] [Google Scholar]
- 3. Tworoger S. S., Hankinson S. E. (2008) Prolactin and breast cancer etiology: an epidemiologic perspective. J. Mammary Gland Biol. Neoplasia 13, 41–53 [DOI] [PubMed] [Google Scholar]
- 4. Fernandez I., Touraine P., Goffin V. (2010) Prolactin and human tumourogenesis. J. Neuroendocrinol. 22, 771–777 [DOI] [PubMed] [Google Scholar]
- 5. Clevenger C. V., Furth P. A., Hankinson S. E., Schuler L. A. (2003) The role of prolactin in mammary carcinoma. Endocr. Rev. 24, 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jacobson E. M., Hugo E. R., Borcherding D. C., Ben-Jonathan N. (2011) Prolactin in breast and prostate cancer: molecular and genetic perspectives. Discov. Med. 11, 315–324 [PubMed] [Google Scholar]
- 7. Bole-Feysot C., Goffin V., Edery M., Binart N., Kelly P. A. (1998) Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr. Rev. 19, 225–268 [DOI] [PubMed] [Google Scholar]
- 8. Hennighausen L., Robinson G. W., Wagner K. U., Liu X. (1997) Developing a mammary gland is a stat affair. J. Mammary Gland Biol. Neoplasia 2, 365–372 [DOI] [PubMed] [Google Scholar]
- 9. Wagner K. U., Krempler A., Triplett A. A., Qi Y., George N. M., Zhu J., Rui H. (2004) Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol. Cell. Biol. 24, 5510–5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peck A. R., Witkiewicz A. K., Liu C., Stringer G. A., Klimowicz A. C., Pequignot E., Freydin B., Tran T. H., Yang N., Rosenberg A. L., Hooke J. A., Kovatich A. J., Nevalainen M. T., Shriver C. D., Hyslop T., Sauter G., Rimm D. L., Magliocco A. M., Rui H. (2011) Loss of nuclear localized and tyrosine phosphorylated Stat5 in breast cancer predicts poor clinical outcome and increased risk of antiestrogen therapy failure. J. Clin. Oncol. 29, 2448–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cotarla I., Ren S., Zhang Y., Gehan E., Singh B., Furth P. A. (2004) Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers. Int. J. Cancer 108, 665–671 [DOI] [PubMed] [Google Scholar]
- 12. Yamashita H., Nishio M., Ando Y., Zhang Z., Hamaguchi M., Mita K., Kobayashi S., Fujii Y., Iwase H. (2006) Stat5 expression predicts response to endocrine therapy and improves survival in estrogen receptor-positive breast cancer. Endocr. Relat. Cancer 13, 885–893 [DOI] [PubMed] [Google Scholar]
- 13. Arendt L. M., Rugowski D. E., Grafwallner-Huseth T. A., Garcia-Barchino M. J., Rui H., Schuler L. A. (2011) Prolactin-induced mouse mammary carcinomas model estrogen resistant luminal breast cancer. Breast Cancer Res. 13, R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arendt L. M., Evans L. C., Rugowski D. E., Garcia-Barchino M. J., Rui H., Schuler L. A. (2009) Ovarian hormones are not required for PRL-induced mammary tumorigenesis, but estrogen enhances neoplastic processes. J. Endocrinol. 203, 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arendt L. M., Rose-Hellekant T. A., Sandgren E. P., Schuler L. A. (2006) Prolactin potentiates transforming growth factor α induction of mammary neoplasia in transgenic mice. Am. J. Pathol. 168, 1365–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carver K. C., Schuler L. A. (2008) Prolactin does not require insulin-like growth factor intermediates but synergizes with insulin-like growth factor I in human breast cancer cells. Mol. Cancer Res. 6, 634–643 [DOI] [PubMed] [Google Scholar]
- 17. Gutzman J. H., Rugowski D. E., Nikolai S. E., Schuler L. A. (2007) Stat5 activation inhibits prolactin-induced AP-1 activity: distinct prolactin-initiated signals in tumorigenesis dependent on cell context. Oncogene 26, 6341–6348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sato H., Seiki M. (1993) Regulatory mechanism of 92 kDa type IV collagenase gene expression which is associated with invasiveness of tumor cells. Oncogene 8, 395–405 [PubMed] [Google Scholar]
- 19. Sultan A. S., Xie J., LeBaron M. J., Ealley E. L., Nevalainen M. T., Rui H. (2005) Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene 24, 746–760 [DOI] [PubMed] [Google Scholar]
- 20. Boyd N. F., Lockwood G. A., Byng J. W., Tritchler D. L., Yaffe M. J. (1998) Mammographic densities and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 7, 1133–1144 [PubMed] [Google Scholar]
- 21. McCormack V. A., dos Santos Silva I. (2006) Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol. Biomarkers Prev. 15, 1159–1169 [DOI] [PubMed] [Google Scholar]
- 22. Vachon C. M., van Gils C. H., Sellers T. A., Ghosh K., Pruthi S., Brandt K. R., Pankratz V. S. (2007) Mammographic density, breast cancer risk and risk prediction. Breast Cancer Res. 9, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keely P. J., Wu J. E., Santoro S. A. (1995) The spatial and temporal expression of the α2β1 integrin and its ligands, collagen I, collagen IV, and laminin, suggest important roles in mouse mammary morphogenesis. Differentiation 59, 1–13 [DOI] [PubMed] [Google Scholar]
- 24. Li T., Sun L., Miller N., Nicklee T., Woo J., Hulse-Smith L., Tsao M.-S., Khokha R., Martin L., Boyd N. (2005) The association of measured breast tissue characteristics with mammographic density and other risk factors for breast cancer. Cancer Epidemiol. Biomarkers Prev. 14, 343–349 [DOI] [PubMed] [Google Scholar]
- 25. Walker R. A. (2001) The complexities of breast cancer desmoplasia. Breast Cancer Res. 3, 143–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keely P. (2011) Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 16, 205–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Butcher D. T., Alliston T., Weaver V. M. (2009) A tense situation: forcing tumour progression. Nat. Rev. Cancer 9, 108–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Provenzano P. P., Inman D. R., Eliceiri K. W., Keely P. J. (2009) Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 28, 4326–4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Acosta J. J., Muñoz R. M., González L., Subtil-Rodríguez A., Dominguez-Caceres M. A., García-Martínez J. M., Calcabrini A., Lazaro-Trueba I., Martín-Pérez J. (2003) Src mediates prolactin-dependent proliferation of T47D and MCF7 cells via the activation of focal adhesion kinase/Erk1/2 and phosphatidylinositol 3-kinase pathways. Mol. Endocrinol. 17, 2268–2282 [DOI] [PubMed] [Google Scholar]
- 30. Gutzman J. H., Rugowski D. E., Schroeder M. D., Watters J. J., Schuler L. A. (2004) Multiple kinase cascades mediate prolactin signals to activating protein-1 in breast cancer cells. Mol. Endocrinol. 18, 3064–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Piazza T. M., Lu J. C., Carver K. C., Schuler L. A. (2009) Src family kinases accelerate prolactin receptor internalization, modulating trafficking and signaling in breast cancer cells. Mol. Endocrinol. 23, 202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Canbay E., Norman M., Kilic E., Goffin V., Zachary I. (1997) Prolactin stimulates the JAK2 and focal adhesion kinase pathways in human breast carcinoma T47-D cells. Biochem. J. 324, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schroeder M. D., Symowicz J., Schuler L. A. (2002) PRL modulates cell cycle regulators in mammary tumor epithelial cells. Mol. Endocrinol. 16, 45–57 [DOI] [PubMed] [Google Scholar]
- 34. Wozniak M. A., Keely P. J. (2005) Use of three-dimensional collagen gels to study mechanotransduction in T47D breast epithelial cells. Biol. Proced. Online 7, 144–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Provenzano P. P., Inman D. R., Eliceiri K. W., Beggs H. E., Keely P. J. (2008) Mammary epithelial-specific disruption of focal adhesion kinase retards tumor formation and metastasis in a transgenic mouse model of human breast cancer. Am. J. Pathol. 173, 1551–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Provenzano P. P., Inman D. R., Eliceiri K. W., Knittel J. G., Yan L., Rueden C. T., White J. G., Keely P. J. (2008) Collagen density promotes mammary tumor initiation and progression. BMC Med. 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kleiner D. E., Stetler-Stevenson W. G. (1994) Quantitative zymography: detection of picogram quantities of gelatinases. Anal. Biochem. 218, 325–329 [DOI] [PubMed] [Google Scholar]
- 38. Costes S. V., Daelemans D., Cho E. H., Dobbin Z., Pavlakis G., Lockett S. (2004) Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 86, 3993–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Denk W., Strickler J. H., Webb W. W. (1990) Two-photon laser scanning fluorescence microscopy. Science 248, 73–76 [DOI] [PubMed] [Google Scholar]
- 40. Mohler W., Millard A. C., Campagnola P. J. (2003) Second harmonic generation imaging of endogenous structural proteins. Methods 29, 97–109 [DOI] [PubMed] [Google Scholar]
- 41. Provenzano P. P., Eliceiri K. W., Campbell J. M., Inman D. R., White J. G., Keely P. J. (2006) Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lahlou H., Muller W. (2011) β1-integrins signaling and mammary tumor progression in transgenic mouse models: implications for human breast cancer. Breast Cancer Res. 13, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Streuli C. H. (2009) Integrins and cell-fate determination. J. Cell Sci. 122, 171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mould A. P., Akiyama S. K., Humphries M. J. (1996) The inhibitory anti-β1 integrin monoclonal antibody 13 recognizes an epitope that is attenuated by ligand occupancy. Evidence for allosteric inhibition of integrin function. J. Biol. Chem. 271, 20365–20374 [DOI] [PubMed] [Google Scholar]
- 45. Akhtar N., Marlow R., Lambert E., Schatzmann F., Lowe E. T., Cheung J., Katz E., Li W., Wu C., Dedhar S., Naylor M. J., Streuli C. H. (2009) Molecular dissection of integrin signalling proteins in the control of mammary epithelial development and differentiation. Development 136, 1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akhtar N., Streuli C. H. (2006) Rac1 links integrin-mediated adhesion to the control of lactational differentiation in mammary epithelia. J. Cell Biol. 173, 781–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Köhrmann A., Kammerer U., Kapp M., Dietl J., Anacker J. (2009) Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer 9, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 49. Conklin M. W., Eickhoff J. C., Riching K. M., Pehlke C. A., Eliceiri K. W., Provenzano P. P., Friedl A., Keely P. J. (2011) Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 178, 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ingman W. V., Wyckoff J., Gouon-Evans V., Condeelis J., Pollard J. W. (2006) Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev. Dyn. 235, 3222–3229 [DOI] [PubMed] [Google Scholar]
- 51. Brunton V. G., Avizienyte E., Fincham V. J., Serrels B., Metcalf C. A., 3rd, Sawyer T. K., Frame M. C. (2005) Identification of Src-specific phosphorylation site on focal adhesion kinase: dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res. 65, 1335–1342 [DOI] [PubMed] [Google Scholar]
- 52. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schlaepfer D. D., Hanks S. K., Hunter T., van der Geer P. (1994) Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 372, 786–791 [DOI] [PubMed] [Google Scholar]
- 54. Boateng L. R., Huttenlocher A. (2012) Spatiotemporal regulation of Src and its substrates at invadosomes. Eur. J. Cell Biol. 91, 878–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tomar A., Schlaepfer D. (2009) Focal adhesion kinase: switching between GAP's and GEF's in the regulation of cell motility. Curr. Opin. Cell Biol. 21, 676–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wehrle-Haller B. (2012) Assembly and disassembly of cell matrix adhesions. Curr. Opin. Cell Biol. 24, 569–581 [DOI] [PubMed] [Google Scholar]
- 57. Wozniak M. A., Desai R., Solski P. A., Der C. J., Keely P. J. (2003) ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J. Cell Biol. 163, 583–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schaller M. D., Hildebrand J. D., Shannon J. D., Fox J. W., Vines R. R., Parsons J. T. (1994) Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 14, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schaller M. D., Otey C. A., Hildebrand J. D., Parsons J. T. (1995) Focal adhesion kinase and paxillin bind to peptides mimicking β integrin cytoplasmic domains. J. Cell Biol. 130, 1181–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chan K. T., Cortesio C. L., Huttenlocher A. (2009) FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol. 185, 357–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gialeli C., Theocharis A. D., Karamanos N. K. (2011) Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 278, 16–27 [DOI] [PubMed] [Google Scholar]
- 62. Deryugina E. I., Quigley J. P. (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 25, 9–34 [DOI] [PubMed] [Google Scholar]
- 63. Singh J., Murata K., Itahana Y., Desprez P.-Y. (2002) Constitutive expression of the Id-1 promoter in human metastatic breast cancer cells is linked with the loss of NF-1/Rb/HDAC-1 transcription repressor complex. Oncogene 21, 1812–1822 [DOI] [PubMed] [Google Scholar]
- 64. Wu Y., Deng J., Rychahou P. G., Qiu S., Evers B. M., Zhou B. P. (2009) Stabilization of snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell 15, 416–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Katz E., Streuli C. H. (2007) The extracellular matrix as an adhesion checkpoint for mammary epithelial function. Int. J. Biochem. Cell Biol. 39, 715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kass L., Erler J. T., Dembo M., Weaver V. M. (2007) Mammary epithelial cell: influence of extracellular matrix composition and organization during development and tumorigenesis. Int. J. Biochem. Cell Biol. 39, 1987–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chan K. T., Bennin D. A., Huttenlocher A. (2010) Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 285, 11418–11426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Luo M., Fan H., Nagy T., Wei H., Wang C., Liu S., Wicha M. S., Guan J.-L. (2009) Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 69, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lahlou H., Sanguin-Gendreau V., Zuo D., Cardiff R. D., McLean G. W., Frame M. C., Muller W. J. (2007) Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc. Natl. Acad. Sci. U.S.A. 104, 20302–20307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Moro L., Venturino M., Bozzo C., Silengo L., Altruda F., Beguinot L., Tarone G., Defilippi P. (1998) Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 17, 6622–6632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Plopper G. E., McNamee H. P., Dike L. E., Bojanowski K., Ingber D. E. (1995) Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 6, 1349–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Margulies S. S., Dembo M., Boettiger D., Hammer D. A., Weaver V. M. (2005) Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254 [DOI] [PubMed] [Google Scholar]
- 73. Gass S., Harris J., Ormandy C., Brisken C. (2003) Using gene expression arrays to elucidate transcriptional profiles underlying prolactin function. J. Mammary Gland Biol. Neoplasia 8, 269–285 [DOI] [PubMed] [Google Scholar]
- 74. Greendale G. A., Huang M.-H., Ursin G., Ingles S., Stanczyk F., Crandall C., Laughlin G. A., Barrett-Connor E., Karlamangla A. (2007) Serum prolactin levels are positively associated with mammographic density in postmenopausal women. Breast Cancer Res. Treat. 105, 337–346 [DOI] [PubMed] [Google Scholar]
- 75. Bremnes Y., Ursin G., Bjurstam N., Rinaldi S., Kaaks R., Gram I. T. (2007) Endogenous sex hormones, prolactin and mammographic density in postmenopausal Norwegian women. Int. J. Cancer 121, 2506–2511 [DOI] [PubMed] [Google Scholar]
- 76. Xu C., Langenheim J. F., Chen W. Y. (2012) Stromal-epithelial interactions modulate cross-talk between prolactin receptor and HER2/Neu in breast cancer. Breast Cancer Res. Treat. 134, 157–169 [DOI] [PubMed] [Google Scholar]
- 77. Gioni V., Karampinas T., Voutsinas G., Roussidis A. E., Papadopoulos S., Karamanos N. K., Kletsas D. (2008) Imatinib mesylate inhibits proliferation and exerts an antifibrotic effect in human breast stroma fibroblasts. Mol. Cancer Res. 6, 706–714 [DOI] [PubMed] [Google Scholar]
- 78. Du J.-Y., Chen M.-C., Hsu T.-C., Wang J.-H., Brackenbury L., Lin T.-H., Wu Y.-Y., Yang Z., Streuli C. H., Lee Y.-J. (2012) The RhoA-Rok-myosin II pathway is involved in extracellular matrix-mediated regulation of prolactin signaling in mammary epithelial cells. J. Cell. Physiol. 227, 1553–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Plotnikov A., Li Y., Tran T. H., Tang W., Palazzo J. P., Rui H., Fuchs S. Y. (2008) Oncogene-mediated inhibition of glycogen synthase kinase 3β impairs degradation of prolactin receptor. Cancer Res. 68, 1354–1361 [DOI] [PubMed] [Google Scholar]