

Background: Sphingolipid ceramide regulates cellular responses to stress stimuli.
Results: Aldh1l1, the enzyme regulating folate metabolism, leads to CerS6 up-regulation and C16-ceramide accumulation in a p53-dependent manner as a proapoptotic signal.
Conclusion: Ceramide mediates the cellular response to nongenotoxic folate stress.
Significance: We have demonstrated the interaction between two major metabolic pathways, folate and sphingolipids, in regulation of cellular homeostasis.
Keywords: Apoptosis, Ceramide, Folate, p53, Signal Transduction, Stress Response, Aldh1l1, CerS6, PUMA
Abstract
We have investigated the role of ceramide in the cellular adaptation to folate stress induced by Aldh1l1, the enzyme involved in the regulation of folate metabolism. Our previous studies demonstrated that Aldh1l1, similar to folate deficiency, evokes metabolic stress and causes apoptosis in cancer cells. Here we report that the expression of Aldh1l1 in A549 or HCT116 cells results in the elevation of C16-ceramide and a transient up-regulation of ceramide synthase 6 (CerS6) mRNA and protein. Pretreatment with ceramide synthesis inhibitors myriocin and fumonisin B1 or siRNA silencing of CerS6 prevented C16-ceramide accumulation and rescued cells supporting the role of CerS6/C16-ceramide as effectors of Aldh1l1-induced apoptosis. The CerS6 activation by Aldh1l1 and increased ceramide generation were p53-dependent; this effect was ablated in p53-null cells. Furthermore, the expression of wild type p53 but not transcriptionally inactive R175H p53 mutant strongly elevated CerS6. Also, this dominant negative mutant prevented accumulation of CerS6 in response to Aldh1l1, indicating that CerS6 is a transcriptional target of p53. In support of this mechanism, bioinformatics analysis revealed the p53 binding site 3 kb downstream of the CerS6 transcription start. Interestingly, ceramide elevation in response to Aldh1l1 was inhibited by silencing of PUMA, a proapoptotic downstream effector of p53 whereas the transient expression of CerS6 elevated PUMA in a p53-dependent manner indicating reciprocal relationships between ceramide and p53/PUMA pathways. Importantly, folate withdrawal also induced CerS6/C16-ceramide elevation accompanied by p53 accumulation. Overall, these novel findings link folate and de novo ceramide pathways in cellular stress response.
Introduction
Ceramide, a central molecule in sphingolipid metabolism, has emerged in the past several decades as a key bioeffector in cellular adaptation to stress (1–4). Stress-induced ceramide accumulation, which is often a proapoptotic signal (5), can occur via either de novo or salvage pathways (2). Ceramides with distinct acyl chain length are synthesized by a family of ceramide synthases, consisting of six members, CerS1–66 (4, 6). Activation of the sphingomyelin pathway has been linked to such apoptosis-inducing stimuli as ionizing radiation (7), FAS ligand (8), and TNF-α (9–11). The de novo pathway, in turn, has emerged as the one activated by treatment of cancer cells with chemotherapeutics including etoposide (12), daunorubicin (13), or gemcitabine (14).
Ceramide accumulation has also been implicated in the cellular response to nutrient deprivation. For example, serum starvation in Molt-4 leukemia cells causes a significant increase in ceramide, the effect contributing to the mechanism of G0/G1 arrest (15). Furthermore, ceramide can activate autophagy as a response mechanism to nutrient starvation (16, 17). Interestingly though, ceramide is able to trigger autophagy in the presence of extracellular nutrients (18) whereas ceramide-related prosurvival sphingolipid, sphingosine 1-phosphate, regulates autophagy in response to nutrient starvation as a protective mechanism against cell death (19).
Folate, one of the essential nutrients, is a water-soluble vitamin required for numerous reactions of one-carbon transfer (20). Several of these reactions are involved in de novo purine and thymidylate pathways and thus are vital for nucleic acid biosynthesis (20, 21). Folate also participates in the regeneration of methionine from homocysteine, a process linked to the biosynthesis of S-adenosylmethionine, a key molecule for methylation reactions in the cell (20, 21). In agreement with the fundamental role of folate for cellular processes, folate deficiency causes dramatic consequences including altered protein expression (22–24), accumulation of DNA damage, increased chromosomal aberrations and fragility (25, 26), reduced growth rate, and impaired cell division (27). Folate availability and efficient folate metabolism are especially crucial for rapidly proliferating cells, including cancer cells that serves as the basis for treatment of cancers with folate antimetabolites (antifolates), compounds inhibiting folate-metabolizing enzymes (28, 29).
One of the folate enzymes, 10-formyltetrahydrofolate dehydrogenase (Aldh1l1 or FDH), converts 10-formyltetrahydrofolate to tetrahydrofolate and CO2 in a NADP+-dependent dehydrogenase reaction (30). This reaction removes carbon groups (as CO2) from the intracellular folate pool, thus fulfilling a catabolic function. It has been proposed that Aldh1l1 plays a regulatory role by controlling the flux of activated one-carbon groups toward biosynthetic processes (30, 31).
Aldh1l1 is an abundant cytosolic protein, which is ubiquitously down-regulated in human cancers (31–33). This down-regulation is achieved through extensive methylation of the ALDH1L1 promoter and apparently serves the purpose of relieving transformed cells from one of the proliferation controlling mechanisms (34). In agreement with this mechanism, reconstitution of Aldh1l1 in Aldh1l1-deficient cancer cells produces strong antiproliferative effects including cell cycle arrest (35, 36), inhibition of motility (37), and apoptosis (35). The antiproliferative effect of Aldh1l1, resulting from the decrease in the intracellular purine levels (38) and impaired folate-dependent homocysteine remethylation (39), is mediated by numerous downstream effectors including p53, p21, PUMA, JNK1/2, c-Jun, caspases 3, 8, and 9, protein phosphatase 1/protein phosphatase 2A, and cofilin (36–38, 40, 41).
Although alterations of either ceramide or folate metabolism can induce stress responses, these two major metabolic pathways have never been linked before in the context of cellular signaling network. In the present study, we investigated the role of ceramide in cellular response to Aldh1l1 and demonstrated that similar response is induced upon folate withdrawal.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Generation of Tet-On A549/Aldh1l1 cells and A549 cells with p53 silenced by shRNA was described previously (35, 38). Cells were grown in F-12 medium (Mediatech) supplemented with 10% (v/v) Tet-On certified (Clontech) or regular (Atlanta Biologicals) fetal bovine serum, respectively, at 37 °C under humidified air containing 5% CO2. HCT116 and HCT116 p53−/− cell lines (a gift from Dr. Bert Vogelstein, The Sidney Kimmel Comprehensive Cancer Center, John Hopkins University School Medicine) were grown in McCoy's medium. Myriocin and fumonisin B1 were purchased from Sigma and Enzo, respectively. For folate depletion experiments, we used folate-free RPMI 1640 medium containing 10% dialyzed fetal bovine serum (both from Invitrogen).
Transient Transfection
Cells (∼1.0 × 106) were transfected with 2 μg of corresponding expression vector using either a Amaxa nucleofector kit V (Lonza) or a Neon transfection system (Invitrogen) according to the manufacturers' protocols. In control experiments, respective “empty” plasmids were used for transfection. The pCEP4-175 vector (for the expression of R175H p53 mutant) was a kind gift from Dr. Jennifer Pietenpol; pCEP4/PUMA (PUMA expression) was obtained from Addgene; pcDNA3.1/Aldh1l1 (Aldh1l1 expression), pCEP4/p53 (wild type p53 expression) and pCMV/CerS6 (CerS6 expression) were described previously (31, 38, 43).
Real Time PCR
Total RNA was purified using RNA Easy® Mini Kit (Qiagen). Reverse transcriptase reaction was performed with an oligo(dT)18 primer using AdvantageTM RT-for-PCR Kit (Clontech). The resulting cDNA was used to measure CerS1–6 mRNA levels using MyiQTM Single-Color Real Time PCR detection system (Bio-Rad) and iQ5 optical system software (Bio-Rad). CerS1–6 quantitative RT-PCR primers are shown in Table 1. β-Actin mRNA levels were used to normalize samples.
TABLE 1.
Primers for real time PCR of ceramide synthases
| Gene | Orientation | Sequence |
|---|---|---|
| β-Actin | Forward | 5′-ATT GGC AAT GAG CGG TTC C-3′ |
| Reverse | 5′-GGT AGT TTC GTG GAT GCC ACA-3′ | |
| CerS1 | Forward | 5′-ACG CTA CGC TAT ACA TGG ACA C-3′ |
| Reverse | 5′-AGG AGG AGA CGA TGA GGA TGA G-3′ | |
| CerS2 | Forward | 5′-CCG ATT ACC TGC TGG AGT CAG-3′ |
| Reverse | 5′-GGC GAA GAC GAT GAA GAT GTT G-3′ | |
| CerS3 | Forward | 5′-ACA TTC CAC AAG GCA ACC ATT G-3′ |
| Reverse | 5′-CTC TTG ATT CCG CCG ACT CC-3′ | |
| CerS4 | Forward | 5′-CTT CGT GGC GGT CAT CCT G-3′ |
| Reverse | 5′-TGT AAC AGC AGC ACC AGA GAG-3′ | |
| CerS5 | Forward | 5′-GTT TCG CCA TCG GAG GAA TC-3′ |
| Reverse | 5′-GCC AGC ACT GTC GGA TGT C-3′ | |
| CerS6 | Forward | 5′-GGG ATC TTA GCC TGG TTC TGG-3′ |
| Reverse | 5′-GCC TCC TCC GTG TTC TTC AG-3′ |
siRNA
Knockdown of CerS6 and CerS2 was performed using siRNA duplexes purchased from Qiagen (Table 2) with the targeting sequences AACGCTGGTCCTTTGTCTTCA and AAGGAACAGATCATCCACCAT, correspondingly (43). Tet-On A549/Aldh1l1 cells (∼1.5 × 105) were transfected with 25 nmol of CerS6 siRNA using 5–10 μl of Lipofectamine 2000 (Invitrogen). Scrambled siRNA with medium GC content (Invitrogen) was used as a control. Transfections were performed following the manufacturer's protocol. Silencing of PUMA by siRNA was carried out as we described previously (36). Tet-On A549/Aldh1l1 cells (1.2 × 105) were transfected with 25 nmol of Stealth RNAi (Invitrogen) using 5–10 μl of Lipofectamine 2000. Scrambled Stealth RNAi was used as a negative control. Transfection was performed following the manufacturer's protocols.
TABLE 2.
siRNA sequences used in this work
| Target | Orientation | Sequence |
|---|---|---|
| CerS2 | Sense | r(GGA ACA GAU CAU CCA CCA U)dTdT |
| Antisense | r(AUG GUG GAU GAU CUG UUC C)dTdT | |
| CerS6 | Sense | r(CGC UGG UCC UUU GUC UUC A)dTdT |
| Antisense | r(UGA AGA CAA AGG ACC AGC G)dTdT |
Western Blot Assays
Cell lysates were prepared in buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, 0.1% SDS, 1 mm PMSF, and mammalian protease inhibitor mixture (Sigma). Lysates were subjected to SDS-PAGE followed by Western blotting with specified antibodies. Expression of Aldh1l1 was verified with an in-house Aldh1l1-specific polyclonal antibody (1:10,000) (31, 35, 38). CerS6 polyclonal antibody (1:1000) was purchased from Novus Biologicals. CerS2 polyclonal antibody (1:200) was from Exalpha Biologicals (Shirley, MA). Monoclonal p53 antibody (0.1 μg/mg) was purchased from Calbiochem. PUMA polyclonal antibody was from Cell Signaling (1:1000). Actin was detected using a monoclonal antibody from Sigma (clone AC-15, 1:5000).
Cell Proliferation and Apoptosis Assays
Cell viability was assessed using an MTT cell proliferation assay (Promega). Cells were plated at a density of 5 × 103 cells/well in 96-well format, and MTT was added at specified time points. Plates were further processed according to the manufacturer's instructions. A570 nm was read using a Wallace 1420 multilabel counter (PerkinElmer Life Sciences). Apoptotic cells were detected by annexin V and PI labeling using an Annexin V-FLUOS staining kit (Roche Applied Science). All cells (floating and attached) were used in these experiments. Samples were analyzed at the Flow Cytometry core facility, Hollings Cancer Center, using a BD Biosciences FACSCalibur and Mod Fit software.
Analysis of Lipids by LC-MS/MS
Approximately 1.5 × 106 cells were trypsinized and washed twice with cold PBS. Samples were centrifuged at 1000 rpm for 5 min at 4 °C, and the final pellet was stored at −80 °C prior to analysis. Further preparation of samples and measurement of endogenous ceramides by LC-MS/MS followed the protocol described previously (44). Briefly, samples were fortified with internal standards, and 2 ml of isopropyl alcohol:water:ethyl acetate (30:10:60; v:v:v) was added to the cell pellet mixtures. Samples were subjected to two rounds of vortex and sonication followed by a 10-min centrifugation at 4000 rpm. The supernatant or top layer was used as lipid extract and subjected to LC-MS/MS for analysis of ceramide species. Samples were normalized to total inorganic phosphate (Pi) levels. Lipid extraction and analyses were performed by the MUSC Lipidomics Core facility.
RESULTS
Expression of Aldh1l1 in A549 Cells Induces Ceramide Accumulation
Our previous studies demonstrated that Aldh1l1 expression in deficient cancer cells induces strong metabolic alterations and apoptosis (35, 36, 38). Because involvement of proapoptotic ceramide signaling in these effects has not been studied previously, we investigated whether Aldh1l1-induced folate stress leads to ceramide generation. We have measured levels of a variety of sphingolipids in A549 cells at different time points after Aldh1l1 induction using LC-MS/MS. The notable and statistically significant effect of Aldh1l1 was observed for C16-, C24-, C24:1-, and dhC16-ceramides, which were elevated up to 3.6-, 2.5-, 2.2-, and 3.7-fold, respectively, 72 h after Aldh1l1 induction (Fig. 1A). The effect of Aldh1l1 on other ceramide species was less profound and not statistically significant (Fig. 1A). Elevation for C16-, C24-, C24:1-, and dhC16-ceramides was also seen at 48 h after Aldh1l1 induction, although to a lesser extent (data not shown). These data suggest that the proapoptotic Aldh1l1 folate stress response includes ceramide accumulation.
FIGURE 1.

Ceramide synthase-dependent accumulation of ceramide in response to Aldh1l1. A, levels of ceramide in Tet-On A549/Aldh1l1 cells 72 h after Aldh1l1 induction. Error bars represent ± S.D., n = 3. Inset, levels of Aldh1l1 in Tet-On A549 cells at different postinduction times (actin is shown as loading control). B, levels of CerS1–6 mRNA (quantitative real time PCR) at 24, 36, and 48 h after Aldh1l1 induction. Error bars represent ± S.D., n = 3. C, levels of total ceramide (LC-MS/MS) in Aldh1l1-expressing Tet-On A549 cells (72 h after induction) pretreated with either MYR (50 nm) or FB1 (50 μm). Untreated cells with and without Aldh1l1 expression (control) are included. Error bars represent ± S.D., n = 3. D, MTT assay of MYR-pretreated Tet-On A549 cells with (Aldh1l1+MYR) or without Aldh1l1 induction (MYR). Negative (control, untreated cells) and positive (Aldh1l1, untreated cells expressing Aldh1l1) controls are shown. Time after Aldh1l1 induction is indicated. Error bars represent ± S.D., n = 4. E, same as D, but FB1 was used instead of MYR. For statistical analysis Student's t test was performed. Statistically significant changes (p < 0.005) are marked with an asterisk (*).
CerS6 Is Up-regulated in Response to Aldh1l1
Ceramide biosynthesis is catalyzed by a set of six ceramide synthase enzymes, CerS1–6 (3, 6). We have examined the mRNA levels of all six enzymes in Aldh1l1-expressing cells by quantitative real time PCR. These experiments revealed a significant (p < 0.005) increase in mRNA levels for two of the enzymes, CerS4 and CerS6, 36 h after Aldh1l1 induction (approximately 3.2- and 5.5-fold, respectively; Fig. 1B). Levels of CerS3 and CerS5 mRNA were only marginally changed between 24 and 48 h after Aldh1l1 induction; the increase in CerS1 and CerS2 mRNA was notable, but not statistically significant (Fig. 1B). Interestingly, the increase in CerS4 and CerS6 mRNA in response to Aldh1l1 was transient: their mRNA levels returned to the values observed in control cells 48 h after Aldh1l1 induction (Fig. 1B). Similarly, an increase in CerS6 protein was also observed at 24 h after Aldh1l1 induction, which persisted up to 72 h, as detected by Western blotting (Fig. 2A). Overall, these data indicate that Aldh1l1-induced CerS6 expression is regulated at mRNA and protein levels.
FIGURE 2.

CerS6 mediates effects of Aldh1l1 in Tet-On A549 cells. A, levels of CerS6 protein (Western blotting) after Aldh1l1 induction. B, CerS6 mRNA (quantitative real time PCR) upon siRNA knockdown. Scrambled (Scr) siRNA was used as a negative control. C, levels (Western blotting) of Aldh1l1 and CerS6 at 48 h after transfection with CerS6 siRNA (siRNA) or scrambled siRNA in Tet-On A549 cells with (+) or without (−) Aldh1l1 expression. Actin is shown as loading control. D, ceramide levels in Tet-On A549 cells transfected with scrambled siRNA or CerS6 siRNA (CerS6) with or without Aldh1l1 (72 h after induction). E, MTT assay of cells transfected with scrambled or CerS6 siRNA in the presence or absence of Aldh1l1. Time after Aldh1l1 induction is shown. Error bars represent ± S.D., n = 3 (B and D), n = 4 (E). #, p < 0.01; *, p < 0.005; **, p < 0.0005. F and G, annexin V/PI assays of control, Aldh1l1-expressing, and Aldh1l1-expressing/CerS6 siRNA-transfected cells (F represents calculations of data from G).
Inhibition of de Novo Ceramide Generation Prevents Aldh1l1-induced Apoptosis
To determine whether the elevated ceramide mediates Aldh1l1-induced cytotoxic effects, we have evaluated cellular proliferation and apoptosis in Aldh1l1-expressing cells treated with inhibitors of ceramide biosynthesis. Two compounds were used: myriocin (MYR, an inhibitor of serine palmitoyl transferase/de novo biosynthesis (45)) and fumonisin B1 (FB1, a ceramide synthase inhibitor (46)). Aldh1l1 was induced immediately following pretreatment of cells for 6 h with either MYR (50 nm) or FB1 (50 μm). After 6 h preincubation inhibitors were washed out, and cells were kept on regular inhibitor-free medium. We observed that both MYR and FB1 prevented the total ceramide elevation in response to Aldh1l1 (Fig. 1C). Furthermore, MTT assays have demonstrated that Aldh1l1-expressing cells pretreated with MYR or FB1 undergo normal proliferation similar to Aldh1l1-deficient cells (Fig. 1, D and E). In agreement with this finding, annexin V/PI analysis showed that inhibition of ceramide pathways almost completely protected Aldh1l1-expressing cells from apoptosis (Fig. 2, F and G). These data indicate that Aldh1l1-induced apoptosis requires increased de novo ceramide generation.
CerS6 Knockdown Rescues Cells from Aldh1l1-induced Apoptosis
The elevation of C16-ceramide and dhC16-ceramide, observed in our experiments, was in agreement with the increased levels of CerS6 mRNA because the enzyme is specifically involved in this ceramide generation (47). To determine whether CerS6 up-regulation was responsible for the increase in C16-ceramide upon Aldh1l1 expression, we knocked down this ceramide synthase in Tet-On A549/Aldh1l1 cells using the siRNA approach. Quantitative real time PCR showed the strong decrease of CerS6 at 36 and 48 h after siRNA transfection compared with transfection with scrambled siRNA (Fig. 2B). The knockdown was confirmed at the protein level by Western blot analysis: very prominent decrease of CerS6 at 48 h was seen in these experiments (Fig. 2C). LC-MS/MS analysis has further shown that CerS6 knockdown prevented the increase in C16-, C24-, and C24:1-ceramide in response to Aldh1l1 (Fig. 2D). MTT proliferation assays revealed a dramatic increase in viable cell number upon CerS6 siRNA compared with scrambled control at 48 h of Aldh1l1 induction (Fig. 2E). This effect was even more profound at 72 h after Aldh1l1 induction (Fig. 2E), indicating a remarkable rescue effect in the absence of CerS6. Annexin V/PI staining has further shown that cells deficient in CerS6 or treated with ceramide synthesis inhibitors myriocin or fumonisin B1 were protected from Aldh1l1-induced apoptosis (Fig. 2, F and G). Thus, these experiments established CerS6/C16-ceramide as an essential mediator of cellular responses to Aldh1l1.
Because in our experiments we also observed the elevation of C24- and C24:1-ceramide, species produced by CerS2 (3), we investigated the role of this enzyme in mediation of Aldh1l1 responses. The knockdown of CerS2 by siRNA did not rescue cells from Aldh1l1 (Fig. 3), indicating that this ceramide synthase is not involved in the Aldh1l1 stress response.
FIGURE 3.

siRNA silencing of CerS2 does not protect A549 cells from the Aldh1l1 antiproliferative effect. MTT assay of cells transfected with scrambled or CerS2 siRNA in the presence or absence of Aldh1l1 is shown, including time after Aldh1l1 induction. Aldh1l1 induction was done simultaneously with siRNA transfection. Error bars represent ± S.D., n = 3. Statistically significant changes (p < 0.005) are marked with an asterisk (*). Inset shows levels (Western blots) of Aldh1l1 and CerS2 at different time points (24–72 h) of Aldh1l1 induction/CerS2 siRNA transfection. Scr, scrambled siRNA. Actin is shown as loading control.
Aldh1l1-induced CerS6 Elevation and Ceramide Accumulation Are p53-dependent
A p53-dependent ceramide accumulation has been shown in response to actinomycin D (48), γ-irradiation (48, 49), and TNF-α (50). Likewise, previous studies from our laboratory demonstrated that Aldh1l1-induced apoptosis in several cell lines is mediated by p53 (38). Using HCT116 isogenic cell lines (p53+/+ and p53−/−), we examined whether Aldh1l1 induces CerS6 and ceramide accumulation in a p53-dependent manner. We observed that in response to the Aldh1l1 transient transfection both CerS6 and C16-ceramide were elevated in p53+/+ but not p53−/− cells (Fig. 4, A and B). Similar to Tet-On A549/Aldh1l1 cells, HCT116 p53+/+ cells have also shown a significant increase in C24-ceramide but not C24:1-ceramide (Fig. 4B). In contrast, HCT116 p53−/− cells did not reveal an increase in any of these ceramides upon Aldh1l1 expression (Fig. 4B). Of note, HCT116 p53−/− cells are resistant to Aldh1l1-induced apoptosis in contrast to HCT116 p53+/+ cells (Fig. 4C). Similar results were obtained in experiments with A549 cell line and its derivative, in which p53 was silenced by shRNA (38): CerS6 and ceramide levels were elevated in p53-proficient but not -deficient cells in response to Aldh1l1 (Fig. 5). Altogether, our data demonstrate that p53 is required for ceramide accumulation and CerS6 induction upon Aldh1l1 stress.
FIGURE 4.

Effects of Aldh1l1 on CerS6 and ceramide levels in HCT116 p53+/+ and p53−/− cells. A, levels of CerS6 in HCT116 p53+/+ and HCT116 p53−/− cells at different time points after Aldh1l1 transfection. Actin was included as loading control. B, levels of C16-, C24-, and C24:1-ceramides in HCT116 p53+/+ and p53−/− cells transfected with Aldh1l1. Error bars represent ± S.D., n = 3. Statistically significant changes (p < 0.005) are marked with an asterisk (*). C, annexin V/PI staining of HCT116 and HCT116 p53−/− cells transfected with empty (control) or Aldh1l1-expressing vector (+Aldh1l1) at 48 h after transfection.
FIGURE 5.

Effects of Aldh1l1 on CerS6 and ceramide levels in A549 p53-proficient and p53-knockdown cells. A, levels of CerS6 at different time points after Aldh1l1 transfection. Actin was included as loading control. B, levels of C16-, C24-, and C24:1-ceramides in the same cells transfected with Aldh1l1. Error bars represent ± S.D., n = 3, p < 0.005 is marked with an asterisk (*).
Transient Expression of CerS6 Produces p53-dependent Antiproliferative Effects
Because in our experiments the CerS6 elevation was a required event in the mediation of the Aldh1l1 cytotoxic effect, we investigated whether CerS6 itself can induce an antiprolioferative effect. Indeed, we observed that transient expression of CerS6 in p53-proficient A549 cells inhibits proliferation and is associated with the elevation of p53 and its downstream target PUMA (Fig. 6A). In contrast, the expression of CerS6 in p53-deficient A549 cells did not inhibit proliferation (Fig. 6B). Of note, PUMA was not elevated in these cells in response to CerS6 (Fig. 6B).
FIGURE 6.

Transient expression of CerS6 produces antiproliferative effects in 53-proficient (A) but not p53-deficient (B) A549 cells. Insets show levels of transiently expressed CerS6 and associated levels of p53 and p53 downstream target PUMA. Actin is shown as loading control. Error bars represent ± S.D., n = 4, p < 0.005 is marked with an asterisk (*).
PUMA Is Involved in Aldh1l1-induced Ceramide Accumulation
To our knowledge, PUMA has not been yet identified as an upstream regulator or downstream target of ceramide. To investigate whether PUMA plays a role in Aldh1l1-induced p53-dependent ceramide accumulation, we evaluated changes in ceramide levels in A549 cells in response to Aldh1l1 upon siRNA-mediated PUMA knockdown. Down-regulation of PUMA (as confirmed by Western blotting, Fig. 7A, inset) significantly (p < 0.05) prevented C16-ceramide accumulation (Fig. 7A), indicating the involvement of PUMA, downstream of p53, in mediation of ceramide generation in response to Aldh1l1 folate stress. Likewise, PUMA down-regulation in our experiments inhibited Aldh1l1-induced apoptosis (Fig. 7B) in agreement with our previous report (36). Furthermore, the silencing of PUMA also rescues the inhibitory effect of CerS6 in p53-proficient cells (Fig. 7C). These findings imply a role for PUMA as both a target of ceramide and a mediator of ceramide effects.
FIGURE 7.
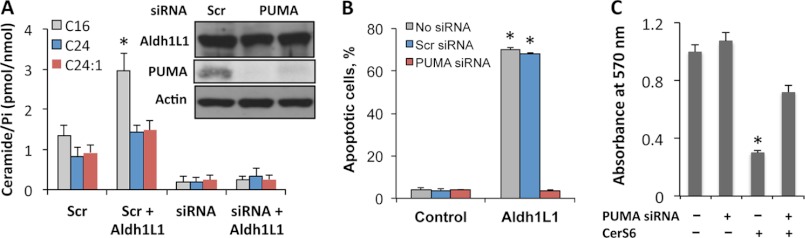
Knockdown of PUMA prevents Aldh1l1-induced ceramide increase in A549 cells. A, levels of ceramide species in cells co-transfected with Aldh1l1-expressing plasmid and scrambled or PUMA siRNA. Inset, levels of PUMA in cells co-transfected with Aldh1l1-expressing vector and scrambled or PUMA siRNA (time after transfection is shown). Cells transfected with scrambled or PUMA siRNA in the absence of Aldh1l1 are shown as negative controls. B, percentage of apoptotic cells (annexin V/PI assays) in the presence or absence (Control) of Aldh1l1 upon transfection with scrambled or PUMA siRNA. C, siRNA silencing of PUMA rescuing cells from CerS6. Error bars represent ± S.D., n = 3; *, p < 0.005.
CerS6 Is Up-regulated by Wild Type p53
Whereas p53 regulates expression of a whole array of genes, CerS6 was not established as a p53 transcriptional target. In our experiments, transient expression of wild type p53 in A549 cells resulted in strong elevation of CerS6 protein (Fig. 8A). At the same time, the expression of the R175H p53 mutant deficient in DNA binding did not increase CerS6 levels (Fig. 8A). Interestingly, silencing of PUMA prevented ceramide accumulation in response to Aldh1l1 (Fig. 7A). However, a transient expression of PUMA did not affect CerS6 levels (Fig. 8B), suggesting the lack of its direct effect on CerS6 expression and implying a different effect of mediation of ceramide levels by PUMA. The direct effect of p53 on CerS6 expression was further demonstrated in our experiments with transient expression of the R175H p53 mutant simultaneously with the induction of Aldh1l1: this dominant negative mutant prevented elevation of CerS6 in response to Aldh1l1 (Fig. 8C).
FIGURE 8.
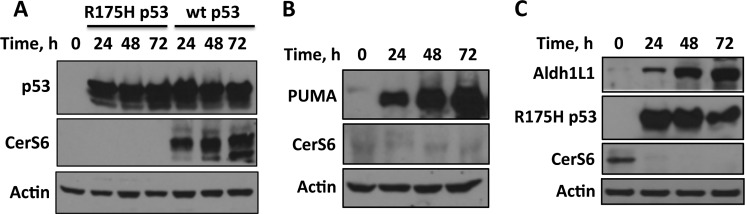
CerS6 is a transcriptional target of p53. A, levels of p53 and CerS6 in A549 cells transiently transfected with wild type p53 or the R175H mutant are shown. B, transient expression of PUMA does not affect CerS6 levels. C, transient expression of the R175H p53 mutant prevents elevation of CerS6 in response to Aldh1l1.
Folate Withdrawal Results in CerS6 Elevation and Ceramide Accumulation
We have also measured levels of sphingolipids in A549 cells kept on folate-free medium. Similar to Aldh1l1 expression, folate withdrawal resulted in elevation of C16-, C24-, C24:1-, and dhC16-ceramides, which were elevated 4.3-, 2.7-, 4.3-, and 11-fold, respectively, after 2 weeks in folate-free medium (Fig. 9A). In addition, C14-, C18-, C20-, and C22-ceramides were strongly elevated (Fig. 9A). The analysis of mRNA levels of six ceramide synthases in folate-depleted cells by quantitative real time PCR has shown a pattern similar to that observed for Aldh1l1-expressing cells with the strongest elevation of CerS4 and CerS6 and marginal changes for CerS3 and CerS5 (Fig. 9B). In line with the data obtained in Aldh1l1-expressing cells, we have seen elevated levels of p53 and CerS6 proteins in folate-depleted cells (Fig. 9C).
FIGURE 9.

Accumulation of ceramide and elevation of ceramide synthases in A549 cells in response to folate withdrawal. A, levels of ceramide in cells kept on normal (control) or folate-free medium. B, levels of CerS1–6 mRNA (quantitative real time PCR) after folate withdrawal. C, elevation of p53 and CerS6 proteins in folate-depleted cells. Actin is shown as loading control. Error bars represent ± S.D., n = 2; *, p < 0.005.
DISCUSSION
Numerous studies have connected ceramide pathways with induction of apoptosis in response to stress stimuli (5). Among others, these stimuli include starvation, such as serum or nutrient deprivation (15, 19). Folate is one of the essential nutrients, which cannot be synthesized by mammalian cells (20). Accordingly, folate deficiency produces dramatic effects on cellular homeostasis, eventually inhibiting proliferation and inducing apoptosis (51). Interestingly, whereas the relationship between ceramide and folate pathways is largely unknown, it has been reported that treatment of Molt-4 human T cell leukemia cells with the thymidylate synthase inhibitor GW1843 results in ceramide accumulation (52). Because the inhibitor affects one of the folate-dependent pathways, this finding indicates that the ceramide signaling could play a role in initiating cell death in response to the disruption of folate metabolism. Effects similar to folate deficiency or antifolate treatment can be also achieved by up-regulation of Aldh1l1, a common folate enzyme, which has been implicated as a metabolic regulator controlling cellular proliferation (31). In agreement with this role, the protein is strongly and ubiquitously down-regulated in human tumors and cancer cell lines with its reexpression evoking strong cytotoxicity by induction of apoptosis (35). The Aldh1l1-induced apoptosis is a complex cellular response involving a variety of downstream mediators (36–38, 40, 41). A possible link between this major folate regulatory enzyme and ceramide metabolism has not been pursued previously.
The increase in levels of C16-, C24-, and C24:1-ceramides upon Aldh1l1 expression, observed in our experiments, suggested that the ceramide signaling pathways are involved in folate stress response. This increase could be a nonspecific postapoptotic accumulation of ceramide species. However, the fact that ceramide synthesis inhibitors MYR and FB1 protected cells from Aldh1l1-induced apoptosis indicates otherwise and points to ceramide as an essential downstream mediator of Aldh1l1 effects. The inhibitory effect of MYR and FB1 on Aldh1l1-induced apoptosis also suggested that ceramide accumulation in response to Aldh1l1 is likely a result of the activation of de novo ceramide biosynthesis because both compounds target enzymes in this pathway. The accumulation of dhC16-ceramide was also in agreement with activation of the de novo pathway. In support of this mechanism, we observed a substantial elevation of CerS6 at both mRNA and protein levels in response to Aldh1l1. Interestingly, this increase was temporal and did not sustain beyond 48 h. Because CerS6 produces preferentially shorter chain ceramide species, including C16 (47), the above data are in agreement with the type of ceramide accumulated in response to Aldh1l1, through CerS6 induction. Surprisingly, we did not observe the elevation of C18- or C20-ceramide as could be expected based on the increase of CerS4 mRNA (3). The increase in C24-ceramide is in agreement with notable (although not statistically significant) increase in CerS2 mRNA. Of note, the increase in CerS2 mRNA (p < 0.05, n = 2) concomitant with the C24-ceramide elevation was also seen upon folate depletion. A recent report highlighted heterodimerization of CerS enzymes as a mechanism affecting ceramide production (53). Relevant to our data, it has been shown that CerS6 activates CerS2 upon their heterodimerization. In this regard, it is likely that such mechanism can regulate specific ceramides in response to folate stress. The finding that silencing CerS6 prevented accumulation of C24- and C24:1-ceramide as well (Fig. 2D) supports this possibility. The lack of the effect of CerS2 siRNA knockdown on the antiproliferative function of Aldh1l1 is in line with this mechanism and further underscores CerS6 as the mediator of the stress response.
In cancer cells, the activation of CerS6 was mainly associated with proapoptotic responses (54–56), although an opposite effect was observed in human head and neck squamous cell carcinomas (43). In our study, the support for the proapoptotic role of CerS6 in cellular response to Aldh1l1 was obtained in siRNA knockdown experiments. Importantly, the knockdown of CerS6 in A549 cells not only eliminated C16-ceramide accumulation upon Aldh1l1 expression but also completely prevented Aldh1l1-induced apoptosis, thus indicating that this ceramide synthase is a part of the Aldh1l1 apoptotic signaling.
Aldh1l1 induces several apoptotic pathways but in p53-proficient cells the activation of p53 is a required event (35, 38). Thus, silencing of p53 in A549 or HCT116 cells completely rescued them from Aldh1l1-induced apoptosis (38). Whereas the relationship between p53 and ceramide accumulation in response to stress perhaps depends on the insult and might be cell type-specific, several studies support the role of ceramide as a downstream effector of p53 in apoptosis induction. For example, the ceramide accumulation following treatment with low concentrations of actinomycin D or γ-irradiation was p53-dependent in Molt-4 leukemia cells (48). Likewise, TNF-α-stimulated ceramide generation does not take place in cells deficient in p53 or expressing mutant protein (57). Results of our study are in line with these observations: no changes in ceramide accumulation in response to Aldh1l1 were observed in p53-deficient A549 or HCT116 cells. Furthermore, Aldh1l1 does not evoke CerS6 accumulation in the absence of p53. Because in p53-proficient cells we observed accumulation of both Cers6 mRNA and protein, these results could be interpreted as the transcriptional activation of the enzyme by p53. Whereas p53 consensus binding motifs were not found in the CerS6 promoter (58), the p53 activation can perhaps result in up-regulation of CerS6 indirectly through other transcription factors, mRNA stability, microRNA, or protein degradation. However, our results indicate that CerS6 is a likely transcriptional target of p53. Indeed, it was up-regulated upon transient expression of wild type p53 but not the R175H p53 mutant, which does not bind DNA (59). Moreover, this dominant negative mutant prevented accumulation of CerS6 in response to Aldh1l1. In further support of this mechanism, our search using SABiosciences Transcription Factor Search Portal revealed the presence of the p53 binding site at approximately 3 kb downstream of the CerS6 transcription start site.
The main downstream mediator of the p53 apoptotic response is PUMA, a proapoptotic BH-3-only member of the Bcl-2 family of proteins (60). It binds antiapoptotic Bcl-2, thereby relieving the inhibition of Bax and/or Bak by Bcl-2 (61). PUMA has been also shown to directly activate Bax and assist its membrane association and oligomerization, independent of Bax interaction with Bcl-2 (61). PUMA is essential for p53-dependent apoptosis in response to a variety of genotoxic stimuli, including adriamycin, fluorouracil, cisplatin, etoposide, and UV radiation, but it can be also activated by nongenotoxic stress (62). One of the examples of the latter is the Aldh1l1-induced apoptosis, which takes place in response to nucleotide deprivation in the absence of detectable DNA damage (36, 38). Of note, PUMA was strongly activated in p53-proficient cells in response to Aldh1l1 whereas its siRNA silencing completely prevented cell death in this system (36). Surprisingly, the present study also demonstrated that the silencing of PUMA prevented ceramide accumulation in response to Aldh1l1, a finding implicating PUMA as an upstream activator of ceramide generation. Whereas precise mechanisms of the PUMA involvement in the ceramide production are not clear at present, several lines of evidence suggested a connection between ceramide and proteins of the Bcl-2 family. Thus, in the case of etoposide-treated C6 glioma cells, ceramide was found to play a role in the increase of the Bax/Bcl-2 ratio (63). Sphingolipid metabolism has been further implicated in co-operation with BAK/BAX activation in the induction of apoptosis (64). Importantly, a recent study demonstrated that, in response to apoptotic stimuli, the proapoptotic Bcl-2 family member Bak mediates an increase in long chain ceramide via the de novo pathway through the activation of CerS enzymes (42). Furthermore, our findings that CerS6 activates PUMA expression in a p53-dependent manner indicate reciprocal relationships between ceramide pathways and this proapoptotic effector.
Overall, our study revealed that ceramide metabolism responds to Aldh1l1-induced folate stress and pointed toward CerS6 as an essential downstream mediator of Aldh1l1-induced p53-dependent apoptosis. Importantly, our experiments with folate-depleted cells further demonstrated that this mechanism is not limited to Aldh1l1 but is a more general response to folate stress. Mechanistically, this study demonstrated that ceramide signaling is regulated by p53 upon nongenotoxic stress and highlighted two downstream effectors, a canonical p53 target, PUMA, and a novel target, CerS6 (schematically depicted in Fig. 10). Of note, PUMA has not been previously implicated in the regulation of ceramide generation. The question of whether these two effectors function independently or co-operatively in ceramide regulation is a matter for future studies. Our finding that proapoptotic functions of CerS6/C16-ceramide are mediated downstream of p53/PUMA signaling implies that cancer cells with altered p53/PUMA axis might respond differently to CerS6/C16-ceramide-induced stress. In this regard, it would be interesting to determine whether the distinct role attributed to CerS6/C16-ceramide in head and neck squamous cell carcinomas (43) is due to alterations of p53/PUMA in these cancers. On a more general notion, both folate and ceramide are involved in fundamental cellular processes such as nucleic acid biosynthesis, methylation, and signaling. Therefore, it is not surprising that the two pathways interplay in the regulation of cellular homeostasis.
FIGURE 10.
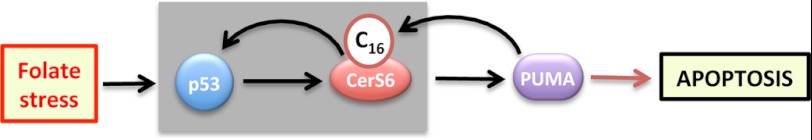
Model for mediation of folate stress by CerS6. Folate stress is sensed by p53 and translated to CerS6 through its transcriptional activation. The elevation of C16-ceramide by CerS6 feeds back to p53 further amplifying stress-response signal through stronger induction of proapoptotic PUMA. PUMA itself positively modulates C16-ceramide generation, which is an additional mechanism to enhance the apoptotic response.
Acknowledgments
We thank Dr. Bert Vogelstein for the HCT116 p53−/− cells, Dr. Jennifer Pietenpol for providing pCEP4-175 vector, and Dr. Can Senkal for valuable advice on CerS6 siRNA.
This work supported, in whole or in part, by National Institutes of Health Grants DK54388 and CA095030 (to S. A. K.). This work was also supported by the South Carolina Lipidomics and Pathobiology Center of Biomedical Research Excellence Grant P20 RR017677 (to N. I. K.) and the U.S. Department of Education Graduate Assistance in Areas of National Need Training Grant P200A100105 (to L. A. H.). The Flow Cytometry and Lipidomics Shared Resource Facilities were supported in part by Cancer Center Grant P30 CA138313 to the Hollings Cancer Center, Medical University of South Carolina.
- CerS
- ceramide synthase
- Aldh1l1
- 10-formyltetrahydrofolate dehydrogenase
- FB1
- fumonisin B1
- MS/MS
- tandem MS
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MYR
- myriocin
- PI
- propidium iodide.
REFERENCES
- 1. Futerman A. H., Hannun Y. A. (2004) The complex life of simple sphingolipids. EMBO Rep. 5, 777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ogretmen B., Hannun Y. A. (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 4, 604–616 [DOI] [PubMed] [Google Scholar]
- 3. Levy M., Futerman A. H. (2010) Mammalian ceramide synthases. IUBMB Life 62, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hannun Y. A., Obeid L. M. (2011) Many ceramides. J. Biol. Chem. 286, 27855–27862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pettus B. J., Chalfant C. E., Hannun Y. A. (2002) Ceramide in apoptosis: an overview and current perspectives. Biochim. Biophys. Acta 1585, 114–125 [DOI] [PubMed] [Google Scholar]
- 6. Mullen T. D., Hannun Y. A., Obeid L. M. (2012) Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 441, 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haimovitz-Friedman A., Kan C. C., Ehleiter D., Persaud R. S., McLoughlin M., Fuks Z., Kolesnick R. N. (1994) Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J. Exp. Med. 180, 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cifone M. G., De Maria R., Roncaioli P., Rippo M. R., Azuma M., Lanier L. L., Santoni A., Testi R. (1994) Apoptotic signaling though CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J. Exp. Med. 180, 1547–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim M. Y., Linardic C., Obeid L., Hannun Y. (1991) Identification of sphingomyelin turnover as an effector mechanism for the action of tumor necrosis factor α and γ-interferon: specific role in cell differentiation. J. Biol. Chem. 266, 484–489 [PubMed] [Google Scholar]
- 10. Dbaibo G. S., Obeid L. M., Hannun Y. A. (1993) Tumor necrosis factor-α (TNF-α) signal transduction through ceramide: dissociation of growth inhibitory effects of TNF-α from activation of nuclear factor-κB. J. Biol. Chem. 268, 17762–17766 [PubMed] [Google Scholar]
- 11. Liu B., Andrieu-Abadie N., Levade T., Zhang P., Obeid L. M., Hannun Y. A. (1998) Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-α-induced cell death. J. Biol. Chem. 273, 11313–11320 [DOI] [PubMed] [Google Scholar]
- 12. Perry D. K., Carton J., Shah A. K., Meredith F., Uhlinger D. J., Hannun Y. A. (2000) Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J. Biol. Chem. 275, 9078–9084 [DOI] [PubMed] [Google Scholar]
- 13. Bose R., Verheij M., Haimovitz-Friedman A., Scotto K., Fuks Z., Kolesnick R. (1995) Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell 82, 405–414 [DOI] [PubMed] [Google Scholar]
- 14. Chalfant C. E., Rathman K., Pinkerman R. L., Wood R. E., Obeid L. M., Ogretmen B., Hannun Y. A. (2002) De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells: dependence on protein phosphatase-1. J. Biol. Chem. 277, 12587–12595 [DOI] [PubMed] [Google Scholar]
- 15. Jayadev S., Liu B., Bielawska A. E., Lee J. Y., Nazaire F., Pushkareva M. Yu., Obeid L. M., Hannun Y. A. (1995) Role for ceramide in cell cycle arrest. J. Biol. Chem. 270, 2047–2052 [DOI] [PubMed] [Google Scholar]
- 16. Bedia C., Levade T., Codogno P. (2011) Regulation of autophagy by sphingolipids. Anticancer Agents Med. Chem. 11, 844–853 [DOI] [PubMed] [Google Scholar]
- 17. Martínez-Borra J., López-Larrea C. (2012) Autophagy and self-defense. Adv. Exp. Med. Biol. 738, 169–184 [DOI] [PubMed] [Google Scholar]
- 18. Scarlatti F., Bauvy C., Ventruti A., Sala G., Cluzeaud F., Vandewalle A., Ghidoni R., Codogno P. (2004) Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 279, 18384–18391 [DOI] [PubMed] [Google Scholar]
- 19. Lavieu G., Scarlatti F., Sala G., Carpentier S., Levade T., Ghidoni R., Botti J., Codogno P. (2006) Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 281, 8518–8527 [DOI] [PubMed] [Google Scholar]
- 20. Bailey L. B., Gregory J. F., 3rd (1999) Folate metabolism and requirements. J. Nutr. 129, 779–782 [DOI] [PubMed] [Google Scholar]
- 21. Fox J. T., Stover P. J. (2008) Folate-mediated one-carbon metabolism. Vitam. Horm. 79, 1–44 [DOI] [PubMed] [Google Scholar]
- 22. Katula K. S., Heinloth A. N., Paules R. S. (2007) Folate deficiency in normal human fibroblasts leads to altered expression of genes primarily linked to cell signaling, the cytoskeleton, and extracellular matrix. J. Nutr. Biochem. 18, 541–552 [DOI] [PubMed] [Google Scholar]
- 23. Jhaveri M. S., Wagner C., Trepel J. B. (2001) Impact of extracellular folate levels on global gene expression. Mol. Pharmacol. 60, 1288–1295 [DOI] [PubMed] [Google Scholar]
- 24. Zhu H., Cabrera R. M., Wlodarczyk B. J., Bozinov D., Wang D., Schwartz R. J., Finnell R. H. (2007) Differentially expressed genes in embryonic cardiac tissues of mice lacking Folr1 gene activity. BMC Dev. Biol. 7, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blount B. C., Mack M. M., Wehr C. M., MacGregor J. T., Hiatt R. A., Wang G., Wickramasinghe S. N., Everson R. B., Ames B. N. (1997) Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. U.S.A. 94, 3290–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim Y. I., Pogribny I. P., Basnakian A. G., Miller J. W., Selhub J., James S. J., Mason J. B. (1997) Folate deficiency in rats induces DNA strand breaks and hypomethylation within the p53 tumor suppressor gene. Am. J. Clin. Nutr. 65, 46–52 [DOI] [PubMed] [Google Scholar]
- 27. Bohnsack B. L., Hirschi K. K. (2004) Nutrient regulation of cell cycle progression. Annu. Rev. Nutr. 24, 433–453 [DOI] [PubMed] [Google Scholar]
- 28. Bertino J. R. (2009) Cancer research: from folate antagonism to molecular targets. Best Pract. Res. Clin. Haematol. 22, 577–582 [DOI] [PubMed] [Google Scholar]
- 29. Goldman I. D., Chattopadhyay S., Zhao R., Moran R. (2010) The antifolates: evolution, new agents in the clinic, and how targeting delivery via specific membrane transporters is driving the development of a next generation of folate analogs. Curr. Opin. Investig. Drugs 11, 1409–1423 [PubMed] [Google Scholar]
- 30. Krupenko S. A. (2009) FDH: an aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem. Biol. Interact. 178, 84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krupenko S. A., Oleinik N. V. (2002) 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ. 13, 227–236 [PubMed] [Google Scholar]
- 32. Rodriguez F. J., Giannini C., Asmann Y. W., Sharma M. K., Perry A., Tibbetts K. M., Jenkins R. B., Scheithauer B. W., Anant S., Jenkins S., Eberhart C. G., Sarkaria J. N., Gutmann D. H. (2008) Gene expression profiling of NF-1-associated and sporadic pilocytic astrocytoma identifies aldehyde dehydrogenase 1 family member L1 (ALDH1L1) as an underexpressed candidate biomarker in aggressive subtypes. J. Neuropathol. Exp. Neurol. 67, 1194–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen X. Q., He J. R., Wang H. Y. (2012) Decreased expression of ALDH1L1 is associated with a poor prognosis in hepatocellular carcinoma. Med. Oncol. 29, 1843–1849 [DOI] [PubMed] [Google Scholar]
- 34. Oleinik N. V., Krupenko N. I., Krupenko S. A. (2011) Epigenetic silencing of ALDH1L1, a metabolic regulator of cellular proliferation, in cancers. Genes Cancer 2, 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oleinik N. V., Krupenko S. A. (2003) Ectopic expression of 10-formyltetrahydrofolate dehydrogenase in A549 cells induces G1 cell cycle arrest and apoptosis. Mol. Cancer Res. 1, 577–588 [PubMed] [Google Scholar]
- 36. Hoeferlin L. A., Oleinik N. V., Krupenko N. I., Krupenko S. A. (2011) Activation of p21-dependent G1/G2 arrest in the absence of DNA damage as an antiapoptotic response to metabolic stress. Genes cancer 2, 889–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oleinik N. V., Krupenko N. I., Krupenko S. A. (2010) ALDH1L1 inhibits cell motility via dephosphorylation of cofilin by PP1 and PP2A. Oncogene 29, 6233–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oleinik N. V., Krupenko N. I., Priest D. G., Krupenko S. A. (2005) Cancer cells activate p53 in response to 10-formyltetrahydrofolate dehydrogenase expression. Biochem. J. 391, 503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anguera M. C., Field M. S., Perry C., Ghandour H., Chiang E. P., Selhub J., Shane B., Stover P. J. (2006) Regulation of folate-mediated one-carbon metabolism by 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 281, 18335–18342 [DOI] [PubMed] [Google Scholar]
- 40. Oleinik N. V., Krupenko N. I., Krupenko S. A. (2007) Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene 26, 7222–7230 [DOI] [PubMed] [Google Scholar]
- 41. Ghose S., Oleinik N. V., Krupenko N. I., Krupenko S. A. (2009) 10-formyltetrahydrofolate dehydrogenase-induced c-Jun-NH2-kinase pathways diverge at the c-Jun-NH2-kinase substrate level in cells with different p53 status. Mol. Cancer Res. 7, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Siskind L. J., Mullen T. D., Romero Rosales K., Clarke C. J., Hernandez-Corbacho M. J., Edinger A. L., Obeid L. M. (2010) The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J. Biol. Chem. 285, 11818–11826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Senkal C. E., Ponnusamy S., Bielawski J., Hannun Y. A., Ogretmen B. (2010) Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 24, 296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bielawski J., Szulc Z. M., Hannun Y. A., Bielawska A. (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39, 82–91 [DOI] [PubMed] [Google Scholar]
- 45. Horvath A., Sütterlin C., Manning-Krieg U., Movva N. R., Riezman H. (1994) Ceramide synthesis enhances transport of GPI-anchored proteins to the Golgi apparatus in yeast. EMBO J. 13, 3687–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang E., Norred W. P., Bacon C. W., Riley R. T., Merrill A. H., Jr. (1991) Inhibition of sphingolipid biosynthesis by fumonisins: implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 266, 14486–14490 [PubMed] [Google Scholar]
- 47. Mizutani Y., Kihara A., Igarashi Y. (2005) Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 390, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dbaibo G. S., Pushkareva M. Y., Rachid R. A., Alter N., Smyth M. J., Obeid L. M., Hannun Y. A. (1998) p53-dependent ceramide response to genotoxic stress. J. Clin. Invest. 102, 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Panjarian S., Kozhaya L., Arayssi S., Yehia M., Bielawski J., Bielawska A., Usta J., Hannun Y. A., Obeid L. M., Dbaibo G. S. (2008) De novo N-palmitoylsphingosine synthesis is the major biochemical mechanism of ceramide accumulation following p53 up-regulation. Prostaglandins Other Lipid Mediat. 86, 41–48 [DOI] [PubMed] [Google Scholar]
- 50. Sawada M., Kiyono T., Nakashima S., Shinoda J., Naganawa T., Hara S., Iwama T., Sakai N. (2004) Molecular mechanisms of TNF-α-induced ceramide formation in human glioma cells: p53-mediated oxidant stress-dependent and -independent pathways. Cell Death Differ. 11, 997–1008 [DOI] [PubMed] [Google Scholar]
- 51. Li G. M., Presnell S. R., Gu L. (2003) Folate deficiency, mismatch repair-dependent apoptosis, and human disease. J. Nutr. Biochem. 14, 568–575 [DOI] [PubMed] [Google Scholar]
- 52. Laethem R. M., Hannun Y. A., Jayadev S., Sexton C. J., Strum J. C., Sundseth R., Smith G. K. (1998) Increases in neutral, Mg2+-dependent and acidic, Mg2+-independent sphingomyelinase activities precede commitment to apoptosis and are not a consequence of caspase 3-like activity in Molt-4 cells in response to thymidylate synthase inhibition by GW1843. Blood 91, 4350–4360 [PubMed] [Google Scholar]
- 53. Laviad E. L., Kelly S., Merrill A. H., Jr., Futerman A. H. (2012) Modulation of ceramide synthase activity via dimerization. J. Biol. Chem. 287, 21025–21033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Novgorodov S. A., Chudakova D. A., Wheeler B. W., Bielawski J., Kindy M. S., Obeid L. M., Gudz T. I. (2011) Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J. Biol. Chem. 286, 4644–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Separovic D., Breen P., Joseph N., Bielawski J., Pierce J. S., Van Buren E., Gudz T. I. (2012) Ceramide synthase 6 knockdown suppresses apoptosis after photodynamic therapy in human head and neck squamous carcinoma cells. Anticancer Res. 32, 753–760 [PubMed] [Google Scholar]
- 56. White-Gilbertson S., Mullen T., Senkal C., Lu P., Ogretmen B., Obeid L., Voelkel-Johnson C. (2009) Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 28, 1132–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aoyama M., Sawada H., Shintani Y., Isomura I., Morita A. (2008) Case of unilateral focal dermal hypoplasia (Goltz syndrome). J. Dermatol. 35, 33–35 [DOI] [PubMed] [Google Scholar]
- 58. Meyers-Needham M., Ponnusamy S., Gencer S., Jiang W., Thomas R. J., Senkal C. E., Ogretmen B. (2012) Concerted functions of HDAC1 and microRNA-574–5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol. Med. 4, 78–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Szak S. T., Pietenpol J. A. (1999) High affinity insertion/deletion lesion binding by p53: evidence for a role of the p53 central domain. J. Biol. Chem. 274, 3904–3909 [DOI] [PubMed] [Google Scholar]
- 60. Yu J., Zhang L. (2003) No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell 4, 248–249 [DOI] [PubMed] [Google Scholar]
- 61. Chipuk J. E., Green D. R. (2009) PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle 8, 2692–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yu J., Zhang L. (2008) PUMA, a potent killer with or without p53. Oncogene 27, S71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sawada M., Nakashima S., Banno Y., Yamakawa H., Hayashi K., Takenaka K., Nishimura Y., Sakai N., Nozawa Y. (2000) Ordering of ceramide formation, caspase activation, and Bax/Bcl-2 expression during etoposide-induced apoptosis in C6 glioma cells. Cell Death Differ. 7, 761–772 [DOI] [PubMed] [Google Scholar]
- 64. Chipuk J. E., McStay G. P., Bharti A., Kuwana T., Clarke C. J., Siskind L. J., Obeid L. M., Green D. R. (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]