

Background: Mitochondrial accumulation of amyloid β (Aβ) promotes organelle dysfunction and Alzheimer disease (AD) neuropathology.
Results: IDE-Met1 localizes in mitochondria, is present in brain, and is regulated by the master regulator of mitochondrial biogenesis.
Conclusion: Mitochondrial biogenesis controls mitochondrial Aβ levels.
Significance: This study identifies a molecular mechanism that links mitochondrial biogenesis with Aβ degradation, suggesting that deregulation of this pathway could induce Aβ-mediated mitochondrial dysfunction.
Keywords: Amyloid, Mitochondrial Metabolism, Neurodegeneration, Protease, Transcription Factors
Abstract
Studies of post-mortem brains from Alzheimer disease patients suggest that oxidative damage induced by mitochondrial amyloid β (mitAβ) accumulation is associated with mitochondrial dysfunction. However, the regulation of mitAβ metabolism is unknown. One of the proteases involved in mitAβ catabolism is the long insulin-degrading enzyme (IDE) isoform (IDE-Met1). However, the mechanisms of its expression are unknown, and its presence in brain is uncertain. We detected IDE-Met1 in brain and showed that its expression is regulated by the mitochondrial biogenesis pathway (PGC-1α/NRF-1). A strong positive correlation between PGC-1α or NRF-1 and long IDE isoform transcripts was found in non-demented brains. This correlation was weaker in Alzheimer disease. In vitro inhibition of IDE increased mitAβ and impaired mitochondrial respiration. These changes were restored by inhibition of γ-secretase or promotion of mitochondrial biogenesis. Our results suggest that IDE-Met1 links the mitochondrial biogenesis pathway with mitAβ levels and organelle functionality.
Introduction
Mitochondrial dysfunction is believed to play an important role in human diseases, including diabetes mellitus type 2, cancer, and neurodegenerative disorders such as Alzheimer (AD)2 and Parkinson diseases (1, 2). The proteolytic system in mitochondria is crucial for the maintenance of protein turnover and integrity of the organelle. Several proteases have been identified in mitochondrial subcompartments among which presequence protease and insulin-degrading enzyme (IDE) through their capacity of degrading amyloid β (Aβ) peptide may be implicated in AD neuropathology. Although the amyloid plaques characteristic of AD consist of extracellular Aβ aggregates, accumulating data suggest that Aβ peptide may exert toxicity within the cell (3).
Interestingly, human PreP (hPreP) is an organellar functional analog of human IDE (hIDE). Both proteins belong to the pitrilysin M16 family of proteases containing an inverted Zn2+-binding motif. In contrast to hPreP, which was described only in mitochondria, hIDE is ubiquitous both at tissue and cellular levels (4, 5) and is considered a multifunctional protein involved in proteolysis-dependent and -independent cellular processes (for a review, see Ref. 6).
hPreP and hIDE are encoded by single genes PITRM1 and IDE, respectively, on chromosome 10. In silico and in vitro studies demonstrated that PreP is a matrix mitochondrial protease (7). The 5′ hIDE promoter region is CpG-rich with multiple transcription start sites with transcripts comprising one or two possible translation start sites (5). Depending on which starting AUG codon is translated, IDE proteins starting at Met1 and/or Met42 may be produced. The N terminus of the long variant (IDE-Met1) has a sequence that predicts mitochondrial importation (8). Mitochondrial localization of IDE was confirmed in vitro (8, 9); however, its relevance in vivo is unknown. By using in vitro degradation assays with synthetic peptides as substrates, significantly lower activity of hPreP isolated from temporal lobes of the brains of AD cases and of transgenic mice as models of AD was recently reported (10), suggesting a crucial role of PreP in the clearance of mitochondrial Aβ (mitAβ) (11). In regard to expression and activity of cytosolic IDE in AD brains, there are still controversies with reports showing decrements (12–15), increments (16, 17), or no changes (18, 19). It has been proposed that part of the loss of IDE and PreP activities in AD brain may be due to oxidative damage to which both proteases are highly sensitive (7, 20). Despite IDE involvement in extracellular Aβ peptide clearance, little is known about the regulation of IDE-Met1 expression and its relevance in mitAβ metabolism. The molecular mechanisms that govern the promoter activity of hPreP and hIDE-Met1 are still unknown. However, recent reports showed that the hIDE promoter may be a direct target of PPARγ (21), HES-1/Hey-1 (key downstream targets of Notch signaling) (15), and NRF-1 (22). NRF-1 interacts with peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) family proteins (23) to induce mitochondrial biogenesis. Upon appreciation that mitochondrial IDE content can be influenced by these coactivators and that the reported impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in AD, we hypothesized that IDE-Met1 impacts mitAβ homeostasis through mitochondrion-related cell signaling mechanisms. Given that many mitochondrion-related events are regulated by PGC-1α and NRF-1 (24), we assessed whether pyrroloquinoline quinone (PQQ), a mitochondrial biogenesis stimulator (25), enhances IDE-Met1 expression and reverts mitochondrial dysfunction in a cellular model of Aβ accumulation. Taken together, our results identify in cell culture experiments and in human brain an unanticipated link between master genes of mitochondrial biogenesis and IDE-Met1 up-regulation that may open avenues to enhance mitAβ clearance and thereby halt disease progression.
EXPERIMENTAL PROCEDURES
In Silico Analysis of hIDE Gene
The genomic sequence of the 5021 base pairs (bp) corresponding to the promoter of the human IDE gene (−5039/−18 upstream of the first translation start site, ATG) was obtained from GenBankTM (accession number NG_013012). Different programs were run to predict transcription factor consensus binding sites: TESS (Transcription Element Search System), TF Search software, Genomatix, Regulatory Sequence Analysis Tool, Signalscan, Jaspar, Transfac, and MitoProt-II v1.101.
RNA Secondary Structure and Stability Predictions
The first 185 nucleotides (from −39 to 146) and 119 nucleotides (from 27 to 146) of IDE mRNA (considering as 1 the adenine of the first translation start site) were analyzed. The minimum free energy structure and the suboptimal folding structures were calculated with Zuker and Turner Mfold software, version 3.2. The single strand probability was estimated from the Mfold results by normalization of the ss-count using a specific Perl script. To compare the stability of the 5′ fraction of the long and short IDE transcripts, the first 100 nucleotides of both RNA species were used. The free energy was calculated with Mfold and used to calculate the stability per nucleotide for both transcripts.
Transcription Start Site (TSS) Identification
TSS was obtained using a rapid amplification of cDNA 5′-end kit, second generation (Roche Applied Science). Final PCR products were analyzed in 8.4% acrylamide/bisacrylamide Tris borate-EDTA gels. The purified DNA was subcloned into TOPO TA cloning vector (Invitrogen), and 20 clones were sequenced for each experiment. The sequences of primers are listed in Table 1.
TABLE 1.
Sequences of oligonucleotides used in this study
The oligonucleotide sequences used for PCR, site-directed mutagenesis, methylation status of hIDE core promoter, and gel retardation experiments (28) are given.
| Method | Primer | Sequence (5′–3′) (restriction site) |
|---|---|---|
| PCRa | OUTER | GCAATATTTGGAGGATCCGACAAT |
| R147 | TCTCTTGATGGCTGGATTATTCAT | |
| R59 | ACTGAGCGGAAGGTGCTGGGCAGT | |
| R98 | TTGGAAAACCCATTCTTGAGG | |
| F (−1209) | GTATGGTACCCTCTGCATTGGGACTG (KpnI) | |
| F (−575) | GTATGGTACCCACGCCGCACTCGA (KpnI) | |
| F (−118) | GTATGGTACCTCTCCGCAGCTCC (KpnI) | |
| F (−82) | GTATGGTACCGCTAGAGCATGCGC (KpnI) | |
| F (−19) | GTATCTCGAGCCGGCGACTGCGCTGG (XhoI) | |
| R (−19) | GACTAAGCTTGATCACCGCAAACGCT (HindIII) | |
| R (−59) | GACTAAGCTTCTGCGCACTGCGCA (HindIII) | |
| Site-directed mutagenesisb | NRF-1 mut1 | GCCGCGGCTAGAAAATGCGCAGTGCGCAGGGCCGGCTCGAAGCGC |
| NRF-1 mut2 | GCCGCGGCTAGAGCATGAACAGTGCGCAGGGCCGGCTCGAAGCGC | |
| Methylation status of hIDE core promoter | CpG-F | GGAAAGTCTGGGTGCTGGCTC |
| CpG-R | TTAAGTGCTGAATCACCACTTTGCAAC | |
| EMSA | IDE WT | CCGCGGCTAGAGCATGCGCAGTGCGCAGGG |
| IDE MUT | CCGCGGCTAGAAAATGAACAGTGCGCAGGG |
a Restriction site sequences are shown in italics and underlined.
b Mutated bases are shown in bold and underlined.
Deletions and Mutations on IDE Promoter Constructs
Deletion constructs of the IDE gene promoter (−1209/−19, −575/−19, −118/−19, and −82/−19) were derived by PCR from bp −5039/−18 of the 5′ upstream region of human IDE gene (kindly provided by Dr. W. Farris, Pittsburgh University). In addition, two different constructs (−118/59 and −19/59) including 59 bp downstream of the first TSS were generated from genomic DNA by PCR. Forward and reverse primers and restriction sites are listed in Table 1. PCR-amplified fragments were inserted upstream of the luciferase (LUC) reporter gene in the pGL3-Basic vector (Promega). The correct sequence and orientation were confirmed by sequencing (Macrogen, Korea). Mutations in the IDE promoter were generated by PCR using sense and antisense primers (Table 1). All of the constructs were verified by DNA sequencing (Macrogen).
Luciferase Reporter Assay
48 h after transfection, cell lysates were obtained, and luciferase activities were measured using the Dual-Luciferase assay kit (Promega) according to the supplier's instructions. Results are expressed as relative luciferase activity normalized (=100) to the construct that showed the highest activity. The light intensity was measured using a luminometer (Tecan Genius).
Plasmid DNA Constructs
The human NRF-1 expression constructs wild type (WT) and dominant negative (DN) (pcDNA3-FLAG-WT-NRF-1 and pcDNA3-FLAG-DN-NRF-1) were provided by Dr. Kimitoshi Kohno (University of Occupational and Environmental Health, Japan) (26). Human myc-PGC1α plasmid was obtained from Addgene.
Cell Cultures and Treatments
U87 (human glioma cells), HeLa (human epithelial cells), SK-N-SH (human neuroblastoma cells), were cultured following conditions described by ATCC. OLN-93 (rat oligodendroglial cells; kindly provided by Dr. Richter-Landsberg, University of Oldenburg, Germany) were cultured as previously reported (60). Human embryonic kidney (HEK293) cells stably transfected with human Swedish mutant APP (HEKSw; a generous gift from Dr. Sisodia, University of Chicago) were cultured in Dulbecco's modified essential medium (DMEM), high glucose supplemented with 10% fetal bovine serum and G418. The impact of intracellular Aβ levels was determined in HEKSw cells treated or not with 1 μm DAPT (Calbiochem), a γ-secretase inhibitor, for 18 h. Mitochondrial biogenesis was stimulated by PQQ (provided as a gift from Mitsubishi Gas and Chemical, Japan) treatment (30 μm for 24 h) (25).
Transient Transfections and Cellular Sensibility Assay
For LUC reporter assay experiments, cells were plated at 4 × 104cells/well in a 24-well plate 18 h prior to transient transfection. Each well was transfected with 420 ng of IDE promoter construct and 80 ng of pCMV Renilla vector. For NRF-1 or PGC-1α expression, cells were plated at 1.5 × 106 cells/100-mm dish 16 h prior to transient transfection. In all cases, transfection was performed with Lipofectamine LTX (Invitrogen). HEKSw cells were transfected with 50 nm IDE siRNA or non-targeting siRNA (Dharmacon) using jetPRIME (Polyplus) reagent. The silencing effect was analyzed 72 h post-transfection by RT-qPCR or ELISA. Cellular viability was assessed in transfected cells through the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide by mitochondria. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was incubated at 0.5 mg/ml for 4 h at 37 °C in darkness, and the products of reaction were dissolved in DMSO. Absorbance was measured at 570 nm, and viability was expressed as the percentage relative to untransfected cells.
Human Tissues
Frozen samples of hippocampus from late onset sporadic AD Braak stage V (n = 7) and non-demented age-matched controls (ND; n = 4) were kindly provided by the Harvard Brain Tissue Resource Center (Boston, MA). Clinical and pathological features of each group were described previously (15). The methylation status of cytosines in the NRF-1 binding site of IDE core promoter was performed using the CpGenome DNA modification kit (Chemicon) followed by PCR amplification with a set of primers (CpG-F and CpG-R) detailed in Table 1. The PCR products were cloned in pGEM-T vector (Promega), and 10 colonies of each subject were sequenced (Macrogen).
Immunoprecipitation of IDE from Rat and Human Brains
Rat brains were obtained from adult Sprague-Dawley animals following protocols approved by the Fundación Instituto Leloir ethics committee. Tissues were homogenized, and IDE was immunoprecipitated using a set of specific anti-IDE mouse IgG1 monoclonal antibodies (1C1 and 3A2) developed in our laboratory as reported previously (17)
“In-gel” Digestion and Mass Spectrometry (MS) Analysis
Immunoprecipitated IDE separated by SDS-PAGE was processed for in-gel digestion and MS analysis as described previously (27). The precision in analyses was equal or less than an error of 0.1%. The negative control included digested fragments from gels corresponding to immunoprecipitations with IgG unrelated to IDE.
Electrophoretic Mobility Shift Assay
The electrophoretic mobility shift assay was performed as described (28). Nuclear extracts were obtained following the rapid protocol for DNA-binding protein (29). Oligonucleotides (Table 1) were biotinylated at their 5′-end and purified by HPLC (Genbiotech). Binding and competition reactions and blots were performed following the LightShift chemiluminescent EMSA (Pierce) protocol.
Chromatin Immunoprecipitation (ChIP) Assay
Cells were cross-linked, harvested in lysis buffer, and sonicated using a sonicator (Bioruptor UCD-200, Diagenode). Chromatin was incubated overnight at 4 °C in a rotation platform with goat anti-NRF-1 serum (kindly provided by Dr. Richard C. Scarpulla, Northwestern University Medical School, Chicago, IL) or normal goat serum as a negative control (30). Primers F-118 and R59 (Table 1) were used to amplify the 177-bp IDE promoter (from −118 to 59). The cycling conditions were as follows: 5 min at 94 °C followed by 35 cycles of 94 °C for 30 s, 54 °C for 1 min, and 72 °C for 30 s. The PCR products were resolved in 8.4% acrylamide/bisacrylamide Tris borate-EDTA gels.
RNA Isolation and RT-qPCR
One microgram of total RNA was reverse transcribed using the IDE-specific primer R98 (sequence detailed in Table 1), oligo(dT) primer, and SuperScript II reverse transcriptase (Invitrogen) (15). cDNA was amplified by SYBR Green RT-qPCR with a Mx3005P cycler (Stratagene). Cycling conditions were as follows: 94 °C for 10 min, 94 °C for 30 s, annealing for 1 min at 58 °C, and 72 °C for 30 s for 40 cycles. Melt curve analysis and agarose gels verified the formation of a single desired PCR product. The relative amount of transcripts to hypoxanthine phosphoribosyltransferase or TATA-binding protein was quantified by the 2−ΔΔCT method using MxPro software. The relative expression of the genes in human brain was determined by comparison of Ct using the following equation: RAD/RNDC = (CtxAD − CthAD) − (CtxNDC − CthNDC) (31).
Mitochondrial Respiration
Mitochondrial oxygen consumption was performed in intact cells polarographically with a Clark-type oxygen electrode in a two-channel respirometer for high resolution respirometry (Hansatech Oxygraph, Hansatech Instruments Ltd., Norfolk, UK). Confluent monolayers were resuspended in 64 μm digitonin for 10 min and centrifuged at 600 × g for 5 min. Pellets were resuspended in PBS and used as samples. Mitochondrial respiratory rates were measured in a reaction medium as described (32). To achieve a resting respiration state, 2 μm oligomycin was used as a F0-F1 ATP synthase inhibitor (state 4o). To evaluate active respiration, the protonophore 2 μm carbonyl cyanide m-chlorophenylhydrazone was added to reach uncoupled respiration (state 3u). Results were expressed as ng-atoms of oxygen/mg of cell protein. Respiratory control ratio (RCR) was calculated as the relationship between state 3u respiration and state 4o respiration (33).
Synchronization of Cells and Measurement of the Mitochondrial Mass
Cells were synchronized in G0 phase as described previously (34), then washed, and cultured in the presence or absence of PQQ for 48 h. After trypsinization, cells were fixed with cold 70% ethanol, washed, and stained with 10 mm 10-N-nonyl acridine orange (35) for 10 min at room temperature in the dark. After washes with cold PBS, cells were immediately analyzed by flow cytometry (FACSCalibur) with the software Cell Quest (BD Biosciences).
Mitochondrial DNA Content
The amount of mitochondrial DNA was estimated by qPCR using KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems). Total DNA was extracted using the GFX genomic blood DNA purification kit (GE Healthcare). PCRs contained 1 ng of DNA and a 0.5 μm concentration of each primer of a subunit of the human electron transport chain as described previously (36).
Quantification of IDE and Aβ40 Levels
Mitochondria were isolated by using supramagnetic microbeads (Miltenyi Biotec). After the application of the cell lysate onto the column, mitochondria were retained, and the flow-through was collected and centrifuged at 100,000 × g to further analyze the cytosolic fraction. Eluted mitochondrial fractions were sonicated in PBS containing a mixture of protease inhibitors. IDE levels were quantified by a sandwich ELISA with a set of specific anti-IDE monoclonal antibodies (3A2 to capture and biotinylated 1C1 to detect IDE, respectively) as reported previously (17), and Aβ40 levels were quantified using a commercial ELISA kit (Invitrogen). Data for each set of duplicate experiments were normalized by the protein content and expressed for IDE as ng/mg or as percentage as compared with mock-transfected cells (=100%) and for Aβ40 as pg/mg.
Statistical Analysis
All experiments were performed at least in triplicate, and values are presented as means ± S.E. Data were analyzed by Student's t test using GraphPad Prism 3.0 software. Statistical significance was set at p < 0.05.
RESULTS
The First AUG of Endogenous hIDE mRNA Is Translated in Vivo
To characterize transcription start sites, total RNA from the U87 human cell line was amplified with an IDE-specific reverse primer anchored in the middle region of exon 1 (bp 36–59 where 1 refers to adenine of the first putative translated ATG expressed as DNA) (Fig. 1A). PCR products (Fig. 1B, upper panel) revealed multiple 5′ start sites at positions between −40 and 28 (Fig. 1B, lower panel) included in a CpG island located at position −53/42 (data not shown) with the following characteristics: observed/expected ratio, >0.6; percent C + percent G, >60.00; length, >200. The CpG island predicted contains all TSSs detected. Although rapid amplification of cDNA 5′-end is not a strictly quantitative method, the abundance of clones obtained suggested that in U87 cells the most abundant TSS lies within this region. Our results were concurrent with information in the Database of Transcriptional Start Sites (Release 8.0; based on UCSC Genome Browser hg19, mm9) in which all mRNA 5′-ends (from bp −75 to 36) were localized within exon 1. As shown in Fig. 1C, the 5′-end of the long IDE (L-IDE) mRNA region containing both putative translation start sites that may generate IDE-Met1 and/or IDE-Met42 is predicted to be highly structured. However, both putative sites have a high probability to be single strand regions and are plausibly recognized by RNA polymerase. Next, we compared the stability predicted for the 5′-end of L-IDE mRNA and short IDE (may produce only IDE-Met42) and showed that the estimated stability for L-IDE mRNA is 10 kcal higher than that predicted for the short IDE (−34.9 versus −24.0 kcal/mol). To determine whether the long isoform of IDE (IDE-Met1) is transcribed in the brain, we immunoprecipitated IDE from human and rat cortical homogenates, and after in-gel digestion with endo-Lys-C, proteolytic fragments were characterized by MALDI-TOF MS (Fig. 2A, lower panel). These peptides were in full agreement with the expected major sites of human IDE-Met1, reinforcing the concept that IDE-Met1 is transcribed and translated in the human brain. Similar results were obtained with rat brain homogenates (Fig. 2B).
FIGURE 1.

Schematic representation of hIDE promoter region. A, primer positions used for the rapid amplification of cDNA 5′-end (5′RACE). B, upper panel, the asterisk indicates the heterogenic composition of the product obtained from the second PCR resolved in an 8.4% Tris borate-EDTA polyacrylamide gel. On the left are markers in bp. Lower panel, arrows indicate different transcription start sites identified in the −40/28 region of the hIDE promoter. The first putative translation start site (DNA) is underlined. C, prediction of RNA secondary structure (minimum free energy) of L-hIDE showing both putative translation start sites (asterisks). The bases are colored according to the calculated probability to be a single strand: red, probability greater than 0.75; orange, probability greater than 0.5 and lower than 0.75; and black, probability lower than 0.5.
FIGURE 2.

In vivo identification of IDE-Met1 isoform. A and B, upper panels, schematic representation of the amino acid sequences (1–56) of hIDE (A) and rat IDE (B) showing two alternative in-frame initiation Met residues, Met1 and Met42 (bold). The arrowheads indicate the predictive cleavage sites in the N terminus of IDE-Met1 isoform by endo-Lys-C. Solid lines below depict the expected peptides obtained after digestion. A and B, lower panels, MALDI-TOF MS analysis of the peptides (P1, P2, and P3) obtained after endo-Lys-C digestion. Insets, hIDE (H-IDE) and rat IDE (R-IDE) species and expected/calculated masses of predicted peptides. Cys-CM, carboxymethyl-Cys; A.U., arbitrary units; Obs., observed; Calc., calculated.
Transcriptional Regulation of L-IDE mRNA Is Mediated by the Mitochondrial Biogenesis Pathway
To define the boundaries of the minimal hIDE promoter, we prepared a series of truncated constructs (Fig. 3A) and tested them in different cell lines by transfection using the LUC reporter system. IDE −118/59 showed the strongest transcriptional activity in different human and rat cell lines (Fig. 3, B–E). As compared with promoter activity of IDE −118/59, a 3′ deletion of 118 bp (IDE −118/−59) or a 5′deletion of 78 bp (IDE −19/59) drastically reduced LUC activity to ∼20 and ∼1%, respectively, in all cell lines analyzed, indicating that this DNA sequence contains the minimal region with hIDE promoter activity. Moreover, this region covers all the IDE TSSs reported so far, which may explain the highest levels of LUC expression obtained with this DNA fragment. By using prediction programs for transcription factor binding sites and multiple sequence alignment, we confirmed that the NRF-1 binding site is the most frequently identified and highly conserved among different species as reported recently (22). We analyzed the relevance of the NRF-1 putative binding site to IDE core promoter activity by introducing disrupting point mutations in the NRF-1 site and testing the mutant constructs in the U87 cell line using a LUC assay. Mutations of the NRF-1 site demonstrated that it played a positive regulatory role in human IDE transcription activity. The putative site of NRF-1 in the human IDE core promoter is a 12-base sequence, 5′-AGAGCATGCGCA-3′, with one cytosine in the context of CpG plausibly methylated.3 When the hemicores AGAGC and GCGCA were replaced by AGAAA and GAACA, a drastic, almost complete loss of promoter activity was observed (Fig. 3F), consistent with the finding that homodimeric NRF-1 binding to DNA is required for full transcriptional activity as reported (24). Similar results were obtained when the construct IDE −118/59 was in vitro methylated (Fig. 3G). These results point to a crucial role for the NRF-1 DNA motif in IDE core promoter activity. To determine whether the NRF-1 binding site on the IDE promoter was effectively recognized by endogenous NRF-1 protein, we performed EMSA (Fig. 4A) and ChIP assays (Fig. 4B). Our results showed the specific binding of NRF-1 to the hIDE promoter region in vivo as was previously suggested in a ChIP-on-chip analysis (37). The requirement of NRF-1 for effective IDE promoter activity was further studied by co-transfection of FLAG-tagged DN-NRF-1 with the reporter construct IDE −118/59. As shown in Fig. 4C the IDE −118/59 promoter activity upon expression of FLAG-DN-NRF-1 was inhibited in a dose-dependent manner (40 and 30% after 0.1 or 0.2 μg, respectively, of DN-NRF-1 plasmid transfection). Then, to analyzed whether NRF-1 regulates transcription of L-IDE mRNA, which comprises the two possible translation start sites, we designed primers to quantify the L-IDE mRNA sequence detected by rapid amplification of cDNA 5′-end (Fig. 1A) and measured its levels by RT-qPCR in cells transfected with DN-NRF-1 or WT-NRF-1. In both cases, the correct expression of constructs was confirmed by immunoblotting with anti-FLAG antibody (38) (Fig. 4D). L-IDE mRNA showed a 15-fold reduction after DN-NRF-1 transfection and an ∼3-fold increment when WT-NRF-1 was expressed as compared with control (Fig. 4E, left panel). To dismiss that cell death might have contributed to the striking reduction observed in DN-NRF-1-transfected cells, we performed a viability assay. Viability greater than 70% was observed in both experimental conditions with no statistical differences between mock- and DN-NRF-1-transfected cells (83.37 ± 0.09% versus 78.46 ± 1.680%).
FIGURE 3.
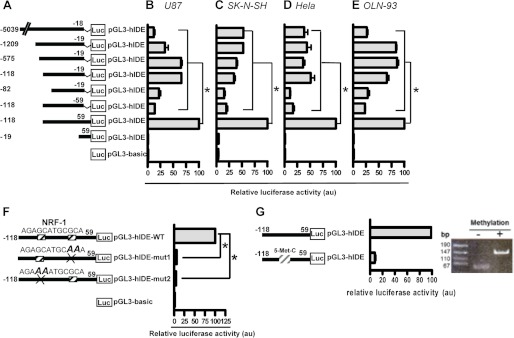
NRF-1 mediates the transcription of the core promoter of human IDE and is sensitive to methylation. A, schematic representation of the deletion mutants derived from the bp −5039/−18 construct and generated by PCR. Bars show the mean ± S.E. of the percentage of LUC activity determined in U87 (B), SK-N-SH (C), HeLa (D), and OLN-93 (E) cells transfected with each construct, respectively. In each experiment (n = 3), the construct that showed the highest LUC activity represents 100%. All of the constructs showed significant differences as compared with the −118/59 hIDE promoter region. *, p < 0.01. F, schematic representation of the NRF-1 mutated sites generated by site-directed mutagenesis from the −118/59 hIDE promoter region. Bars show the mean ± S.E. of relative LUC activity determined in U87 cells transfected with each construct, respectively (n = 4). *, p < 0.01 versus WT construct. G, left panel, the promoter activity of construct −118/59 was measured after treatment without (−) or with (+) methyltransferase. Right panel, products of DNA digestion to test the methylation state for each condition. Error bars represent S.E. au, arbitrary units.
FIGURE 4.

Transcriptional regulation of L-IDE mRNA is mediated by the mitochondrial biogenesis pathway. A, the labeled oligonucleotides containing the canonical NRF-1 binding sites were used as probes and incubated with nuclear extracts from HeLa cells alone (lane 2) or in competition with 50- or 100-fold excess amounts of unlabeled probe (lanes 3 and 4, respectively). Specific DNA-protein complexes formed are indicated (arrow). The mutant (mut.) probes failed to form specific DNA-protein complexes (lane 6). To identify the DNA-binding proteins in an antibody (Ab) disruption assay, polyclonal antibodies specific to NRF-1 or normal rabbit control IgG were added to the binding reactions and incubated for 20 min at room temperature before probes were added (lanes 9 and 10). B, the association of the transcription factor and the NRF-1 motif on the hIDE promoter was examined in intact HeLa cells by PCR amplification of the sequence between −118 and 59 after chromatin immunoprecipitation with specific antiserum (anti-NRF-1). As a positive control, the NRF-1 binding region of the hIDE promoter was PCR-amplified from genomic DNA. The fragment of interest is 190 bp. Goat IgG was used as a negative control. bp, base pairs; MK, marker. C, U87 cells were transfected with different amounts of DN-NRF-1 construct (n = 3). Bars show the mean ± S.E. of LUC activity in arbitrary units. ***, p < 0.001 as compared with mock-transfected cells. D, upper panel, representative Western blot of homogenates of transfected cells probed with anti-FLAG antibody showing the expression of WT-NRF-1 or DN-NRF-1. DN-NRF-1 is a deletion of WT-NRF-1 lacking the transcription activation domain (∼250 bp). Lower panel, representative Western blot of loading control with anti-tubulin (38) antibody. Left, molecular mass markers in kDa. E, left panel, HeLa cells were transiently transfected with DN-NRF-1 or WT-NRF-1, respectively (n = 3). Bars show the mean ± S.E. of endogenous L-IDE mRNA levels normalized by TATA-binding protein (TBP) content. Right panel, HeLa cells were transiently transfected with PGC-1α (n = 3). Bars show the mean ± S.E. of endogenous NRF-1 and L-IDE mRNA levels normalized by TATA-binding protein content. F, bars show the mean ± S.E. of the percentage of IDE levels as compared with mock-transfected cells (=100%). The amount of IDE was determined by ELISA in isolated mitochondrial (Mito.) and cytosolic fractions of NRF-1- (left panel) (n = 4) and PGC-1α (right panel) (n = 4)-transfected HeLa cells, respectively, and normalized by mg of total protein. *, p < 0.05 WT-NRF-1 versus DN-NRF-1. G, HEKSw cells were exposed to 30 μm PQQ, and the endogenous PGC-1α, NRF-1, and L-IDE transcript levels were determined by RT-qPCR after 30 min, 1 h, 2 h, and 4 h, respectively (n = 3). White, PGC-1α; dashed, NRF-1; and black, L-IDE. Bars show the mean ± S.E. of the expression of the genes normalized by TATA-binding protein content.*, p < 0.05 as compared with mock-transfected cells at each time point; ≠, PGC-1α versus L-IDE. Error bars represent S.E.
Taking into account that PGC-1α is a mitochondrial regulator that induces NRF-1 overexpression, we assessed whether PGC-1α is involved in L-IDE expression. In accordance with our prediction, NRF-1 levels were increased 2-fold and L-IDE levels were increased ∼3-fold in PGC-1α as compared with mock-transfected cells (Fig. 4E, right panel), showing the positive activation of PGC-1α on NRF-1 and L-IDE transcription, respectively. To assess whether the PGC-1α/NRF-1 pathway has a restricted effect on IDE-Met1 or not, we determined the levels of IDE protein in mitochondria (IDE-Met1) and cytosol (IDE-Met42) of WT-NRF-1, DN-NRF-1, and PGC-1α transiently transfected cells, respectively. Our results showed a significant reduction in the percentage of IDE in the mitochondrial fraction of DN-NRF-1 as compared with mock-transfected cells (=100%) (Fig. 4F, left panel), suggesting that NRF-1 has a direct impact on mitochondrial IDE levels. By contrast, we did not detect any significant difference in mitochondrial IDE protein levels after WT-NRF-1 transient transfection (Fig. 4F, left panel). These results may be the consequence of at least two factors: the moderate increment of the target genes both in vivo (39) and in vitro (40) after the overexpression of NRF-1 as reported previously and the extended half-life of IDE (∼33 h) (41) that may mask the slight increments that are a consequence of the 3-fold increase in the L-IDE mRNA. Moreover, our results suggest that NRF-1 had no detectable effect on cytosolic IDE because neither WT-NRF-1 nor DN-NRF-1 modulated cytosolic IDE protein expression. In addition, in cells transfected with PGC-1α, the levels of IDE in both subcellular compartments increased as compared with mock-transfected cells (145.6% ± 42.05% and 200 ± 69.9%) without a significant difference between mitochondria and cytosol (p = 0.5). Considering that in addition to NRF-1 overexpression PGC-1α also greatly increases the transcriptional activity of PPARγ (42) and that PPARγ has a direct impact on the IDE 5′-UTR upstream of the minimal promoter (21), our results suggest that PGC-1α up-regulates both IDE isoforms.
A time course analysis of PGC-1α, NRF-1, and L-IDE transcript levels in HEKSw cells showed that PGC-1α increased its expression significantly at 30 min after PQQ treatment (Fig. 4G) as compared with mock-transfected cells (p < 0.01) and then decreased from 30 min to 4 h with no significant differences as compared with mock-transfected cells (p > 0.05), whereas the transcripts of NRF-1 and L-IDE showed a significant increment after 1 (p < 0.05) and 2 h (p < 0.01) of exposure, respectively, suggesting that L-IDE is a target of the mitochondrial biogenesis transcriptional pathway. To assess whether Aβ40 accumulation affects mitochondrial biogenesis in HEKSw cells, we determined mitochondrial DNA levels in PQQ-treated cells in the presence or absence of a γ-secretase inhibitor (DAPT). Our results showed that mitochondrial DNA content increased ∼57% when HEKSw cells were preincubated with DAPT, suggesting that Aβ40 accumulation affects mitochondrial biogenesis (data not shown).
To determine whether the PGC-1α/NRF-1/L-IDE pathway is altered in AD brain, we evaluated the methylation status of seven CpGs in the context of the NRF-1 binding site of the IDE promoter in hippocampal samples from ND and AD brains and found that the percentage of methylation was similar (97.1 versus 98.5%). However, we found that in ND there was a positive strong correlation between the expression of PGC-1α (r2 = 0.83) or NRF-1 (r2 = 0.94) and L-IDE (Fig. 5, upper panels) mRNAs. By contrast, in AD brains, the correlation was weaker with values of r2 = 0.012 and r2 = 0.32 for PGC-1α or NRF-1 and L-IDE, respectively (Fig. 5, lower panels). These results agree with the recent reported lower levels of PGC-1α and NRF-1 in AD as compared with ND (36). However, because of the low number of brains analyzed when the test of comparison of correlation coefficients was applied, the values of the statistic were 0.894 and 0.857 for PGC-1α/L-IDE and NRF-1/L-IDE, respectively, suggesting that there is not sufficient evidence to affirm that both populations (AD and ND) have different correlation coefficients for each of the variables analyzed.
FIGURE 5.

The mitochondrial biogenesis pathway is altered in AD brains. mRNA from frozen hippocampal samples was isolated, and endogenous PGC-1α, NRF-1, and L-IDE transcript levels were determined by RT-qPCR. A strong positive correlation was observed in controls (ND) (upper panels) between PGC-1α or NRF-1 and L-IDE in contrast to AD (lower panels) in which the correlation decreased.
The Enhancement of L-IDE Expression Reverts the Mitochondrial Dysfunction Mediated by mitAβ Accumulation
To evaluate the impact of IDE-Met1 expression on mitAβ homeostasis, mitochondria were isolated from HEKSw (Fig. 6A) transfected with control or IDE siRNA. After down-regulation of IDE (Fig. 6B), mitAβ was increased (6.4 ± 2.4 versus 17.12 ± 2.62 pg/mg) (Fig. 6C), suggesting that mitAβ is regulated by IDE. The analysis of oxygen consumption by isolated mitochondria is the classical approach to characterize mitochondrial function. The HEKSw cells showed a decrement of ∼22% in the RCR value compared with HEK cells (1.73 ± 0.04 versus 2.22 ± 0.02; p < 0.05), reinforcing the concept that intracellular accumulation of Aβ40 promotes mitochondrial dysfunction. In addition, HEKSw cells treated with γ-secretase inhibitor (DAPT) diminished mitAβ levels as compared with DMSO-treated cells (3.51 versus 5.16 pg/mg) and reverted the RCR impairment (1.73 ± 0.04 versus 2.09 ± 0.04; p < 0.05). These results are in agreement with the mitochondrial enzyme dysfunctionality due to Aβ40 accumulation as reported (43). Interestingly, after PQQ treatment, a significant increase in the RCR value (∼29%) was observed as compared with HEKSw untreated cells (2.24 ± 0.03 versus 1.73 ± 0.04; p < 0.05) in accordance with increments in L-IDE mRNA levels (Fig. 4G) and mitochondrial content determined by flow cytometry (Fig. 7A). The impact of L-IDE on RCR was determined by treatment of HEKSw cells with control siRNA or IDE siRNA. Our results showed an impairment of ∼25% after IDE siRNA treatment without changes in cell viability (more that 90%). Taking into account that RCR is calculated as state 3u/state 4o, the return to control values observed after PQQ treatment and the decrements observed after IDE siRNA treatment, respectively, could be due to an enhancement (13.9 ± 1.18 versus 6 ± 1.3.; p < 0.05) or inhibition (7.73 ± 0.16 versus 9.3 ± 1.08.; p < 0.05) of state 3u as compared with HEKSw cells, respectively (Fig. 7B). Our results suggest that IDE-Met1 impacts mitAβ40 accumulation and mitochondrial functionality. Overall, we showed that the PGC-1α/NRF-1 pathway modulates mitAβ through the regulation of L-IDE and that this pathway may be changed in AD brain.
FIGURE 6.

Mitochondrial IDE expression impacts Aβ catabolism and mitochondrial functionality. A, representative Western blot showing the purity of mitochondria (Mito.) isolated from cellular extracts (Cell. Ext.) (8 μg/lane) of transfected HEKSw cells probed with anti-actin (38) (upper panel) and with anti-TOM20 (Santa Cruz Biotechnology) (lower panel) antibodies, respectively. Left, molecular mass markers in kDa. IDE (B) and Aβ40 (C) levels were determined by ELISA in mitochondria isolated from HEKSw cells transfected with control siRNA (si-RNA-CNT) or siRNA specific for IDE (si-RNA-IDE). Bars show the mean ± S.E. of mitochondrial IDE and Aβ40 amounts normalized by mg of total protein. *, p < 0.01, control siRNA versus IDE siRNA (n = 3). Error bars represent S.E.
FIGURE 7.

L-IDE expression modulates the RCR in HEKSw cells. A, increment in cellular mitochondrial content after PQQ exposure detected after 10-N-nonyl acridine orange staining and flow cytometry assay. Representative histograms (cell counts at each fluorescence intensity, 101–102) for control (white) and PQQ (black) are shown. Significant differences were detected in fluorescence values between control and PQQ-treated cells (mean values in arbitrary units 28.74 ± 0.20 versus 30.31 ± 0.23; p < 0.001). B, mitochondrial respiration was determined by oxygen electrode measurement in HEKSw cells exposed or not to PQQ and treated with control siRNA (si-RNA-CNT) or IDE siRNA. *, p < 0.05, control (CNT) versus PQQ and control siRNA versus IDE siRNA. Inset, mean ± S.E. of state 4, state 3, and RCR (n = 5). Error bars represent S.E.
DISCUSSION
NRF-1 acts as a central transcription factor that regulates IDE transcription (22). By using DAVID, which allows in silico analysis of regulatory sequences of 14,821 human genes, we realized that 471 genes (3.17%) have the NRF-1 DNA-binding motif in the proximal region (−250/1), and nine of those are mitochondrial genes, suggesting that at least part of hIDE transcriptional regulation is similar to that of this specific subset of genes. The NRF-1 site is located in a CpG-rich island. In normal cells, methylation occurs predominantly in CG-poor regions, whereas the CpG island remains unmethylated. The exceptions are the extensive methylation of CpG islands associated with transcriptional inactivation of regulatory regions of genes. Methylation of DNA is an important regulator of gene expression, and early studies of genomic DNA methylation reported it to decrease with age (44, 45). However, recent evidence indicates that this relationship is more complex. A recent study reported positive correlations between CpG island methylation and aging (46). The change in the epigenetic signature with age is likely to be significantly determined by environmental exposures, including diet, tobacco smoke, alcohol, and previous illnesses and therapies (47). The biological consequence of the methylation status in the cluster of the NRF-1 binding site is not clear. Although the reporter gene experiments underlined the potential importance of promoter methylation in IDE expression, the assay determined the effects of methylation of the whole promoter fragment rather than individual or clusters of CpGs. However, one of the CpGs analyzed corresponds to the polymorphism C/T at position −51 of the IDE promoter; this haplotype with the G/T at position −1002 was described as a protective (−1002G/−51T) or risk (1002T/−51C) factor for sporadic AD in the Chinese population (48). In this context, methylation of C-51 may result in a down-regulation of IDE transcription and a potential risk factor for AD.
We have described in AD brains a poor correlation between the expression of genes involved in mitochondrial biogenesis and L-IDE, supporting the concept that impairments in mitochondrial biogenesis due to repression of PCG-1α-dependent genes preclude mitAβ degradation and that this mechanism may be an upstream event responsible for the mitochondrial dysfunction, a prominent and early feature of AD brain (49) mainly characterized by reduced energy metabolism (50), lower expression of mitochondrial DNA (51), excessive mitochondrial fragmentation, and abnormal mitochondrial distribution (52, 53).
The link described here among mitochondrial biogenesis, IDE promoter activation, and L-IDE expression supports a concerted action of NRF-1 together with PPARγ (co-activated by PGC-1α) (21) to enhance hIDE transcription. The role of PCG-1α in gluconeogenesis and insulin resistance (54); the relevance of the insulin-PI3K-akt pathway in PPARγ (21), NRF-1 activity (55), and IDE expression (56); and the metabolic phenotype of insulin resistance described in AD and diabetes mellitus patients confirm the relevance of our results in understanding the role of hIDE in the pathogenesis of both human diseases (Fig. 8). Indeed, the study of the transcriptional regulation of hIDE by mutated NRF-1 associated with the pathogenesis of type 2 diabetes mellitus (57, 58) may be of significance to molecular mechanisms underlying the disease.
FIGURE 8.

Schematic representation of the effect of mitochondrial biogenesis on hIDE transcriptional activation and mitochondrial functionality. Activation of PGC1-α by the effect of mitochondrial biogenesis stimuli promotes NRF-1 expression, which impacts proximal hIDE-specific DNA binding domains, promoting the transcription of L-IDE and short IDE (S-IDE) mRNAs, which further translate the long (IDE-Met1) and the short (IDE-Met42) IDE isoforms. Mitochondrial Aβ can be degraded by IDE-Met1-originating non-toxic peptides, or it can interact with cyclophilin D (CypD), Aβ-binding alcohol dehydrogenase (ABAD), or cytochrome c oxidase (COX), causing elevated reactive oxygen species (ROS). The balance between both processes results in functional or dysfunctional mitochondria, respectively.
This study favors the notion that IDE has an “eclipsed” distribution (59) with huge amounts of the cytosolic isoform (IDE-Met42) that mask mitochondrial IDE (IDE-Met1). The high levels of cytosolic IDE may be explained by the multiplicity and abundance of the “short” transcripts. Taking into account that mitochondrial accumulation of Aβ may not be a physiologic process, the role of IDE-Met1 may be to degrade free mitochondrial targeting peptides and other small peptide substrates that may be toxic to organellar function. The identification that IDE-Met1 isoform is expressed in human brain, that the PGC-1α/NRF-1 pathway modulates mitAβ through the regulation of L-IDE mRNA expression, and that this pathway has a functional significance in mitochondrial function may provide a starting point for examining novel prophylactic strategies in the treatment of mitochondrial dysfunction in AD.
Acknowledgments
We acknowledge Dr. Fernando Pitossi and Dr. Osvaldo Podhajcer (Fundación Instituto Leloir) for providing personal equipment, technical assistance, and molecular reagents. We thank Leonardo Sganga for cooperation in the measurement of the mitochondrial mass by flow cytometry. The brain tissues were provided by the Harvard Brain Tissue Resource Center, which is supported in part by United States Public Health Service Grant R24-MH 068855.
This work was supported by grants from the Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET)-PIP693 and Florencio Fiorini Fundation (to L. M.), the International Society for Neurochemistry-Committee for Aid and Education in Neurochemistry (to M. C. L.), and the John Simon Guggenheim Memorial Foundation and Alzheimer's Association Grant IIRG-11-205127) (to E. M. C.).
L.-C. Li, personal communication.
- AD
- Alzheimer disease
- Aβ
- amyloid β
- mitAβ
- mitochondrial Aβ
- HEKSw
- human embryonic kidney (HEK293) cells stably transfected with human Swedish mutant APP
- IDE
- insulin-degrading enzyme
- hIDE
- human IDE
- L-IDE
- long IDE
- LUC
- luciferase
- ND
- non-demented age-matched controls
- PreP
- presequence protease
- hPreP
- human PreP
- PGC-1α
- peroxisome proliferator-activated receptor γ coactivator 1
- PQQ
- pyrroloquinoline quinone
- RT-qPCR
- real time quantitative PCR
- TSS
- transcription start site
- PPARγ
- peroxisome proliferator-activated receptor γ
- DN
- dominant negative
- DAPT
- N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester
- RCR
- respiratory control ratio.
REFERENCES
- 1. Chan C. B., Harper M. E. (2006) Uncoupling proteins: role in insulin resistance and insulin insufficiency. Curr. Diabetes Rev. 2, 271–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin M. T., Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 [DOI] [PubMed] [Google Scholar]
- 3. Gouras G. K., Almeida C. G., Takahashi R. H. (2005) Intraneuronal Aβ accumulation and origin of plaques in Alzheimer's disease. Neurobiol. Aging 26, 1235–1244 [DOI] [PubMed] [Google Scholar]
- 4. Kuo W. L., Montag A. G., Rosner M. R. (1993) Insulin-degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 132, 604–611 [DOI] [PubMed] [Google Scholar]
- 5. Farris W., Leissring M. A., Hemming M. L., Chang A. Y., Selkoe D. J. (2005) Alternative splicing of human insulin-degrading enzyme yields a novel isoform with a decreased ability to degrade insulin and amyloid β-protein. Biochemistry 44, 6513–6525 [DOI] [PubMed] [Google Scholar]
- 6. Fernández-Gamba A., Leal M. C., Morelli L., Castaño E. M. (2009) Insulin-degrading enzyme: structure-function relationship and its possible roles in health and disease. Curr. Pharm. Des. 15, 3644–3655 [DOI] [PubMed] [Google Scholar]
- 7. Falkevall A., Alikhani N., Bhushan S., Pavlov P. F., Busch K., Johnson K. A., Eneqvist T., Tjernberg L., Ankarcrona M., Glaser E. (2006) Degradation of the amyloid β-protein by the novel mitochondrial peptidasome, PreP. J. Biol. Chem. 281, 29096–29104 [DOI] [PubMed] [Google Scholar]
- 8. Leissring M. A., Farris W., Wu X., Christodoulou D. C., Haigis M. C., Guarente L., Selkoe D. J. (2004) Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem. J. 383, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahuja N., Schwer B., Carobbio S., Waltregny D., North B. J., Castronovo V., Maechler P., Verdin E. (2007) Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 282, 33583–33592 [DOI] [PubMed] [Google Scholar]
- 10. Alikhani N., Guo L., Yan S., Du H., Pinho C. M., Chen J. X., Glaser E., Yan S. S. (2011) Decreased proteolytic activity of the mitochondrial amyloid-β degrading enzyme, PreP peptidasome, in Alzheimer's disease brain mitochondria. J. Alzheimers Dis. 27, 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caspersen C., Wang N., Yao J., Sosunov A., Chen X., Lustbader J. W., Xu H. W., Stern D., McKhann G., Yan S. D. (2005) Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 19, 2040–2041 [DOI] [PubMed] [Google Scholar]
- 12. Cook D. G., Leverenz J. B., McMillan P. J., Kulstad J. J., Ericksen S., Roth R. A., Schellenberg G. D., Jin L. W., Kovacina K. S., Craft S. (2003) Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-ϵ4 allele. Am. J. Pathol. 162, 313–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao Z., Xiang Z., Haroutunian V., Buxbaum J. D., Stetka B., Pasinetti G. M. (2007) Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer's disease. Neurobiol. Aging 28, 824–830 [DOI] [PubMed] [Google Scholar]
- 14. Pérez A., Morelli L., Cresto J. C., Castaño E. M. (2000) Degradation of soluble amyloid β-peptides 1–40, 1–42, and the Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem. Res. 25, 247–255 [DOI] [PubMed] [Google Scholar]
- 15. Leal M. C., Surace E. I., Holgado M. P., Ferrari C. C., Tarelli R., Pitossi F., Wisniewski T., Castaño E. M., Morelli L. (2012) Notch signaling proteins HES-1 and Hey-1 bind to insulin degrading enzyme (IDE) proximal promoter and repress its transcription and activity: implications for cellular Aβ metabolism. Biochim. Biophys. Acta 1823, 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miners J. S., Baig S., Tayler H., Kehoe P. G., Love S. (2009) Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J. Neuropathol. Exp. Neurol. 68, 902–914 [DOI] [PubMed] [Google Scholar]
- 17. Morelli L., Llovera R. E., Mathov I., Lue L. F., Frangione B., Ghiso J., Castaño E. M. (2004) Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid β vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J. Biol. Chem. 279, 56004–56013 [DOI] [PubMed] [Google Scholar]
- 18. Miners J. S., van Helmond Z., Kehoe P. G., Love S. (2010) Changes with age in the activities of β-secretase and the Aβ-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 20, 794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang S., Wang R., Chen L., Bennett D. A., Dickson D. W., Wang D. S. (2010) Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer's brain. J. Neurochem. 115, 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shinall H., Song E. S., Hersh L. B. (2005) Susceptibility of amyloid β peptide degrading enzymes to oxidative damage: a potential Alzheimer's disease spiral. Biochemistry 44, 15345–15350 [DOI] [PubMed] [Google Scholar]
- 21. Du J., Zhang L., Liu S., Zhang C., Huang X., Li J., Zhao N., Wang Z. (2009) PPARγ transcriptionally regulates the expression of insulin-degrading enzyme in primary neurons. Biochem. Biophys. Res. Commun. 383, 485–490 [DOI] [PubMed] [Google Scholar]
- 22. Zhang L., Ding Q., Wang Z. (2012) Nuclear respiratory factor 1 mediates the transcription initiation of insulin-degrading enzyme in a TATA box-binding protein-independent manner. PLoS One 7, e42035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andersson U., Scarpulla R. C. (2001) Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 21, 3738–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gleyzer N., Vercauteren K., Scarpulla R. C. (2005) Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 25, 1354–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chowanadisai W., Bauerly K. A., Tchaparian E., Wong A., Cortopassi G. A., Rucker R. B. (2010) Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1α expression. J. Biol. Chem. 285, 142–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Izumi H., Ohta R., Nagatani G., Ise T., Nakayama Y., Nomoto M., Kohno K. (2003) p300/CBP-associated factor (P/CAF) interacts with nuclear respiratory factor-1 to regulate the UDP-N-acetyl-α-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase-3 gene. Biochem. J. 373, 713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Llovera R. E., de Tullio M., Alonso L. G., Leissring M. A., Kaufman S. B., Roher A. E., de Prat Gay G., Morelli L., Castaño E. M. (2008) The catalytic domain of insulin-degrading enzyme forms a denaturant-resistant complex with amyloid β peptide: implications for Alzheimer disease pathogenesis. J. Biol. Chem. 283, 17039–17048 [DOI] [PubMed] [Google Scholar]
- 28. Chan C. B., Kashemsant N. (2006) Regulation of insulin secretion by uncoupling protein. Biochem. Soc. Trans. 34, 802–805 [DOI] [PubMed] [Google Scholar]
- 29. Andrews N. C., Faller D. V. (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuo M. H., Allis C. D. (1999) In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19, 425–433 [DOI] [PubMed] [Google Scholar]
- 31. Pfaffl M. W., Horgan G. W., Dempfle L. (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boveris A., Costa L. E., Cadenas E., Poderoso J. J. (1999) Regulation of mitochondrial respiration by adenosine diphosphate, oxygen, and nitric oxide. Methods Enzymol. 301, 188–198 [DOI] [PubMed] [Google Scholar]
- 33. Brand M. D., Nicholls D. G. (2011) Assessing mitochondrial dysfunction in cells. Biochem. J. 435, 297–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prat M. I., Adamo A. M., González S. A., Affranchino J. L., Ikeda M., Matsubara E., Shoji M., Smith M. A., Castaño E. M., Morelli L. (2002) Presenilin 1 overexpressions in Chinese hamster ovary (CHO) cells decreases the phosphorylation of retinoblastoma protein: relevance for neurodegeneration. Neurosci. Lett. 326, 9–12 [DOI] [PubMed] [Google Scholar]
- 35. Tanabe T., Tsushima K., Yasuo M., Urushihata K., Hanaoka M., Koizumi T., Fujimoto K., Kubo K., Uehara T., Shigematsu S., Hamano H., Kawa S. (2006) IgG4-associated multifocal systemic fibrosis complicating sclerosing sialadenitis, hypophysitis, and retroperitoneal fibrosis, but lacking pancreatic involvement. Intern. Med. 45, 1243–1247 [DOI] [PubMed] [Google Scholar]
- 36. Sheng B., Wang X., Su B., Lee H. G., Casadesus G., Perry G., Zhu X. (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J. Neurochem. 120, 419–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cam H., Balciunaite E., Blais A., Spektor A., Scarpulla R. C., Young R., Kluger Y., Dynlacht B. D. (2004) A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell 16, 399–411 [DOI] [PubMed] [Google Scholar]
- 38. Perera R., Sono M., Sigman J. A., Pfister T. D., Lu Y., Dawson J. H. (2003) Neutral thiol as a proximal ligand to ferrous heme iron: implications for heme proteins that lose cysteine thiolate ligation on reduction. Proc. Natl. Acad. Sci. U.S.A. 100, 3641–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baar K., Song Z., Semenkovich C. F., Jones T. E., Han D. H., Nolte L. A., Ojuka E. O., Chen M., Holloszy J. O. (2003) Skeletal muscle overexpression of nuclear respiratory factor 1 increases glucose transport capacity. FASEB J. 17, 1666–1673 [DOI] [PubMed] [Google Scholar]
- 40. Scarpulla R. C. (2006) Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 97, 673–683 [DOI] [PubMed] [Google Scholar]
- 41. Bulloj A., Leal M. C., Surace E. I., Zhang X., Xu H., Ledesma M. D., Castaño E. M., Morelli L. (2008) Detergent resistant membrane-associated IDE in brain tissue and cultured cells: relevance to Aβ and insulin degradation. Mol. Neurodegener. 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 [DOI] [PubMed] [Google Scholar]
- 43. Walls K. C., Coskun P., Gallegos-Perez J. L., Zadourian N., Freude K., Rasool S., Blurton-Jones M., Green K. N., LaFerla F. M. (2012) Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and β-amyloid. J. Biol. Chem. 287, 30317–30327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Golbus J., Palella T. D., Richardson B. C. (1990) Quantitative changes in T cell DNA methylation occur during differentiation and ageing. Eur. J. Immunol. 20, 1869–1872 [DOI] [PubMed] [Google Scholar]
- 45. Wilson V. L., Jones P. A. (1983) DNA methylation decreases in aging but not in immortal cells. Science 220, 1055–1057 [DOI] [PubMed] [Google Scholar]
- 46. Christensen B. C., Houseman E. A., Marsit C. J., Zheng S., Wrensch M. R., Wiemels J. L., Nelson H. H., Karagas M. R., Padbury J. F., Bueno R., Sugarbaker D. J., Yeh R. F., Wiencke J. K., Kelsey K. T. (2009) Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 5, e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fraga M. F., Ballestar E., Paz M. F., Ropero S., Setien F., Ballestar M. L., Heine-Suñer D., Cigudosa J. C., Urioste M., Benitez J., Boix-Chornet M., Sanchez-Aguilera A., Ling C., Carlsson E., Poulsen P., Vaag A., Stephan Z., Spector T. D., Wu Y. Z., Plass C., Esteller M. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zuo X., Jia J. (2009) Promoter polymorphisms which modulate insulin degrading enzyme expression may increase susceptibility to Alzheimer's disease. Brain Res. 1249, 1–8 [DOI] [PubMed] [Google Scholar]
- 49. Zhu X., Perry G., Moreira P. I., Aliev G., Cash A. D., Hirai K., Smith M. A. (2006) Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J. Alzheimers Dis. 9, 147–153 [DOI] [PubMed] [Google Scholar]
- 50. Blass J. P. (2000) The mitochondrial spiral. An adequate cause of dementia in the Alzheimer's syndrome. Ann. N.Y. Acad. Sci. 924, 170–183 [DOI] [PubMed] [Google Scholar]
- 51. de la Monte S. M., Luong T., Neely T. R., Robinson D., Wands J. R. (2000) Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer's disease. Lab. Invest. 80, 1323–1335 [DOI] [PubMed] [Google Scholar]
- 52. Hirai K., Aliev G., Nunomura A., Fujioka H., Russell R. L., Atwood C. S., Johnson A. B., Kress Y., Vinters H. V., Tabaton M., Shimohama S., Cash A. D., Siedlak S. L., Harris P. L., Jones P. K., Petersen R. B., Perry G., Smith M. A. (2001) Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 21, 3017–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X., Su B., Lee H. G., Li X., Perry G., Smith M. A., Zhu X. (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 29, 9090–9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liang H., Ward W. F. (2006) PGC-1α: a key regulator of energy metabolism. Adv. Physiol. Educ. 30, 145–151 [DOI] [PubMed] [Google Scholar]
- 55. Piantadosi C. A., Suliman H. B. (2006) Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J. Biol. Chem. 281, 324–333 [DOI] [PubMed] [Google Scholar]
- 56. Zhao L., Teter B., Morihara T., Lim G. P., Ambegaokar S. S., Ubeda O. J., Frautschy S. A., Cole G. M. (2004) Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J. Neurosci. 24, 11120–11126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cho Y. M., Shin H. D., Park B. L., Kim J. H., Park K. S., Kim S. Y., Lee H. K. (2005) Association between polymorphisms in the nuclear respiratory factor 1 gene and type 2 diabetes mellitus in the Korean population. Diabetologia 48, 2033–2038 [DOI] [PubMed] [Google Scholar]
- 58. Liu Y., Niu N., Zhu X., Du T., Wang X., Chen D., Wu X., Gu H. F. (2008) Genetic variation and association analyses of the nuclear respiratory factor 1 (nRF1) gene in Chinese patients with type 2 diabetes. Diabetes 57, 777–782 [DOI] [PubMed] [Google Scholar]
- 59. Regev-Rudzki N., Pines O. (2007) Eclipsed distribution: a phenomenon of dual targeting of protein and its significance. BioEssays 29, 772–782 [DOI] [PubMed] [Google Scholar]
- 60. Fernandez-Gamba A., Leal M. C., Maarouf C. L., Richter-Landsberg C., Wu T., Morelli L., Roher A. E., Castano E. M. (2012) Collapsin response mediator protein-2 phosphorylation promotes the reversible retraction of oligodendrocyte processes in response to non-lethal oxidative stress. J. Neurochem. 121, 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]