

Background: Bacteriophage fd is activated for infection by partial unfolding and prolyl isomerization.
Results: NMR spectroscopy localized Pro-213-coupled unfolding to regions of the interdomain hinge of the phage gene-3-protein.
Conclusion: Pro-213 regulates phage infectivity by a specific long-range effect on the conformational stability of the gene-3-protein.
Significance: A proline switch controls the biological function in a remote part of a protein.
Keywords: Nuclear Magnetic Resonance, Protein Folding, Protein Stability, Protein Structure, Signal Transduction, Gene-3-protein, Molecular Timer, Phage Infection, Prolyl Isomerization
Abstract
Infection of Escherichia coli by the filamentous phage fd starts with the binding of the N2 domain of the phage gene-3-protein to an F pilus. This interaction triggers partial unfolding of the gene-3-protein, cis → trans isomerization at Pro-213, and domain disassembly, thereby exposing its binding site for the ultimate receptor TolA. The trans-proline sets a molecular timer to maintain the binding-active state long enough for the phage to interact with TolA. We elucidated the changes in structure and local stability that lead to partial unfolding and thus to the activation of the gene-3-protein for phage infection. Protein folding and TolA binding experiments were combined with real-time NMR spectroscopy, amide hydrogen exchange measurements, and phage infectivity assays. In combination, the results provide a molecular picture of how a local unfolding reaction couples with prolyl isomerization not only to generate the activated state of a protein but also to maintain it for an extended time.
Introduction
The filamentous phage fd initiates the infection of Escherichia coli cells by two sequential binding events. First, it attaches to the tip of an F pilus, and then it interacts with the membrane-bound protein TolA at the cell surface (1–4). Both interactions are mediated by the phage gene-3-protein, which exists in 3–5 copies at one end of the phage. The protein consists of three domains. The C-terminal or CT8 domain anchors the gene-3-protein in the phage coat, the middle domain N2 establishes the initial contact with the F pilus, and the N-terminal domain N1 binds to the C-terminal domain of TolA (TolA-C) at the cell surface (2) (Fig. 1A).
FIGURE 1.

Function of G3P during the phage infection process. A, top, arrangement of the domains of G3P at the pilus tip. Bottom, opening of G3P during infection and interaction of domain N2 (blue)) with the pilus (1), partial unfolding of the hinge subdomain (green), and binding of domain N1 (red) to the C-terminal domain of TolA (shown in light blue: N, M, and C, N-terminal, middle, and C-terminal domains, respectively, of TolA) (2). B, tertiary structure of wild-type G3P showing the amino acids that were substituted in this work. Domain coloring is as in panel A. The binding sites for the F pilus and the C-terminal domain of TolA are indicated. Strand β6 of the hinge, which is deleted in G3P Δβ6, is depicted in light green. Positions 13, 101, 129, and 209 of the stabilizing substitutions in G3P IIHY as well as Gln-212, cis-Pro-213 and Pro-214 are shown in stick representation. The figure was prepared by using PyMOL (31) and the Protein Data Bank (PDB) file 2g3p (5).
The N1-N2 entity of the gene-3-protein (called “G3P” in this article) protrudes from the phage tip. In the fully folded form of G3P, N1 and N2 are tightly associated with each other (5, 6) (Fig. 1B), which renders G3P robust under varying environmental conditions. However, this form is autoinhibited, and the phage is noninfectious because the binding site for TolA-C is buried at the domain interface. To overcome this intramolecular inhibition, the interdomain contacts between N1 and N2 must be broken during the infection process.
In fact, after the initial binding of the N2 domain to the F pilus, the interactions between N1, N2, and the interdomain hinge are weakened, and the TolA binding site of N1 becomes accessible (7, 8). The domain disassembly allows Pro-213 in the hinge (Fig. 1B) to isomerize from the strained cis to the more favorable trans conformation (9, 10). trans-Pro-213 maintains fd-G3P in the open, infection-competent conformation for an extended time and allows the N1 domain to bind to its receptor TolA-C (7, 8). Pro-213 thus functions as a molecular switch. The infectious form of G3P is turned on by cis → trans and turned off by trans → cis isomerization of the Gln-212–Pro-213 peptide bond.
During refolding of G3P, Pro-213 trans → cis isomerization is the final and rate-limiting step and controls domain assembly (9, 10). The structure of the folding intermediate that accumulates before this final folding step was analyzed by NMR spectroscopy for a variant of G3P, termed G3P IIHY, which carries the four stabilizing substitutions T13I, T101I, Q129H, and D209Y (11, 12). Due to its high thermodynamic stability and robust folding, this variant has been used to study the folding mechanism of G3P. Pro-213 trans → cis isomerization in the final folding step was shown to be accompanied by the tightening of a hydrogen-bonding network between the hinge region and the N1 domain and by a strong stabilization of the N2 domain.
It is our aim to understand how changes in the domain interactions of G3P, partial unfolding, and prolyl isomerization activate the phage for binding to its receptor TolA. We modulated the stability of G3P, the domain interactions, and the isomerization rate of Pro-213 by targeted amino acid substitutions, in particular in the contact regions between the domains and around Pro-213. Then, we analyzed the consequences of these substitutions for the folding of G3P, for its interaction with the receptor TolA, and for the infectivity of phage variants carrying the same substitutions in their G3P. High-resolution NMR techniques were employed to reveal, at residue resolution, the differences in structure and stability between the fully folded autoinhibited form of G3P and the partially unfolded activated form.
EXPERIMENTAL PROCEDURES
Expression, Purification, and Modification of the Proteins
To generate the different variants of the N1-N2 fragment of the gene-3-protein (G3P), site-directed mutagenesis was performed by blunt end PCR using primers with the corresponding mutations. The G3P variants, the isolated N1 domain (residues 1–67 of mature G3P, extended by an Ala(His)6 tag), the isolated N2 domain (residues 102–205 of mature G3P followed by (His)6), and TolA-C (residues 295–421 of TolA, with a Cys (Ala)3(His)6 tag) were expressed and purified as described (7, 11, 13). TolA-C was labeled with 5-((((2-iodoacetyl)amino)ethyl)amino) naphthalene-1-sulfonic acid (iodo-Aedans)9 at the cysteine residue introduced at the C terminus of TolA-C as described (7), resulting in TolA-C-Aedans.
Heat-induced Unfolding Transitions
Heat-induced unfolding transitions of isolated N1, domain N1 in wild-type G3P, and its variants were measured by the change in circular dichroism (CD) at 230 nm with 4.0 μm protein in 0.1 m potassium phosphate (pH 7.0) using a JASCO J-600A spectropolarimeter equipped with a PTC-348 WI Peltier device. The unfolding transition of isolated N2 (4.0 μm) was monitored by the increase in CD at 222 nm. For N2 in the N1-N2 fragment of G3P (1.0 μm), the decrease in CD at 210 nm was used as a probe (11). All measurements were performed in 10-mm cells with a 1-nm bandwidth and a heating rate of 60 K/h. The data were analyzed as described before (14) using nonlinear regression and the program GraFit (Erithacus Software, Staines, UK).
Complex Formation between TolA-C-Aedans and Different G3P Variants
The kinetics of binding of TolA-C-Aedans to isolated N1 or to N1 in the G3P variants were followed at 25 °C after stopped-flow mixing in a DX.17MV stopped-flow fluorometer (Applied Photophysics) by the change in fluorescence above 460 nm after excitation at 280 nm. To absorb scattered light from the excitation beam, a 0.5-cm cell with p-nitroaniline in ethanol was placed between the observation chamber and the emission photomultiplier. Association was initiated by 1:1 dilution, resulting in final concentrations of 0–14 μm G3P and 0.2 μm TolA-C-Aedans in 0.1 m potassium phosphate (pH 7.0), 0.1 m GdmCl. Individual kinetics were measured eight times, averaged, and analyzed using monoexponential functions. The resulting amplitudes and apparent rates were plotted as a function of the G3P concentration. The plot of the rate constants was analyzed using linear regression. The rate constant of association kon(app) was derived from the slope, and the rate constant of dissociation koff was derived from the ordinate intercept of this plot.
To measure the kinetics of interaction between TolA-C-Aedans and the active forms with a trans-Pro-213 of wild-type G3P and of G3P IIHY, the active form was populated in an interrupted refolding experiment. Denatured G3P (in 5.0 m GdmCl) was diluted 25-fold with 0.1 m potassium phosphate (pH 7.0) to initiate refolding at 25 °C. After 10 min, the samples were cooled to 4 °C to retard trans → cis isomerization of Pro-213 and transferred into the stopped-flow apparatus. The kinetics of binding were measured as described above.
Phage Infection Experiments
For all experiments, a derivative of the fCKCBS phage was used, which contains the gene for chloramphenicol acetyl transferase and G3P with a modified linker between the N2 and C-terminal domains (15, 16). Phage were isolated from E. coli culture medium as described before, resulting in 50-μl aliquots (11). Phage concentration was determined by isolating the phage DNA from 10 μl of the phage suspension and measuring its absorption at 260 nm.
To compare the infectivity of wild-type phage with phage containing modified G3P toward pilus bearing (F+) E. coli XL1 Blue cells, 5 μl of the phage suspensions were mixed individually with 495 μl of a corresponding cell culture in dYT medium (5 g l−1 NaCl, 10 g l−1 yeast extract, 16 g l−1 caseine peptone, A600 ∼ 0.7) and incubated at 25 °C for 5 min. Then, the samples were centrifuged, and the remaining phage were removed with the supernatant. The cell pellets were washed, resuspended in 500 μl of dYT medium, and incubated at 25 °C for 25 min. Serial dilutions were plated on dYT agar containing 25 μg/ml chloramphenicol to determine the number of infectious phage.
F pili-deficient E. coli HB2156 cells were infected with the same phage suspensions in the presence of 50 mm CaCl2. 25 μl of phage suspension and 25 μl of 1 m CaCl2 were added to 450 μl of an E. coli HB2156 cell culture (A600 ∼ 0.7) and incubated for 5 min at 25 °C. The remaining phage were removed by centrifugation, and infected cells were washed and resuspended in 100 μl of dYT medium. The number of infectious phage was again determined by plating serial dilutions on dYT agar containing 25 μg/ml chloramphenicol.
NMR Experiments
All NMR spectra were recorded in 50 mm sodium phosphate pH 7.0 (or pD 6.6) at 25 °C, containing 10 or 100% D2O. Spectra were processed using NMRPipe (17) and analyzed using NMRView (18). For the backbone resonance assignments (Fig. 2 and supplemental Table 1), trHNCACB and trHN (CO)CACB spectra of a 15N/13C/2H sample were acquired with a Bruker Avance 800 spectrometer.
FIGURE 2.

1H15N-TROSY-HSQC of G3P* recorded at 25 °C in 50 mm sodium phosphate pH 7. 0 with the assignments of the amide cross-peaks.
A 3.6 mm solution of TolA-C was titrated to 0.3 mm [15N]G3P in 14 steps up to a 12-fold excess of TolA-C (1.8 mm TolA-C, 0.15 mm G3P) and monitored by 15N-TROSY-HSQC spectra with a Bruker Avance 900 spectrometer. Free G3P and bound G3P were in slow exchange on the NMR timescale; therefore, binding was analyzed by the decrease of 1H15N cross-peaks of G3P. Intensities were corrected by the dilution factor. The analysis of the intensities suggested that the complex was fully formed when a slight molar excess of TolA-C was reached.
Amide hydrogen/deuterium exchange (HX) of native G3P was followed by dissolving lyophilized protein in D2O buffer and measuring 60 15N-TROSY-HSQC spectra over 120 h with a Bruker Avance 800 spectrometer (Fig. 3, A—C). Amide HX of the IIHY variant of G3P followed the EX2 exchange mechanism (12), and we assume that the wild-type form shows EX2 exchange as well. To measure amide HX of the complex between G3P and TolA-C, 0.15 mm G3P and 0.60 mm TolA-C were dissolved in D2O buffer and the exchange was monitored by collecting 46 15N-TROSY-HSQC spectra over 91 h (Fig. 3, A, B, and D).
FIGURE 3.

Native state amide exchange of G3P*. A and B, section of the 1H15N-TROSY-HSQC in D2O buffer immediately after start (A) and after 600 min (B). The inset shows the same experiment in the presence of TolA-C. C and D, amide exchange kinetics of Ala-179 in the absence (C) and in the presence of TolA-C (D) including the single exponential fit. E and F, competition between refolding and amide exchange. E, section of the 1H15N-TROSY-HSQC in D2O buffer of native G3P. F, same section as in E but recorded after G3P was refolded in D2O buffer and the residual denaturant was removed by rapid gel filtration. Amide cross-signals are labeled and color-coded as in Fig. 8. B, D, and F, residues with protection factors larger than 10,000 are shown in dark green, values between 10,000 and 100 are in green, and values smaller than 100 are in pale green. The buffer was 50 mm sodium phosphate, pH 7.0 (or pD 6.6) in all cases.
For real-time NMR experiments, refolding of 4 mm [15N]G3P was started by a 4-fold dilution of the unfolded protein (in 4.0 m GdmCl, 50 mm sodium phosphate, pH 7.0) to a final GdmCl concentration of 1.0 m in the same buffer. After a dead time of 30 min, two identical 15N-TROSY-HSQC spectra were acquired successively by a Bruker Avance 900 spectrometer. The first (kinetic) spectrum was obtained during the folding reaction; the second spectrum was measured after the completion of refolding and served as the reference for fully folded G3P. The spectra were measured for 8 h each. Subtraction of the kinetic spectrum from the reference spectrum resulted in the difference spectrum. This experiment was repeated in the same way with 1.5 mm [15N]G3P in the presence of 2 mm TolA-C in the refolding buffer.
For the competition experiment between refolding and amide HX, refolding of [15N]G3P (in 4.0 m GdmCl, 50 mm sodium phosphate, pH 7.0) was performed as described above, except that all buffers contained 100% H2O. After 5 min, when the refolding reactions of the individual domains were complete, the solvent was exchanged to a D2O-containing buffer without GdmCl using a NAP10 gel filtration column (GE Healthcare, Uppsala, Sweden). The competition between folding and amide HX was allowed to proceed for 10 h at 25 °C, and then a 15N-TROSY-HSQC spectrum was measured over 8 h at a Bruker Avance 900 spectrometer (Fig. 3, E and F). For referencing, an identical amide HX experiment was performed starting with the same amount of folded G3P in 1.0 m GdmCl.
RESULTS
Experimental Strategy
Amino acid substitutions that strengthen or weaken the domain interactions in G3P were employed to examine how alterations in the stability and the folding mechanism of G3P affect its function during phage infection (Fig. 1B). As a variant with stronger domain interactions, we used the IIHY form of G3P. It contains four substitutions, all identified in the course of an in vitro selection for stabilized phage variants (11): T13I in the N1 domain, Q129H in the N2 domain, and T101I and D209Y in the hinge region. The T101I substitution, in particular, improves the interactions between the hinge region and the N1 domain.
To weaken or abolish the domain interactions, we used two strategies. First, we substituted the residues 212 and 214 before and after the Pro-213 switch by Ala (in the G3P APA variant, Fig. 1B) or combined these two substitutions with the replacement of Pro-213 by Gly (in G3P AGA). Amino acid substitutions in this region decrease the stability of G3P and increase the rate of prolyl isomerization (7, 10). The residues 212–214 thus determine the lifetime of the active, infectious form of G3P.
In an alternative approach, we abolished the domain interactions by deleting strand β6 (residues 96–103) of the hinge region in G3P IIHY. This strand establishes key interactions within the hinge itself and with the N1 domain in the fully folded form of G3P (Fig. 1B). For the corresponding variant (G3P Δβ6), G3P IIHY was used as the parent protein as the wild-type form of G3P without strand β6 started to unfold already at 25 °C (19).
Amino acid substitutions were made in both the gene encoding the N1-N2 fragment (G3P) for the in vitro studies and the phage genome used for the in vivo infection experiments. The isolated domains N1 and N2 served as reference proteins. Unfolding experiments were used to examine the strength of the domain interactions within the G3P variants. The interaction with TolA was analyzed by measuring the kinetics of binding with a labeled fluorescent variant of TolA-C (TolA-C-Aedans). The infectivities of the corresponding phage variants were determined by using cells with or without F pili (F+ or F−).
Modulating the Strength of the Domain Interactions
The thermal unfolding transition of the N2 domain served as an indicator for the strength of the domain interactions in the G3P variants. As an isolated domain, N2 is only marginally stable and starts to unfold at 30 °C already (8, 13). In the fully folded form of G3P, N2 is stabilized by the strong interdomain interactions, and it unfolds only when these interactions are abolished. N1 is more stable and remains folded after domain disassembly. The unfolding of G3P, monitored by CD at 230 nm, thus shows two transitions (Fig. 4A) (8, 11). The minor, first transition, reflects domain disassembly coupled with the unfolding of N2 and is used to probe the strength of the domain interactions in G3P. Substitutions that improve the domain interactions increase the midpoint (the Tm value) of this transition, and substitutions that loosen the domain interactions lead to a decrease. The first transition could also be followed by the change in CD at 210 nm (Fig. 4B) because N1 contributes little to the CD change at this wavelength. The substitutions in the hinge and in N2 led to large shifts in the Tm values of the first transition, whereas the stability of the N1 domain was affected only by the T13I substitution, which resides in this domain.
FIGURE 4.
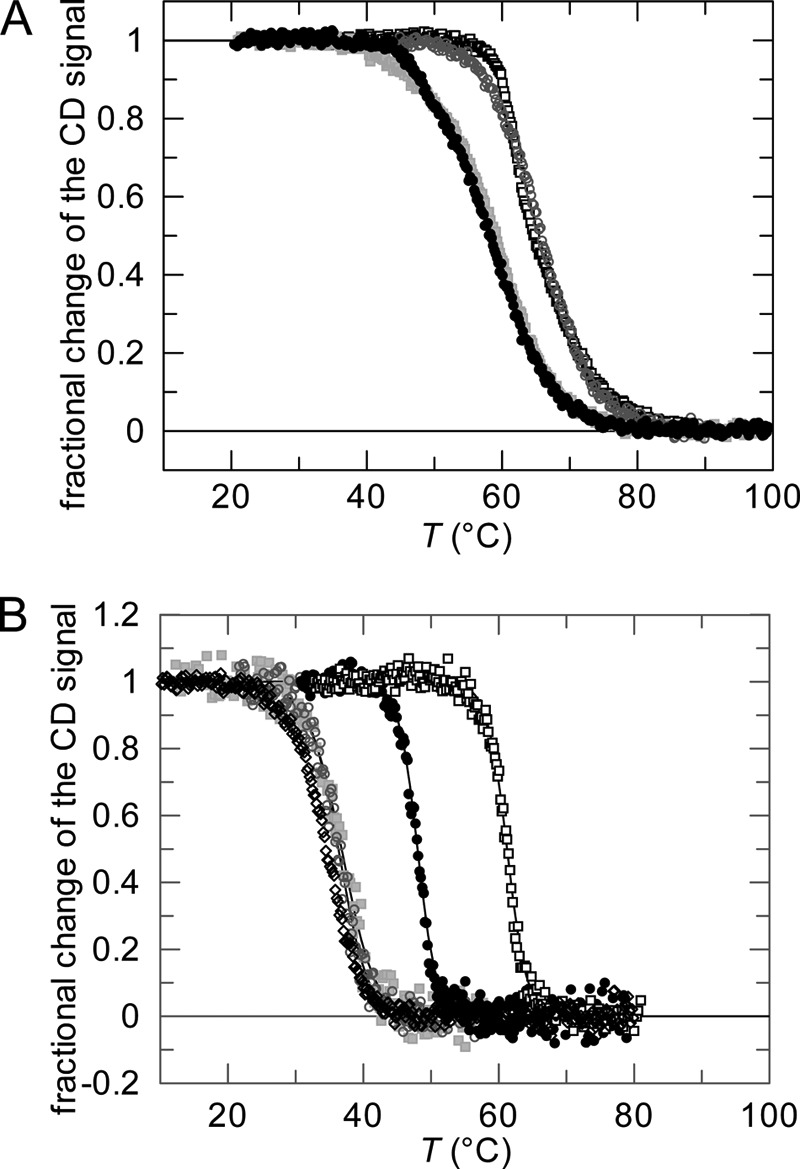
Thermal stabilities of wild-type G3P and of variants with modulated domain interactions.
A, thermal transitions measured by CD at 230 nm for the wild-type protein (●) and the variants G3P IIHY (□), G3P APA ( ), and G3P Δβ6 (○). B, thermal transitions measured by CD at 210 nm for the wild-type protein (●) and the variants G3P IIHY (□), G3P APA (), and G3P Δβ6 (○) and measured by CD at 222 nm for the isolated N2 domain (♢). Fractional changes of the signals as obtained after three-state or two-state analyses, respectively, are shown as a function of temperature. The continuous lines represent the results of the data analyses; the corresponding parameters are given in Table 1. The transitions were measured with 1.0 μm (CD at 210 nm) or 4.0 μm (CD at 230 nm) protein in 0.1 m potassium phosphate (pH 7.0) at a path length of 10 mm.
), and G3P Δβ6 (○). B, thermal transitions measured by CD at 210 nm for the wild-type protein (●) and the variants G3P IIHY (□), G3P APA (), and G3P Δβ6 (○) and measured by CD at 222 nm for the isolated N2 domain (♢). Fractional changes of the signals as obtained after three-state or two-state analyses, respectively, are shown as a function of temperature. The continuous lines represent the results of the data analyses; the corresponding parameters are given in Table 1. The transitions were measured with 1.0 μm (CD at 210 nm) or 4.0 μm (CD at 230 nm) protein in 0.1 m potassium phosphate (pH 7.0) at a path length of 10 mm.
The two stabilizing substitutions T101I and D209Y in the hinge increased the Tm of the first transition from 48.0 °C (G3P) to 61.2 °C (G3P IIHY) (Fig. 4, Table 1), which indicates that the domain interactions are strongly improved in this variant. In contrast, two other substitutions in the hinge, Q212A and P214A, adjacent to cis-Pro-213 (in G3P APA), decreased the Tm value to 36.8 °C (Fig. 4), which is only 2.2 °C higher than the Tm of the isolated N2 domain (Table 1). Thus, the domain interactions are severely weakened but not fully abolished in this variant. A similar destabilization was found for the G3P AGA variant, in which Pro-213 is substituted by Gly (Table 1).
TABLE 1.
Stability data for the variants of G3P and the isolated domains N1 and N2 as derived from thermal unfolding transitions
For the proteins, the melting temperature Tm, the enthalpy of denaturation at Tm (ΔH), and the Gibbs free energy of denaturation (ΔG) at 55 and 65 °C, respectively, are listed. The parameters for domain N2 were obtained from a two-state analysis of the change in CD at 222 nm (isolated N2 domain) or at 210 nm (N2 domain in the G3P variants). The parameters for domain N1 were calculated after a three-state analysis from the second transition, as observed by CD at 230 nm. For the analysis, the heat capacity change (ΔCp) of domain N2 within G3P was fixed at 10,000 J mol−1 K−1, ΔCp of domain N1 at 1000 J mol−1 K−1 (11). For the isolated N2 domains and the domain N2 within G3P Δβ6, a ΔCp of 8000 J mol−1 K−1 was used (14). The transitions were measured in 0.1 m K phosphate (pH 7.0) at a path length of 10 mm with 4.0 μm (CD at 230 nm) or 1.0 μm protein (CD at 210 nm).
| Variant | Unfolding of domain N2 |
Unfolding of domain N1 |
||||
|---|---|---|---|---|---|---|
| Tm | ΔH | ΔG55 °C | Tm | ΔH | ΔG65 °C | |
| °C | kJ mol−1 | kJ mol−1 | °C | kJ mol−1 | kJ mol−1 | |
| Wild-type G3P | 48.0 | 612 | −14.1 | 60.2 | 230 | −3.3 |
| G3P IIHY | 61.2 | 644 | 11.4 | 67.5 | 205 | 1.5 |
| G3P APA | 36.8 | 331 | −24.6 | 59.9 | 227 | −3.5 |
| G3P-AGA | 37.9 | 470 | −30.4 | 58.6 | 189 | −3.7 |
| G3P Δβ6 | 36.4 | 368 | −21.1 | 65.9 | 250 | −0.5 |
| Isolated domain N2 | 34.6 | 304 | −25.4 | |||
| Isolated domain N2 Q129H | 38.8 | 344 | −21.2 | |||
| Isolated domain N1 | 62.3 | 234 | −1.9 | |||
| Isolated domain N1 T13I | 67.5 | 235 | 1.7 | |||
The deletion of strand β6 in the hinge region (in G3P Δβ6) eliminated the domain interactions. The first unfolding transition of this variant shows a midpoint at 36.4 °C (Fig. 4), which is slightly lower than the value of 38.8 °C of the isolated N2 domain of G3P IIHY (N2 Q129H, Table 1). G3P Δβ6 thus provides a good model for a permanently active G3P without domain interactions.
Affinity of the G3P Variants for TolA-C
After the activation by domain disassembly, the N1 domain of G3P binds to TolA-C. We followed this binding reaction by the strong increase in the Förster resonance energy transfer (FRET) between the Trp residues of the N1 domain and a variant of TolA-C that is labeled at its carboxyl terminus by an Aedans group (TolA-C-Aedans) (7, 8). The kinetics of interaction between 0.2 μm TolA-C-Aedans and 0–14 μm of the G3P variants were measured in a stopped-flow apparatus. Binding followed pseudo first-order kinetics under all conditions (Fig. 5A), the amplitudes (Fig. 5B) showed saturation behavior, and the measured rates (Fig. 5C) depended linearly on the concentration of the G3P variants. The rates of association, kon(app), and of dissociation, koff, were obtained from the slopes and the intercepts, respectively, of the plots in Fig. 5C (Table 2). The apparent dissociation constants, KD(app), of the complexes between TolA-C-Aedans and the G3P variants were derived from the ratio of the rate constants kon(app) and koff, as well as from fitting binding curves to the amplitude data in Fig. 5B. These two KD(app) values agree well with each other for the individual G3P variants (Table 2).
FIGURE 5.
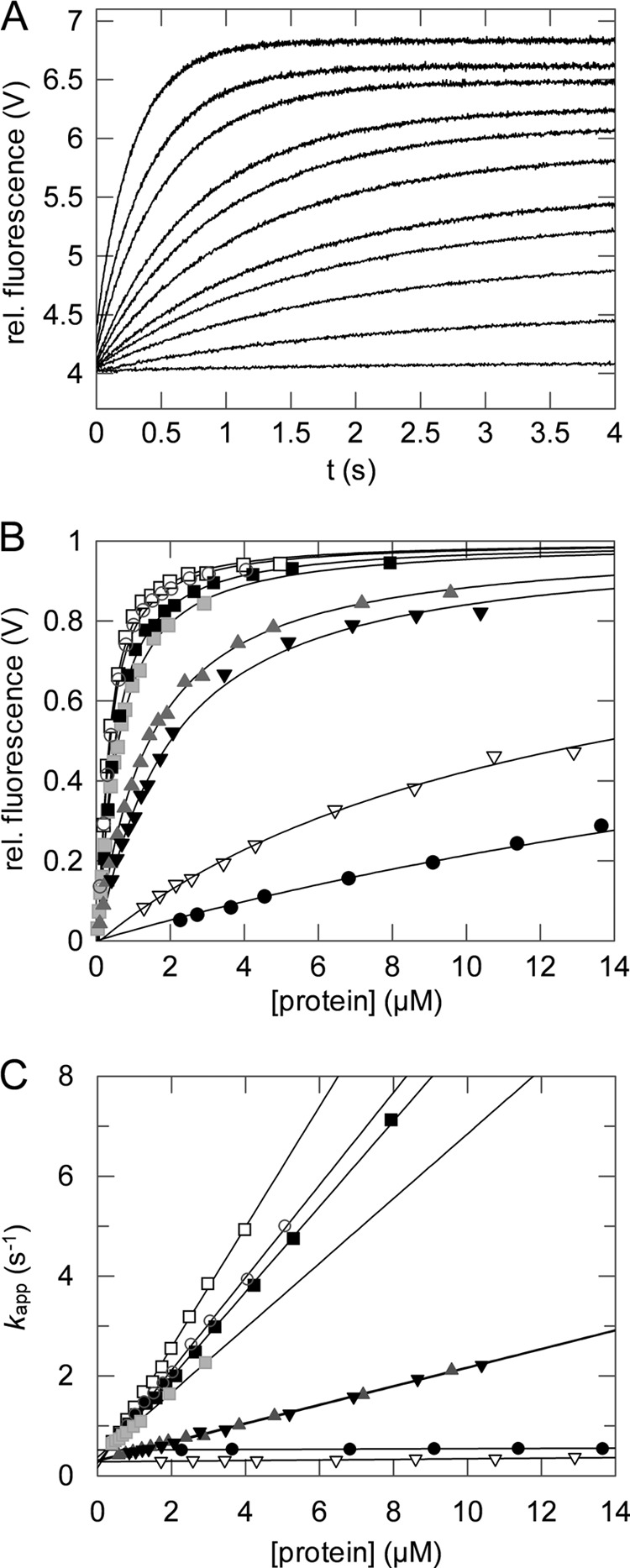
Interaction of TolA-C-Aedans with the G3P variants.
A, association reactions of 0.2 μm TolA-C-Aedans and increasing concentrations of domain N1 T13I (0–3.0 μm, from bottom to top). Binding was measured by FRET between the Trp residues of N1 and TolA-C-Aedans and monitored by fluorescence above 460 nm (excitation at 280 nm) after stopped-flow mixing at 25 °C. rel. fluorescence, relative fluorescence. B and C, the normalized amplitudes (B) and the rates (C) of complex formation are shown as a function of the concentration for isolated N1 (■), isolated N1 T13I (□), native wild-type G3P (●), the folding intermediate of wild-type G3P (▾), the folding intermediate of G3P IIHY (▿), G3P AGA ( ), G3P APA (), and G3P Δβ6 (○). The continuous lines represent analyses of the data assuming a single-step binding mechanism. The derived rate constants kon(app) and koff, and KD(app), as calculated from the amplitudes in B, are listed in Table 2. All experiments were performed in 0.1 m potassium phosphate (pH 7.0), 0.1 m GdmCl. The intermediate forms of wild-type G3P and G3P IIHY were populated as described under “Experimental Procedures.”
), G3P APA (), and G3P Δβ6 (○). The continuous lines represent analyses of the data assuming a single-step binding mechanism. The derived rate constants kon(app) and koff, and KD(app), as calculated from the amplitudes in B, are listed in Table 2. All experiments were performed in 0.1 m potassium phosphate (pH 7.0), 0.1 m GdmCl. The intermediate forms of wild-type G3P and G3P IIHY were populated as described under “Experimental Procedures.”
TABLE 2.
Binding of G3P variants to TolA-C AEDANS
The rate constants kon(app) and koff for the different G3P variants were derived from the slopes and intercepts, respectively, in Fig. 5C, and the dissociation constant KD(app) was derived from the analysis of the amplitudes of association (Fig. 5B). Binding experiments were performed as described in the legend for Fig. 5. The data were analyzed assuming a single-step binding mechanism. The stability constant Kcl was calculated using Equation 4, the KD(app) value from the amplitude analysis, and the KD value of the isolated domain N1. The long-lived folding intermediates of wild-type G3P and G3P IIHY were generated as described under “Experimental Procedures.” The measurements were performed in 0.1 m potassium phosphate (pH 7.0), 0.1 m GdmCl at 25 °C. ND, binding kinetics not detectable.
| Variant | kon(app) | koff | koff/kon | KD(app) | Kcl |
|---|---|---|---|---|---|
| μm−1s−1 | s−1 | μm | μm | ||
| Isolated domain N1 | 0.86 ± 0.01 | 0.26 ± 0.03 | 0.3 | 0.31 ± 0.02 | 0 |
| Isolated domain N1 T13I | 1.21 ± 0.02 | 0.16 ± 0.03 | 0.13 | 0.19 ± 0.01 | 0 |
| Wild-type G3P | 0.003 ± 0.001 | 0.51 ± 0.01 | 171 | 22.1 ± 6.6 | 70 |
| G3P IIHY | ND | ND | ND | ND | ND |
| G3P-APA | 0.65 ± 0.01 | 0.37 ± 0.01 | 0.57 | 0.44 ± 0.02 | 0.4 |
| G3P-AGA | 0.19 ± 0.01 | 0.31 ± 0.01 | 1.68 | 1.26 ± 0.05 | 3.1 |
| G3P Δβ6 | 0.93 ± 0.01 | 0.26 ± 0.03 | 0.28 | 0.21 ± 0.01 | 0.1 |
| G3P folding intermediate (trans Pro213) | 0.19 ± 0.01 | 0.29 ± 0.01 | 1.54 | 1.85 ± 0.12 | 5.0 |
| G3P IIHY folding intermediate (trans-Pro-213) | 0.006 ± 0.001 | 0.28 ± 0.01 | 48 | 13.6 ± 1.8 | 71 |
TolA-C binds most strongly to the isolated N1 domain because the binding site is permanently accessible when the N2 domain and the hinge are absent. For G3P and its variants, the apparent affinity for TolA-C-Aedans (Table 2) decreases when the strength of the interactions between N1 and N2 increases, as reflected in the Tm values of the N2 domain (Table 1). In the stabilized variant G3P IIHY, the domain interactions are particularly tight, and therefore, an interaction with TolA-C-Aedans could not be detected.
In the analysis of the binding data in Table 2, we assumed that the G3P variants exist in a domain-closed form (Gcl) and in a domain-open form (Gop) and that only the open form binds to TolA-C-Aedans (T), as described by the scheme in Reaction 1.
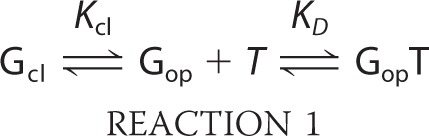 |
Kcl is the equilibrium constant between the closed (Gcl) and the open (Gop) form (Equation 1), and KD (= koff/kon) is the dissociation constant of the complex between the open form and TolA-C (Equation 2).
 |
 |
The measured overall equilibrium constant KD(app) is given by Equation 3.
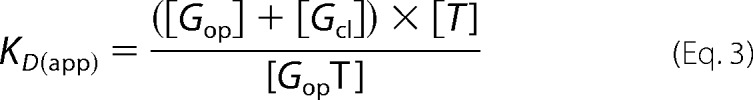 |
The combination of Equations 1–3 results in Equation 4, which relates the stability constants (Kcl) of the closed forms with the measured overall KD(app) values (Table 2) and the KD value of the open form.
 |
In the isolated N1 domain, the TolA binding site is fully accessible, and its KD (KD(N1) = 0.31 μm) served as the reference KD in the calculations based on Equation 4. A very similar KD value was observed for the isolated N1 domain with the stabilizing substitution T13I (Table 2).
The values calculated with Equation 4 for the stability constant Kcl are given in Table 2. Kcl is used for evaluating the interdomain interactions in the G3P variants. For fully folded wild-type G3P, Kcl is 70, indicating that it exists mostly in the closed form. For the destabilized Δβ6 and APA variants, it is smaller than 1, and their kon(app) values approach the kon value of the isolated N1 domain, confirming that the deletion of a β strand in the hinge or the substitutions around Pro-213 virtually abolished the domain interactions in G3P.
The formation of the native, fully folded form of G3P in the final step of its folding is limited in rate by the very slow trans → cis isomerization of Pro-213. Consequently, the state with trans-Pro-213 accumulates as a long-lived refolding intermediate in which the two domains are loosely associated. This form is considered to be a good representative of the physiologically active form of G3P (12). We produced this folding intermediate by a 10-min refolding pulse, measured its binding to TolA-C, and used Equation 4 to calculate the Kcl value from the binding kinetics. When Pro-213 is trans, the domain interactions are not abolished completely, but the Kcl value is 14-fold decreased from 70 to 5 (Table 2). The folding intermediate of the stabilized variant G3P IIHY was populated in the same way. Its stability constant (Kcl) is 71, which is in the same range as the Kcl value of fully folded wild-type G3P (Table 2). This suggests that in this stabilized variant, the interactions between the domains are improved so strongly that they persist most of the time even when Pro-213 is in trans.
Our model in Reaction 1 suggests that once a complex between a G3P variant and TolA-C has formed, the rate of dissociation (koff) should be the same for all G3P variants. In fact, all measured koff values fall into a narrow range between 0.16 and 0.51 s−1 (Table 2).
The measured rates of association, kon(app), are expected to decrease when the stability of the closed form, and thus Kcl, increases. Indeed, the measured kon(app) values vary about 400-fold, between 1.21 and 0.003 s−1 (Table 2). The microscopic association rate constants kon of the open forms of the G3P variants can be calculated by using the measured values kon(app) and the relation kon = kon(app) × (1 + Kcl). These calculated kon values agree well with the value derived for the isolated N1 domain. The reaction scheme in Reaction 1 therefore provides an adequate description for the coupling between the opening and closing reaction in the G3P variants and the binding of the open form to TolA-C. The reactivity of a particular G3P variant toward TolA-C is thus determined by the accessibility of its binding site.
Infectivities of the G3P Variants
To examine how the infectivity of phage fd is changed when the strength of the domain interactions in G3P is modulated, we generated phage variants that carried the same substitutions as the purified G3P proteins. First, cells with F pili (F+ cells) were infected (Fig. 6A). The F pilus is recognized by the N2 domain of G3P. Phage without N2 (fd ΔN2) cannot bind to the F pilus, and therefore, they are virtually noninfectious under the conditions of this assay. They served as a negative control. The initial contact of N2 with the pilus opens the domains of G3P to expose the binding site for the subsequent interaction with TolA (Fig. 1A). The wild-type phage showed the highest activity in these infection experiments (Fig. 6A). The phage variants with G3P IIHY and with G3P APA showed 60 and 25% of this activity, respectively, and the phage variant that lacks strand β6 in the hinge showed only 8% of the infectivity of the wild-type phage (Fig. 6A). In this variant, the domain interactions are virtually abolished, and the N1 domain is accessible for TolA-C during infection. However, the Δβ6 modification also eliminated the protection provided by the stable N1 domain for the marginally stable N2 domain in the fully folded form of G3P. This probably reduced the robustness of the phage. In fact, at 37 °C, the phage with G3P Δβ6 showed only 2% of the infectivity of the wild-type phage.
FIGURE 6.
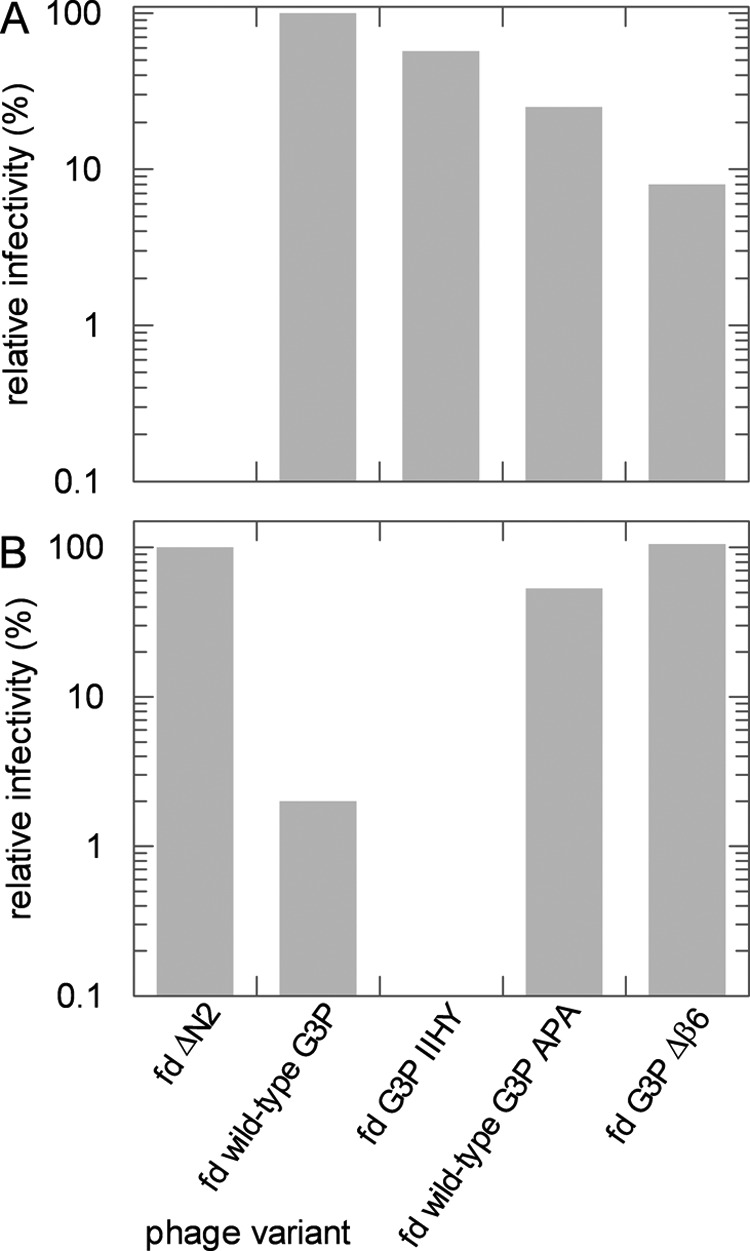
Infectivities of the phage variants. A, infection of F pilus bearing (F+) E. coli XL1 Blue cells by fd phages with different G3P variants. The infectivities are shown relative to the wild-type phage fd. B, infection of pilus-free (F−) E. coli HB2156 cells (2) by variants of phage fd. The infectivities are depicted relative to phage fd ΔN2. Infectivities were determined at 25 °C as described under “Experimental Procedures.” The precision of the infectivity values is about ±10%.
Cells without F pili (F− cells) can be infected by phage fd as well (20), but the efficiency of infection is at least 1000-fold reduced and requires the presence of 50 mm CaCl2. The initial targeting by pilus binding and pilus-triggered domain disassembly is no longer possible, and therefore, a phage can infect E. coli cells without F pili only when the TolA-C binding site of its G3P becomes accessible as a consequence of spontaneous domain disassembly. Infection assays with pilus-free cells are thus well suited to examine whether the strength of the domain interactions in its G3P and thus the accessibility of its N1 domain control the infectivity of a phage variant. The phage without a N2 domain (fd ΔN2) is used here as a positive reference because its N1 domain is permanently accessible for the interaction with TolA-C. In all other phage variants, the domains of G3P must disassemble spontaneously to expose the TolA binding site on the N1 domain. The infectivity of the wild-type phage is reduced to 2% in this assay, evidently because its G3P is mostly in the closed form. For the APA phage variant with the destabilizing alanine substitutions around Pro-213, the infectivity increased more than 20-fold to 53%, and for the phage variant with the β6 deletion (fd G3P Δβ6), the infectivity was as high as in the absence of N2 (Fig. 6B). The phage with the stabilized IIHY variant of G3P was virtually inactive in this infection assay, evidently because its domain interactions are too strong.
The infectivities of the phage variants thus correlate inversely with the strength of the domain interactions in the corresponding variants of purified G3P, as reflected in the Tm values of their first transition taken from Table 1 (Fig. 7A). Also, they correlate with the strength of the interaction between G3P and its receptor TolA, leading to an inverse relationship between the infectivity of the phage and the apparent dissociation constant of the complexes between the G3P* variants and TolA-C-Aedans (Fig. 7B). The phage infectivity thus depends on how easily the final, proline-limited step in the folding of its G3P can be reverted.
FIGURE 7.
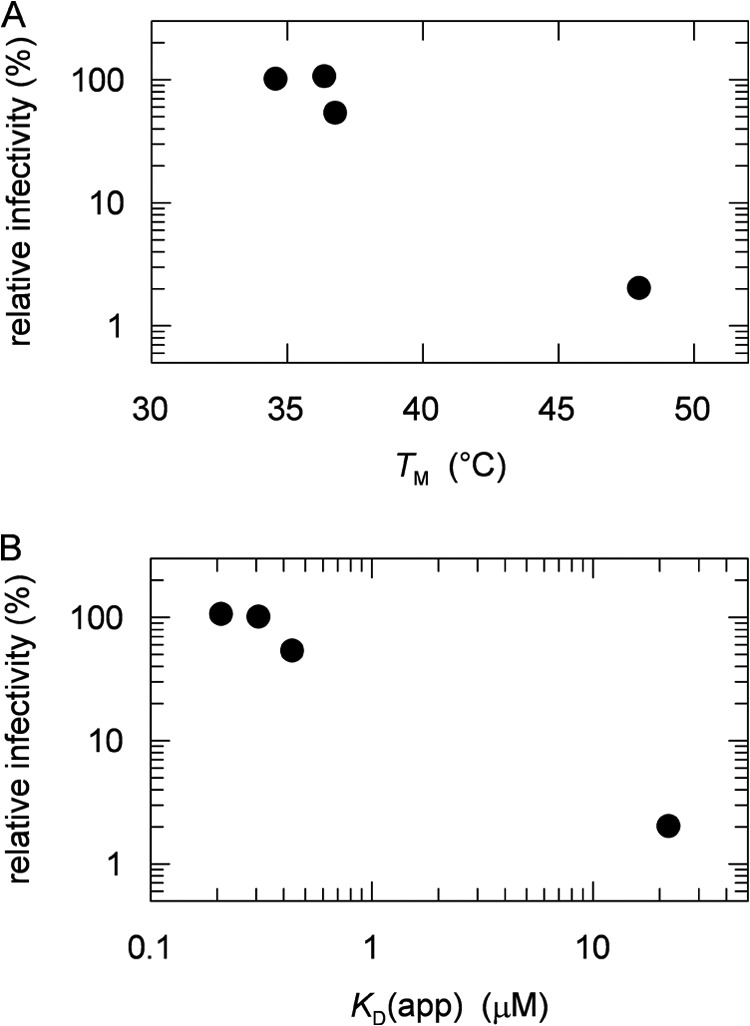
A and B, correlation between the relative infectivities of the phage variants fd ΔN2, fd G3P Δβ6, fd wild-type G3P APA, and fd wild-type G3P toward pilus-free (F−) E. coli HB2156 cells with the midpoints of the thermal transitions, Tm, of the N2 domains of the respective G3P* variants (taken from Table 1) (A) and the apparent dissociation constants, KD(app), of the complexes between the G3P* variants and TolA-C-Aedans (taken from Table 2) (B).
Structural Investigations of the Infectious Form of G3P by Two-dimensional NMR
After the backbone resonance assignments of G3P* (Fig. 2), we used heteronuclear two-dimensional NMR spectroscopy to elucidate the changes in the structure of the phage G3P during the transition from the fully folded inactive form to the partially unfolded active form. Three different strategies were employed to produce the active form.
In the first approach, we exploited the fact that the active form is populated transiently as a long-lived intermediate during the refolding of G3P from the GdmCl-denatured state. The molecules with a trans-Pro-213 and incompletely assembled domains exist for an extended time (>8 h) during refolding because the final rate-limiting trans → cis isomerization at Pro-213 shows a time constant of about 6200 s (at 25 °C) (7, 9). Therefore, kinetic real-time NMR monitoring (12) could be employed to identify residues of the partially folded active G3P that differ from the fully folded inactive form in the chemical shifts of their amide cross-peaks.
In the second approach, a refolding experiment was performed and monitored by real-time NMR spectroscopy as described above, although now in the presence of 30% excess of TolA-C. After the initiation of refolding, the open form of G3P is populated and binds to TolA-C, and thus, folding is arrested at the stage of the binding-competent form.
In the third approach, the fully folded form of G3P was incubated with a 12-fold excess of TolA-C. Here, TolA-C binding was used to transform G3P into its active form and to inhibit the reversion back to the inactive conformation. Under these conditions, about 90% of G3P molecules were converted to and remained in the active, TolA-C bound form.
In all three cases, two-dimensional 15N-TROSY-HSQC spectra were measured for the activated form and compared with a spectrum measured for fully folded inactive G3P under identical conditions (see Fig. 8, right side, for examples). The results are mapped onto the structure of folded G3P in a color-coded fashion in Fig. 8. Residues with native-like cross-peaks in the inactive, fully folded form and in the active, partially folded form are shown in blue; residues that lack native-like cross-peaks are shown in red. Residues colored in gray could not be analyzed. The chemical shift values for individual residues are listed in supplemental Table 1.
FIGURE 8.

Structural characteristics of the activated form of G3P with trans-Pro-213 determined by real-time NMR spectroscopy as described under “Experimental Procedures.” Residues with native-like cross-peaks in the 15N-TROSY-HSQC spectrum are depicted in blue. They identify amide NH in a native environment in the activated form of G3P. Residues that lack native-like cross-peaks are depicted in red. They identify amide NH that are not in a native-like environment. The side chain of Pro-213 is shown in stick representation. Gray residues could not be analyzed. A–C, the activated state of G3P was produced by a 30-min refolding pulse (A), by refolding in the presence of TolA-C (B), and by incubation of fully folded G3P with a 12-fold molar excess of TolA-C (C) as described under “Experimental Procedures.” Sections of reference, kinetic, and difference real-time NMR spectra are depicted on the right-hand side of panels A and B. The signals are labeled in red or blue as in the structure representation. Positive signals are shown in black, and negative signals are in red. The right side of panel C shows the superposition of the NMR spectra of G3P in the absence (black) and in the presence (red) of TolA-C.
The three approaches to populate and characterize the activated form of G3P led to similar results for residues of the N1 domain and the hinge region (Fig. 8, A–C). Most of them experience changes in their chemical shifts upon activation of G3P, presumably because the interdomain interactions in the fully folded inactive form involve mostly residues from these two structural elements. Regions of N1 that are not affected by activation are distant from the domain interface and locate to the upper rim of N1 (in the representations in Fig. 8).
The chemical shift changes in Fig. 8A reflect the activation of G3P in the absence of TolA-C, whereas those in Figs. 5C and 8B represent the activation coupled with the binding to TolA-C. The similarity of the results for N1 confirms that this domain uses similar surface areas for establishing the intramolecular contacts between N1 and N2 in the fully folded form of G3P (Fig. 8A) and between TolA-C and N1 after activation and domain opening (Fig. 8, B and C). The residues of the isolated N1 domain that mediate the interaction with TolA-C have been identified previously by NMR titration (21). The same residues experienced chemical shift changes during the final step of the NMR refolding experiments presented here (Fig. 8A). In summary, for N1 and the hinge region, only minor differences were observed between the active forms of G3P produced as a transient refolding intermediate or by the binding of TolA-C (Fig. 8).
Different results were obtained for the N2 domain. When the activated form of G3P is produced as a long-lived folding intermediate, the N2 domain shows native-like chemical shifts for virtually all residues (Fig. 8A), indicating that its structure is already native-like before domain assembly. This is a remarkable result, regarding the fact that the stability of N2 increases strongly during this final folding reaction (cf. Table 1). In the fully folded form of G3P, N2 presumably gains in folding Gibbs free energy because its chain ends are held together by the folded hinge. This favorable entropic contribution to stability is lost when the hinge unfolds. In fact, the chain regions that connect N2 with the hinge show chemical shift changes during the transition from the folding intermediate to the fully folded inactive form (Fig. 8A).
In the presence of TolA-C, many residues of N2 differ in chemical shift between the fully folded inactive form and the two TolA-bound active forms (Fig. 8, B and C). These residues are located in the area of N2 that faces N1 in the crystal structure of G3P (3, 5). The origin of these major differences in N2 is not known. Possibly, the N1 and N2 domains of G3P are loosely associated in the folding intermediate already. The stability of N2 is likely to be low in this form, but it is able to attain a native-like conformation already with native-like chemical shifts (Fig. 8A). Binding of TolA-C to the N1 domain disrupts this loose association, and thus, the native-like character of the corresponding surface of N2 is lost (Fig. 8, B and C).
Changes in Local Stability upon G3P Activation
The domain interactions between N1 and N2 in the partially folded active and in the fully folded inactive form of G3P were investigated at residue resolution using NMR-detected amide HX experiments (12). Representative exchange kinetics are shown in Fig. 3. Figs. 8B and 9A show the protection factors of the backbone amides of G3P in the fully folded state. They were derived at pH 7.0 by recording a series of two-dimensional 15N-TROSY-HSQC spectra for 120 h after dissolving protonated G3P in D2O. In the fully folded but inactive form of G3P, N1 and N2 show equally high protection for residues in ordered secondary structure elements. The two domains stabilize each other by their tight interactions and thus form a single cooperative unit. In the hinge region, most of the assigned NH showed protection factors between 104 and 105. This confirms that in the inactive form of G3P, the hinge region is stable and folded. Its maximal protection factors are only about 10-fold lower than those of N1 and N2 (Fig. 9A). In the stabilized form (G3P IIHY) (12), residues in the hinge region showed about 50-fold higher protection factors than in the wild-type form of G3P, indicating that the domain interactions are strengthened in this variant. The decreased affinity for TolA-C (Table 2) and the decreased infectivity of the phage with the IIHY variant of G3P (Fig. 6) are presumably a direct consequence of this increased local stability.
FIGURE 9.

Amide hydrogen exchange protection factors of G3P in the fully folded inactive form (A and B), in the activated form with a trans-Pro-213 (C and D), and in the complex of G3P with TolA-C (E and F). In A, C, and E, the protection factors are plotted as a function of the residue number. Red bars represent residues from the N1 domain, blue bars represent residues from the N2 domain, and green bars represent residues from the hinge region. In B, D, and F, the values of the protection factors are mounted on the backbone structure of the fully folded form in a color-coded fashion. Residues with protection factors larger than 10,000 are shown in dark green, values between 10,000 and 100 are in green, and values smaller than 100 are in pale green. Backbone amides that could be analyzed are marked by a yellow background in A, C, and E and correspond to the green shaded residues in B, D, and F. Residues that could not be analyzed or for which amide HX was too rapid have a white background in A, C, and E and are shown in gray in B, D, and F. Bars marked in A by an asterisk indicate amides that did not exchange to a significant extent during the 120 h of the NMR experiment. The protection factor of 5 × 105 thus represents a lower limit.
The NH protection factors of the active form of G3P were determined in an NMR-detected competition experiment between refolding and amide proton exchange. The active form is generated transiently as a long-lived folding intermediate in this experiment (12), and the analysis is restricted to NH that do not exchange measurably from the fully folded state during 8 h in a reference experiment. In the folding intermediate, the protection factors were strongly reduced, in particular for the N2 domain. Its maximal protection factors were more than 10-fold lower than those of the N1 domain, presumably because the two domains are uncoupled when the hinge region is not yet properly folded (Fig. 9, C and D). Accordingly, protected amides could not be detected in the hinge region.
In the active form of G3P that was produced by binding of TolA-C, uncoupling of N1 and N2 was even more pronounced (Fig. 9, E and F). Most amide protons of N2 were protected less than 100-fold in this form, and only a few regions showed protection factors between 103 and 104. Again, no protection could be observed for NH in the hinge region, mostly because the NH resonances changed upon TolA-C binding and domain opening. In the N1 domain, binding of TolA-C led to changes in the chemical shifts of about half of the amide protons (Fig. 8, B and C), and therefore, they could not be analyzed. Several of the assigned NH signals showed an ∼30-fold increase in protection against exchange, which reflects the stabilization of N1 by the binding to TolA-C. TolA-C binding thus disassembles the two domains of G3P. This interaction stabilizes the N1 domain; N2, however, becomes destabilized because the domain interactions are lost.
DISCUSSION
The function of the phage G3P during infection is closely related to its folding and to the isomeric state of Pro-213. The trans → cis reaction at Pro-213 in the final folding step is coupled with structure formation in the hinge region and the assembly of the domains N1 and N2. The fully folded form of G3P is inactive because the binding site for the phage receptor TolA-C on N1 is inaccessible. Consequently, the domain must be disassembled, and Pro-213 isomerization must be reversed to activate G3P for infection.
The thermodynamic coupling between the cis/trans reaction at Pro-213, the folding of the hinge, and the binding of N1 to the receptor TolA forms the structural and energetic basis of the infection mechanism. cis-Pro-213 stabilizes the folded hinge, and the folded hinge stabilizes the cis form of Pro-213. The coupling energy between the two processes is sufficient to shift the cis/trans equilibrium at Pro-213 more than 100-fold from <0.1 in the unfolded state to >10 in the folded state of G3P. This agrees well with the observed more than 100-fold increase in amide protection in the hinge region upon trans → cis isomerization at Pro-213.
In the wild-type form of G3P, the conformational stability of the hinge is optimally balanced. The coupling energy between hinge folding and Pro-213 isomerization is high enough to maintain the phage G3P in a stably locked form with a cis-Pro-213 in the absence of a target cell. On the other hand, it is so low that the interaction of the N2 domain with the F pilus in the initial infection step is sufficient to destabilize the hinge, which in turn exposes the binding site of G3P for TolA and allows Pro-213 to isomerize from the strained cis to the more favorable trans state. It is unknown how the pilus binding signal is communicated through the N2 domain and converted into an unfolding signal for the hinge. Possibly, the intrinsically low conformational stability of the N2 domain facilitates this signal transduction.
The amide NH exchange experiments revealed how the local stability of G3P changes during the transition between the inactive form (with cis-Pro-213) and the active form (with trans-Pro-213). In the inactive cis form, the hinge region is stably folded and forms a cooperative folding unit with the domains N1 and N2. In the active trans form, the protection factors of the hinge residues drop to values below the limit of detection, indicating that activation of phage fd for infection involves a locally well defined unfolding reaction in the hinge of its G3P, coupled with Pro-213 cis → trans isomerization. Apparently, the two-stranded β-sheet of the hinge gains conformational stability from interactions with the N1 domain that can only be established when Pro-213 is cis.
In the IIHY variant of G3P, the local stability of the hinge is increased (12). Several of its amide NH remain protected from exchange with the solvent even when Pro-213 is trans, and the domains N1 and N2 remain partially coupled in their amide proton exchange pattern in this state. This explains at the molecular level the reduced infectivity of phage with G3P IIHY.
Previously, we viewed Pro-213 and its cis → trans isomerization as a molecular timer of phage infection (7). Based on the present results, we suggest that the function of Pro-213 might be better described as a molecular capacitor for the transient storage of energy. In the first step of infection, energy that originates from the binding of N2 to the pilus tip is used to unfold the hinge and thus to expose the binding site for the ultimate receptor TolA. This energy remains available even when the phage leaves the pilus tip because it is stored in the trans form of Pro-213, which prevents refolding of the hinge and thus the reassembly of the domains of G3P. Species with an alternative prolyl isomer are ideally suited for such a transient energy storage function because the cis/trans interconversion is an inherently slow reaction.
It has long been known that prolyl isomerizations play important roles as rate-determining steps in protein folding, but more recently, evidence has accumulated that prolyl isomerizations in folded proteins are involved in a multitude of regulatory processes (22–27). Also, it was proposed that the time course of proline-controlled signaling events is modulated by prolyl isomerases such as cyclophilin or Pin1 (28, 29). The results obtained for the phage gene-3-protein provide a first glimpse on how a prolyl isomerization signal can be propagated to a spatially remote site and, in particular, on how the signal can be stored for a specified time before it is switched off. In the absence of a prolyl isomerase, this time is set by the intrinsic rate of reisomerization, which is determined by the local sequence around the proline (30).
Acknowledgments
We thank the members of our groups for many fruitful discussions and Hartmut Oschkinat, Forschungsinstitut für Molekulare Pharmakologie Berlin, for NMR time at the 900-MHz instrument. Significant investments into the NMR infrastructure from the European Regional Development Fund by the European Union are also gratefully acknowledged.
This work was supported by the Deutsche Forschungsgemeinschaft (SCHM 444/17-3 and BA 1821/5-2) and the German Bundesministerium fuer Forschung und Entwicklung (ProNet-T3).
This article was selected as a Paper of the Week.

This article contains supplemental Table 1.
The following designations are used throughout this study: N1, N2, and CT, the two N-terminal and the C-terminal domains of the gene-3-protein, respectively; G3P, a fragment of the gene-3-protein that consists of domains N1 and N2; G3P APA, G3P variant with the substitutions Q212A and P214A; G3P AGA, G3P variant with the substitutions Q212A, P213G, and P214A; G3P IIHY, G3P variant with the four stabilizing mutations T13I, T101I, Q129H, and D209Y; G3P Δβ6, G3P variant, in which strand 6 (residues 96–102) is deleted; F+ and F−, strain of E. coli with and without F pili, respectively; TolA-C, C-terminal domain of the TolA protein; TolA-C-Aedans, variant of TolA-C, in which a C-terminal Cys was modified by reaction with iodo-Aedans.
- Aedans
- 5-(((acetylamino)ethyl)amino) naphthalene-1-sulfonate
- HX
- hydrogen/deuterium exchange
- GdmCl
- guanidinium chloride
- TROSY
- transverse relaxation optimized spectroscopy
- HSQC
- heteronuclear single quantum correlation.
REFERENCES
- 1. Click E. M., Webster R. E. (1997) Filamentous phage infection: required interactions with the TolA protein. J. Bacteriol. 179, 6464–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Riechmann L., Holliger P. (1997) The C-terminal domain of TolA is the coreceptor for filamentous phage infection of E. coli. Cell 90, 351–360 [DOI] [PubMed] [Google Scholar]
- 3. Lubkowski J., Hennecke F., Plückthun A., Wlodawer A. (1999) Filamentous phage infection: crystal structure of g3p in complex with its coreceptor, the C-terminal domain of TolA. Structure 7, 711–722 [DOI] [PubMed] [Google Scholar]
- 4. Rakonjac J., Bennett N. J., Spagnuolo J., Gagic D., Russel M. (2011) Filamentous bacteriophage: biology, phage display, and nanotechnology applications. Curr. Issues Mol. Biol. 13, 51–76 [PubMed] [Google Scholar]
- 5. Holliger P., Riechmann L., Williams R. L. (1999) Crystal structure of the two N-terminal domains of g3p from filamentous phage fd at 1.9 Ä: evidence for conformational lability. J. Mol. Biol. 288, 649–657 [DOI] [PubMed] [Google Scholar]
- 6. Lubkowski J., Hennecke F., Plückthun A., Wlodawer A. (1998) The structural basis of phage display elucidated by the crystal structure of the N-terminal domains of G3P. Nat. Struct. Biol. 5, 140–147 [DOI] [PubMed] [Google Scholar]
- 7. Eckert B., Martin A., Balbach J., Schmid F. X. (2005) Prolyl isomerization as a molecular timer in phage infection. Nat. Struct. Mol. Biol. 12, 619–623 [DOI] [PubMed] [Google Scholar]
- 8. Eckert B., Schmid F. X. (2007) A Conformational unfolding reaction activates phage fd for the infection of Escherichia coli. J. Mol. Biol. 373, 452–461 [DOI] [PubMed] [Google Scholar]
- 9. Martin A., Schmid F. X. (2003) The folding mechanism of a two-domain protein: folding kinetics and domain docking of the gene-3-protein of phage fd. J. Mol. Biol. 329, 599–610 [DOI] [PubMed] [Google Scholar]
- 10. Martin A., Schmid F. X. (2003) A proline switch controls folding and domain interactions in the gene-3-protein of the filamentous phage fd. J. Mol. Biol. 331, 1131–1140 [DOI] [PubMed] [Google Scholar]
- 11. Martin A., Schmid F. X. (2003) Evolutionary stabilization of the gene-3-protein of phage fd reveals the principles that govern the thermodynamic stability of two-domain proteins. J. Mol. Biol. 328, 863–875 [DOI] [PubMed] [Google Scholar]
- 12. Weininger U., Jakob R. P., Eckert B., Schweimer K., Schmid F. X., Balbach J. (2009) A remote prolyl isomerization controls domain assembly via a hydrogen bonding network. Proc. Natl. Acad. Sci. U.S.A. 106, 12335–12340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jakob R. P., Schmid F. X. (2008) Energetic coupling between native-state prolyl isomerization and conformational protein folding. J. Mol. Biol. 377, 1560–1575 [DOI] [PubMed] [Google Scholar]
- 14. Mayr L. M., Schmid F. X. (1993) Stabilization of a protein by guanidinium chloride. Biochemistry 32, 7994–7998 [DOI] [PubMed] [Google Scholar]
- 15. Krebber C., Spada S., Desplancq D., Krebber A., Ge L., Plückthun A. (1997) Selectively-infective phage (SIP): a mechanistic dissection of a novel in vivo selection for protein-ligand interactions. J. Mol. Biol. 268, 607–618 [DOI] [PubMed] [Google Scholar]
- 16. Sieber V., Plückthun A., Schmid F. X. (1998) Selecting proteins with improved stability by a phage-based method. Nat. Biotechnol. 16, 955–960 [DOI] [PubMed] [Google Scholar]
- 17. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 18. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 19. Lorenz S. H., Jakob R. P., Weininger U., Balbach J., Dobbek H., Schmid F. X. (2011) The filamentous phages fd and IF1 use different mechanisms to infect Escherichia coli. J. Mol. Biol. 405, 989–1003 [DOI] [PubMed] [Google Scholar]
- 20. Russel M., Whirlow H., Sun T. P., Webster R. E. (1988) Low-frequency infection of F− bacteria by transducing particles of filamentous bacteriophages. J. Bacteriol. 170, 5312–5316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holliger P., Riechmann L. (1997) A conserved infection pathway for filamentous bacteriophages is suggested by the structure of the membrane penetration domain of the minor coat protein g3p from phage fd. Structure 5, 265–275 [DOI] [PubMed] [Google Scholar]
- 22. Pletneva E. V., Sundd M., Fulton D. B., Andreotti A. H. (2006) Molecular details of Itk activation by prolyl isomerization and phospholigand binding: the NMR structure of the Itk SH2 domain bound to a phosphopeptide. J. Mol. Biol. 357, 550–561 [DOI] [PubMed] [Google Scholar]
- 23. Schwartz T. U., Schmidt D., Brohawn S. G., Blobel G. (2006) Homodimerization of the G protein SRβ in the nucleotide-free state involves proline cis/trans isomerization in the switch II region. Proc. Natl. Acad. Sci. U.S.A. 103, 6823–6828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sarkar P., Reichman C., Saleh T., Birge R. B., Kalodimos C. G. (2007) Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol. Cell 25, 413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou X. Z., Kops O., Werner A., Lu P. J., Shen M., Stoller G., Küllertz G., Stark M., Fischer G., Lu K. P. (2000) Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell 6, 873–883 [DOI] [PubMed] [Google Scholar]
- 26. Fischer G., Aumüller T. (2003) Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev. Physiol. Biochem. Pharmacol. 148, 105–150 [DOI] [PubMed] [Google Scholar]
- 27. Thiele A., Krentzlin K., Erdmann F., Rauh D., Hause G., Zerweck J., Kilka S., Pösel S., Fischer G., Schutkowski M., Weiwad M. (2011) Parvulin 17 promotes microtubule assembly by its peptidyl-prolyl cis/trans isomerase activity. J. Mol. Biol. 411, 896–909 [DOI] [PubMed] [Google Scholar]
- 28. Andreotti A. H. (2003) Native state proline isomerization: An intrinsic molecular switch. Biochemistry 42, 9515–9524 [DOI] [PubMed] [Google Scholar]
- 29. Lu K. P., Finn G., Lee T. H., Nicholson L. K. (2007) Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3, 619–629 [DOI] [PubMed] [Google Scholar]
- 30. Reimer U., Scherer G., Drewello M., Kruber S., Schutkowski M., Fischer G. (1998) Side-chain effects on peptidyl-prolyl cis/trans isomerisation. J. Mol. Biol. 279, 449–460 [DOI] [PubMed] [Google Scholar]
- 31. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]