

Background: Kinetic discrimination is essential in antibody·antigen recognition.
Results: An otherwise fast second-order reaction is limited by a slow proline isomerization of the epitope.
Conclusion: The antibody recognizes the less populated isoform, suggesting presentation of the epitope as a non-native conformer.
Significance: The conformational diversity of an intrinsically disordered viral epitope slows down the reaction with the antibody without specificity cost.
Keywords: Antigen, Biophysics, Intrinsically Disordered Proteins, Pre-steady-state Kinetics, Protein Folding, E7 Oncoprotein, Antigen Recognition, Conformational Selection, Prolyl Isomerization
Abstract
Conformational rearrangements in antibody·antigen recognition are essential events where kinetic discrimination of isomers expands the universe of combinations. We investigated the interaction mechanism of a monoclonal antibody, M1, raised against E7 from human papillomavirus, a prototypic viral oncoprotein and a model intrinsically disordered protein. The mapped 12-amino acid immunodominant epitope lies within a “hinge” region between the N-terminal intrinsically disordered and the C-terminal globular domains. Kinetic experiments show that despite being within an intrinsically disordered region, the hinge E7 epitope has at least two populations separated by a high energy barrier. Nuclear magnetic resonance traced the origin of this barrier to a very slow (t½ ∼4 min) trans-cis prolyl isomerization event involving changes in secondary structure. The less populated (10%) cis isomer is the binding-competent species, thus requiring the 90% of molecules in the trans configuration to isomerize before binding. The association rate for the cis isomer approaches 6 × 107 m−1 s−1, a ceiling for antigen-antibody interactions. Mutagenesis experiments showed that Pro-41 in E7Ep was required for both binding and isomerization. After a slow postbinding unimolecular rearrangement, a consolidated complex with KD = 1.2 × 10−7 m is reached. Our results suggest that presentation of this viral epitope by the antigen-presenting cells would have to be “locked” in the cis conformation, in opposition to the most populated trans isomer, in order to select the specific antibody clone that goes through affinity and kinetic maturation.
Introduction
Specific recognition of foreign antigens by antibodies is the primary event in the immune system's fight against disease. Antibodies are at the center of humoral, cellular, and innate responses. The first molecular understanding of antibody·antigen recognition came from the x-ray structure of antibody molecules (1). To date, there are nearly 800 antigen-antibody structures (2), a large body of information from bioinformatics analyses has been accumulated (3), and a number of general rules were extracted together with some thermodynamic information on binding interfaces (4). It was recognized early that a large number of antigen-antibody complexes underwent substantial conformational changes upon formation (5). Notwithstanding the fact that these structures provide exquisite detail for those changes, the comparison of structures of liganded and unliganded antibodies and epitopes cannot discriminate changes that occur after interaction between the molecules (induced fit) from those resulting of a preexisting equilibrium (conformational selection). The discrimination between these two scenarios from either the antibody or the antigen side is of utmost importance in immunological terms and requires detailed kinetic mechanistic investigation.
Conformational heterogeneity in antibodies has long been known. The idea that antibodies produced by a given B-cell from a single sequence could present more than one fold or conformer was first proposed by Pauling in 1940 (6) and later demonstrated by Foote and Milstein in 1994 (7). Different conformations adopted by a single antibody sequence may react with different antigens, which is the basis for cross-reactivity, a fundamental phenomenon behind autoimmunity and allergy (8, 9). In addition, antibody conformational isomerism has direct implications for antibody diversity and the expansion of the immune repertoire. James et al. (8, 10) later carried out a comprehensive and integrative analysis of another antibody-hapten interaction using the Fv heterodimer SPE7 IgE model. Crystallographic and pre-steady-state kinetic analyses showed that the antibody adopted at least two different conformations (8, 10). Antigen recognition starts by selection of one of these conformers, and this first complex subsequently undergoes an induced fit conformational transition (8, 10).
Multiple conformations and conformational selection events on the antigen side have also been reported. In a leucine zipper model, the antibody selected a pre-existing monomeric conformer and shifted the equilibrium from a native coiled-coil to a monomeric, unfolded species (11, 12). A somehow different example comes from an epitope within the folded dimeric transcriptional regulator E2 from human papillomavirus (HPV).5 In this case, the rate constant for antibody association was rather slow (104 m−1 s−1), indicative of a conformational rearrangement of the antigen as the rate-limiting step (13).
Peptide epitopes are mostly unstructured but show conformational tendencies in solution, and a correlation between these structural tendencies and antigenicity was observed (14). In the absence of stable structure, Xaa-Pro peptide bonds can populate both the cis and trans isomers to measurable degrees. Cis-trans isomerization thus provides a molecular switch that works in the minute time scale and can expand the conformational repertoire of proteins. Recent evidence shows that proline isomerization in both disordered and globular domains can regulate the kinetics of processes such as phage infection (15), antibody folding (16), estrogen receptor signaling (17), self-inhibition of a signaling protein (18), and aggregation dynamics of the intrinsically disordered Tau protein (19). It has been proposed that some antibodies do recognize the less populated cis isomer of the peptide bond (20, 21). However, in many of these cases, the evidence is indirect, and in none of them was the kinetic mechanism of recognition addressed.
The HPV E7 oncoprotein is the main transforming factor of this pathogen, involved in many types of neoplasias, cervical cancer being the most significant (see Ref. 22 for a review). E7 is also a tumor virus-transforming protein and a model protein targeting the retinoblastoma tumor suppressor and cell cycle regulator (Rb). By targeting Rb for proteasomal degradation, E7 causes the release of the transcription factor E2F and forces the cells into S phase in order to use the cell machinery for DNA replication and further transcription of other early and late genes required for the completion of the virus life cycle (23). From the immunological point of view and as the major transforming protein, E7 is constitutively expressed at high levels in carcinoma tissues, and antibodies are found in sera at higher frequency in patients with cervical cancer (24). Thus, E7 and derivatives are being exploited both in diagnostics and therapeutic vaccination (25, 26).
E7 also emerges as a paradigmatic example of an intrinsically disordered protein (IDP) (27). IDPs are fully functional, most often “promiscuous binders” related to cell signaling pathways (28, 29), characterized by extended conformations without canonical secondary structure and by the lack of tertiary structure. IDPs are enriched in particular among viral proteins, which are often multifunctional, because viruses are required to have a minimal amount of genes (30). HPV16 E7 was shown to be an extended, non-globular protein (31), characterized by an intrinsically disordered nature that maps to its N-terminal domain (E7N) (32) and by a globular C-terminal domain (Fig. 1, A and C). E7N contains several linear motifs that mediate binding to different ligands (33). The E7 protein can also form oligomers with chaperone activity that are present in HPV-transformed cells and cancerous tissues (34, 35).
FIGURE 1.

Mapping of the M1 recognition site. A, amino acid sequence of the HPV16 E7 protein. The location of the E7 N-terminal (E7N) and the E7 C-terminal (E7C) domains (solid line), the linker region (dashed line), and the conserved regions CR1 and CR2 are shown above the sequence. Conserved proline residues in the linker region are shown in boldface type. The E7 fragments used in this study are shown below the sequence, with the amino acids spanned by each fragment shown in parenthesis. B, M1 IgG reactivity against different HPV16 E7 fragments assayed by indirect ELISA. E7, full-length E7 protein (amino acids 1–98). Peptides from different regions of the protein are indicated. Control, non-related E2 peptide. Black bars, M1-purified IgG. White bars, non-related IgG. C, schematic representation of the HPV E7 dimer (Protein Data Bank entry 2F8B) and its E7N and E7C domains. Monomers are depicted in violet, and the M1 epitope (E7Ep) is shown in red. Orange spheres, zinc atoms.
In this work, we present a detailed mechanistic study of the interaction between an immunodominant peptide epitope of HPV-16 E7 (36, 37) and the monoclonal antibody M1, raised by immunization with the full-length E7 protein (34). The reaction was dissected into a very slow pre-equilibrium isomerization followed by a fast association reaction and, finally, by a unimolecular rearrangement. Kinetic association experiments and nuclear magnetic resonance measurements of the free epitope showed that the antibody specifically recognizes a cis proline isomer in the interdomain “hinge” region of E7. Thus, by making use of an important viral disease and cancer-related antigen, we reveal that binding of an epitope within an intrinsically disordered protein may depend on a slow proline isomerization process.
EXPERIMENTAL PROCEDURES
Proteins and Peptides
M1 IgG was purified from ascitic fluids, and the derived Fab fragments were prepared following standard procedures with minor modifications. Briefly, after clarification of ascitic fluids, the sample was exchanged into formate buffer and loaded onto a high performance cationic exchange ceramic S-HyperD column (Pall Corp.). The desired fractions were pooled, exchanged into TBS buffer, and concentrated. Fab fragments were prepared by papain hydrolysis of the IgG molecules and purified by ionic exchange in a Q-HyperD column (Pall Corp.). M1 Fab purity was >95% as judged by SDS-PAGE. Recombinant HPV16 and HPV18 E7 proteins were expressed and purified as described previously (31, 38).
Synthetic peptides corresponding to the N terminus of HPV16 E7 and proline mutants were obtained from the Keck facility (Yale University, New Haven, CT), purified by reverse phase HPLC, and submitted to mass spectrometry. The peptides were quantified by tyrosine absorption at 273 nm in 0.1 m NaOH and UV absorption at 220 nm in HCl for the peptides without tyrosine. The different E7 synthetic peptides used in this study are shown in Fig. 1A.
Fluorescein Isothiocyanate (FITC) Labeling
HPV16 E7 and synthetic peptides were labeled with FITC (Sigma catalogue no. F-3651). The coupling reaction was conducted as described previously (39).
ELISAs
The epitope mapping was measured by a conventional ELISA. Briefly, 96-well plates (Greiner bio-one, high binding) were sensitized overnight at 4 °C with 0.5 μg/well of E7 and E7 C-terminal domain (E7(40–98)) proteins or 1 μg/well of the different E7 fragments in Tris-buffered saline (25 mm Tris-HCl and 600 mm NaCl, pH 7.4). The ionic force of the buffer was set to 0.6 m NaCl. After blocking with 1% bovine serum albumin (BSA) (BSA/TBS), ascitic fluids and control serum samples were diluted in BSA/TBS, and their reactivity was revealed by incubation with peroxidase-conjugated polyclonal antibodies to murine IgG (Jackson). The final color was read at 492 nm in an ELISA reader (SLT Spectra).
Equilibrium Binding
Measurements were performed in an Aminco-Bowman Series 2 spectrofluorimeter assembled in L geometry. A fixed amount of FITC-E7 protein was incubated with increasing concentrations of M1 in different tubes for 30 min prior to fluorescence measurements. A similar protocol was used to titrate FITC-E7(36–48), FITC-E7(36–53), and FITC-E7(36–48) proline mutant peptides with M1Fab. The dissociation constant (KD) of the complex was calculated by fitting the plot of observed fluorescence anisotropy (r) change of FITC-E7 or FITC-E7 peptides versus different M1 concentrations to the following equation assuming a 1:1 stoichiometry (40),
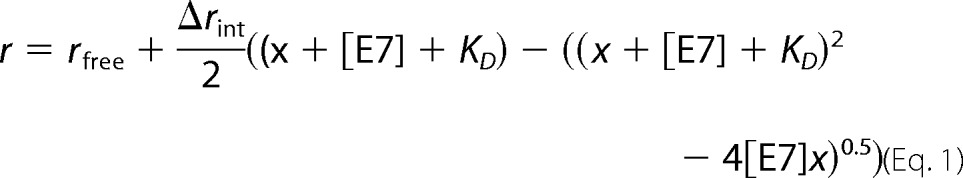 |
where x represents the variable total concentration of M1Fab, Δrint is the difference in intrinsic fluorescence anisotropy between the free and complexed fragments, and rfree is the fluorescence anisotropy of the free fragment.
Association and Dissociation Kinetics
Fluorescence stopped-flow kinetic experiments were performed using an SX.18MV stopped-flow apparatus (Applied Photophysics, Leatherhead, UK). All reported concentrations are those resulting from mixing equal volumes of each syringe. The association and dissociation reactions were monitored by following the change in fluorescence intensity or fluorescence anisotropy of the FITC-E7Ep peptide. For fluorescence measurements, the excitation monochromator was set to 495 nm with a 4.6-nm band pass, and emission was collected through a 515-nm cut-off filter (Schott, Duryea, PA). For anisotropy measurements, two photomultiplier tube detectors were assembled in T-geometry, and vertically and horizontally polarized emissions were collected through 530-nm cut-off filters.
The bimolecular association reaction was measured under second-order conditions by mixing equimolar amounts of M1Fab and FITC-E7Ep. Under these conditions, the data can be analyzed by the following second-order equation (41),
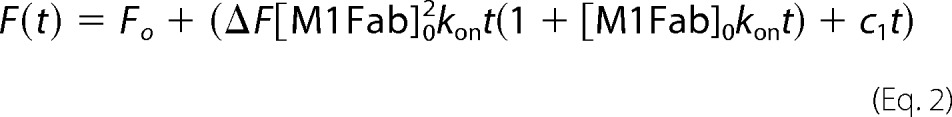 |
where F represents the fluorescence at time t, ΔF is the change in fluorescence upon binding, Fo is the fluorescence at t = 0, [M1Fab]o is the initial concentration of M1Fab, and kon is the second-order rate constant. Signal drift was taken into account by a linear time-dependent term (c1).
Dissociation traces were measured by displacing a stoichiometric FITC-E7Ep·M1Fab complex by adding a 20-fold excess of unlabeled E7Ep peptide. The traces were fit to the sum of two exponential functions to obtain the koff values,
where A1 and A2 are the amplitudes, Fo is the initial fluorescence value, and k1 and k2 are the dissociation rate constants for each phase.
Concentration Dependence
Slow association phases were measured by manual mixing using a fluorimeter. The change in anisotropy was measured after the addition of increasing concentrations of M1Fab to a cuvette containing 50 nm FITC-E7Ep or 50 nm FITC-P47A E7Ep. The individual data traces were fit to a single exponential function to obtain the observed rate constant for the reaction (supplemental Tables S1 and S6). The dependence of the observed rate constant on M1Fab concentration was fit to a previously described pre-equilibrium model (10),
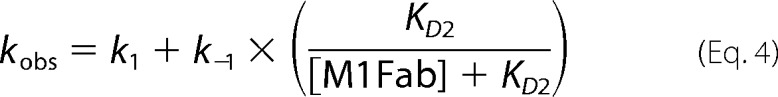 |
where kobs is the observed association rate constant at each M1Fab concentration, k1 and k−1 are the forward and reverse rate constants for the pre-equilibrium reaction, and KD2 is the dissociation equilibrium constant for the bimolecular association reaction. The concentrations used yielded errors in free M1Fab concentration of less than 20 and 5% for the lowest and highest M1Fab concentrations tested, respectively (100 nm and 1 μm), allowing a reliable estimation of the fitting parameters. Using these parameters, we obtained the equilibrium association constants for the pre-equilibrium and association reactions: K1 = k1/k−1 and K2 = 1/KD2, respectively. From this, it is possible to calculate the global equilibrium dissociation constant KD(global) = (1 + K1)/(K1 × K2). Finally, using the equilibrium dissociation constant for the bimolecular association reaction (KD2) and the association rate constant obtained in the second order stopped-flow experiments (k2), one can calculate the dissociation rate constant for the bimolecular association reaction as k−2 = KD2 × k2.
We considered the existence of a second unimolecular reaction with an equilibrium association constant K3, corresponding to rearrangement of the complex. The value of K3 can be estimated from the equilibrium dissociation constant KD, the equilibrium constant for the preassociation equilibrium K1, and the dissociation constant for the bimolecular association reaction K2 and the following relationship (42).
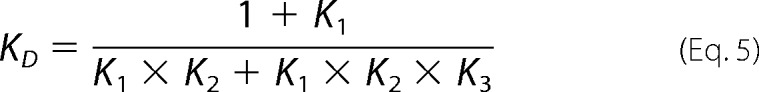 |
Temperature Dependence
The slow association phase was measured at different temperatures in the same conditions described for the concentration dependence experiments. The change in anisotropy was measured after the addition of 600 nm M1Fab to a cuvette containing 100 nm FITC-E7Ep. The individual traces were fit to a single exponential function to obtain the observed rate constant for the reaction (supplemental Table S2). Activation parameters were determined from the Eyring plot of ln(k (h/k*T)) versus 1/T, where h is the Planck constant, k* is the Boltzman constant, T is expressed in Kelvin, and k corresponds to the observed rate constants determined at the different temperatures. The enthalpy of activation was obtained from the slope −ΔH‡/R of this plot, where R is the universal gas constant.
Circular Dichroism
Far-UV CD measurements were conducted on a Jasco J-810 spectropolarimeter using a Peltier temperature-controlled sample holder and a 0.1-cm path length cell. All measurements were carried out in 10 mm Tris-HCl (pH 7.4) and 5 mm NaCl at 20 ± 0.1 °C. For the association kinetic experiment, the ellipticity change at 203 nm was measured after mixing equal amounts of E7Ep and M1Fab (5 μm). Traces were fit to single exponential functions to obtain the kobs values.
Data Analysis and Fitting
Fitting of the data was carried out using the software programs Profit (Quantumsoft) and Kaleidagraph version 4.0 (Synergy Software) to obtain parameters and their S.D. values.
NMR Spectroscopy
NMR experiments were carried out at 600.13 MHz on a Bruker Avance 600-MHz spectrometer equipped with an UltraShield Plus magnet and a triple resonance cryoprobe with gradient unit. The NMR data were processed on Linux workstations with NMRView/NMRPipe software (43, 44). The NMR samples contained 3 mm E7Ep peptide dissolved in water, pH 7.5 (10 or 100% D2O (v/v)). Proton and carbon chemical shift assignments were achieved by analyzing the following experiments at natural abundance: 1H-13C HMQC, 1H-13C HMQC-TOCSY (70-ms mixing time), 1H-1H TOCSY (70-ms mixing time), and 1H-1H NOESY (250-ms mixing time). A series of NOESY experiments of the E7Ep peptide with variable mixing times (0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, and 1.2 s) were performed at 30 °C to obtain the kinetic rates of the proline cis-trans isomerization. At 40 °C, 0.15-, 0.3-, 0.5-, 0.7-, and 0.9-s mixing times were used. The spectra were acquired with 8K*800 complex data points in the t1 × t2 dimensions and with 24 scans. A 1H-13C HMQC spectrum was performed at each temperature to follow the chemical shift changes with temperature. In the NOESY spectrum, the intensity of the diagonal peaks and of the chemical exchange cross-peaks are related to the mixing time, τm, by the following equations,
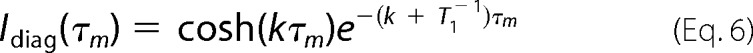 |
where cosh and sinh are the hyperbolic cosine and sine, respectively, k is the kinetic rate for the chemical exchange process, and T1 is the spin-lattice relaxation time of the relevant protons. Assuming that the T1 relaxation rates are the same for the respective protons in the cis and trans isomerizations, we obtain the following.
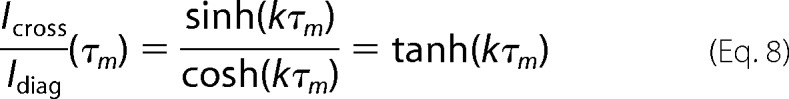 |
For short mixing times, Equation 8 simplifies to the following.
 |
Equation 9 was used for the analysis of the kinetics of the cis-trans proline interconversion. Exchange rate constants were obtained by fitting the diagonal and cross-peaks intensities of δ protons of prolines. The thermodynamic equilibrium cis-trans isomerization constants were also obtained from the mole fractions xcis and xtrans, which were calculated by integrating the volumes of the cross-peaks corresponding to cis and trans proline isomers at each temperature in the a 1H-13C HMQC spectrum.
RESULTS
M1 Monoclonal Antibody Epitope Mapping and Affinity Determination
Papillomavirus E7 proteins show a high degree of sequence conservation across over 200 different virus types (45). The two most clinically relevant types infecting humans are HPV16 and HPV18, which account for over 70% of cervical cancers. We have previously reported the generation of a monoclonal antibody, M1, raised against HPV16 E7 (34). IgG M1 presents a very high reactivity toward HPV16 E7 (1:120,000 titer) and a low reactivity toward HPV18 E7 (1:250 titer), which yields a 500-fold discrimination factor (supplemental Fig. S1).
In order to map the epitope within E7, we analyzed IgG M1 recognition of different fragments encompassing various regions of the HPV16 E7 protein (Fig. 1A) by standard ELISAs. We found no reactivity for either the complete E7 N-terminal (E7(1–40)) or C-terminal (E7(40–98)) domains or for the fragments E7(1–20) and E7(16–40) spanning the conserved regions CR1 and CR2 from the N-terminal domain (Fig. 1B). This result suggested that the epitope spans sequences belonging to both domains. In fact, IgG M1 presented the highest reactivity for the full-length E7 protein but an almost identical reactivity for fragment E7(36–53), which lies in the linker region connecting both domains (Fig. 1, B and C), and a significant reactivity for the smaller fragment E7(36–48) (Fig. 1B).
ELISA is an effective method for reactivity screening and epitope mapping but does not provide accurate quantitative measurements of affinities in solution. Therefore, we decided to test the affinity of the M1-E7 interaction using an alternative technique that could overcome these limitations. We made use of fluorescence spectroscopy by performing titration experiments using FITC-labeled E7 species and the Fab fragment of M1 antibody (M1Fab). By measuring fluorescence anisotropy changes in titrations performed at high (1 μm) protein concentration, we found that M1Fab bound to full-length E7 with a 1:1 stoichiometry (Fig. 2A, inset), expressed as one monomer of E7 per monomer of M1Fab. A titration experiment for the E7-M1Fab interaction under dissociating conditions yielded a KD of 20 ± 3 nm (Fig. 2A). A similar experiment yielded a KD of 122 ± 2 and 129 ± 2 nm for the E7(36–53) and E7(36–48) fragments, respectively (Fig. 2B). Although there was a 6-fold difference in affinity between the E7 fragments and full-length E7, these results indicated that the affinities of the two fragments tested were similar and that the E7(36–48) constituted the minimal binding epitope, which we named E7Ep.
FIGURE 2.

Equilibrium dissociation of M1Fab×antigen complexes. A, FITC fluorescence anisotropy change measured upon adding increasing amounts of M1Fab to a cuvette containing 50 nm FITC-E7′protein. Inset, equilibrium titration performed at 1 μm FITC-E7, showing a 1:1 FITC-E7·M1Fab binding stoichiometry. B, FITC fluorescence anisotropy change measured upon adding increasing amounts of M1Fab to a cuvette containing 100 nm FITC-E7(36–48) (open circles) or 100 nm FITC-E7(36–53) (filled circles) peptides. The different M1Fab·antigen complexes were incubated in separate tubes for 30 min prior to measurements. Lines are fits of the data to a 1:1 binding model, from which the following KD values were obtained for each complex (see “Experimental Procedures”). For FITC-E7·M1Fab, KD = 20 ± 3 nm; for FITC-E7(36–48)·M1Fab, KD = 122 ± 2 nm; for FITC-E7(36–53)·M1Fab, KD = 129 ± 2 nm.
Dissociation Kinetics of the M1Fab·E7Ep Complex
Antibody affinity maturation is based on the modification of the relative association and dissociation rates of antibody·antigen complexes (46). Therefore, as an essential part of elucidating the M1Fab-E7Ep interaction mechanism, we addressed the kinetics of dissociation and association. Full-length E7 gave poor anisotropy and fluorescence signal changes upon M1Fab binding, precluding kinetic analyses (not shown). This was probably because the fluorescent label was coupled at the intrinsically disordered N terminus, 30 residues away from the binding epitope. We thus focused our kinetic studies on the minimal E7Ep epitope using FITC-E7Ep, which reported larger fluorescence changes upon binding.
We first tackled the dissociation kinetics by forming a stoichiometric FITC-E7Ep·M1Fab complex. Once stabilized, we displaced FITC-E7Ep with a 20-fold excess of unlabeled peptide. We monitored the time course of the change in fluorescence anisotropy in a stopped-flow apparatus and observed that two exponential phases were required to explain the shape of the anisotropy signal decrease. This was indicative of at least two reactions in the dissociation process (Fig. 3). The two observed kinetic dissociation rate constants were koff1 = 0.63 ± 0.05 s−1 and koff2 = 0.026 ± 0.001 s−1. Similar values were obtained by fits of the change in fluorescence intensity (koff1 = 0.56 ± 0.02 s−1 and koff2 = 0.015 ± 0.001 s−1, supplemental Fig. S2). The final anisotropy value in the stopped-flow experiments (r = 0.03; Fig. 3) was in excellent agreement with the initial anisotropy value from the equilibrium titration experiments (r = 0.033; Fig. 2B), indicating that complete dissociation of the FITC-E7Ep·M1Fab complex had taken place. The relative amplitudes obtained from the anisotropy change were 90% for the faster and 10% for the slower phases, respectively (Fig. 3).
FIGURE 3.

Dissociation kinetics of E7Ep·M1Fab complex. FITC fluorescence anisotropy time trace measured upon the addition of a 20-fold excess of unlabeled E7Ep peptide (E7(36–48)) to a preformed stoichiometric 400 nm FITC-E7Ep·M1Fab complex. Full line, fit of the time trace to a double exponential function. The residuals from fits to a single (top) and double (bottom) exponential function are shown below the graph. The fitted rate constants were koff,1 = 0.63 ± 5.2 × 10−2 s−1 and koff,2 = 2.62 ± 0.1 × 10−2 s−1, and the corresponding amplitudes were Amp1 = 1.8 ± 0.2 × 10−2 and Amp2 = 3.5 ± 0.1 × 10−3.
Association Kinetics of the E7Ep·M1Fab Complex Followed by Fluorescence
We monitored the association kinetics in stopped-flow experiments by following the fluorescence intensity signal change after mixing equimolar amounts of FITC-E7Ep and M1Fab at 50, 200, and 430 nm (Fig. 4A). The reaction showed clear concentration dependence and proceeded faster at higher protein concentrations, in accordance with a bimolecular association. The data were fitted to a second-order model to obtain the kon values. Similar kon values were obtained at the different protein concentrations tested, with an average value of kon = 6.4 ± 1.8 × 107 m−1 s−1 (Fig. 4A).
FIGURE 4.

Association kinetics of the E7Ep·M1Fab complex. A, bimolecular association phase. FITC fluorescence intensity change after mixing FITC-E7Ep and M1Fab at equimolar concentrations. Light gray, 430 nm; dark gray, 200 nm; black, 50 nm. The fits to a second-order association model for each trace were used to obtain the value of kon for each condition and are shown as full lines (see Equation 2). Residuals from the fits are shown below the graph (430 nm (top), 200 nm (middle), and 50 nm (bottom)). The value obtained by averaging all measurements was kon = 6.4 ± 1.8 × 107 m−1 s−1. B, slow association phase. Change in FITC fluorescence intensity was measured after mixing 100 nm FITC-E7Ep with 600 nm M1Fab. The addition of M1Fab antibody (black arrow) was established as time 0. Traces were fit to a single exponential function to obtain the observed rate constants at each concentration (supplemental Fig. S3). C, concentration dependence of the slow association phase. The observed rate constants were measured at 50 nm E7Ep and increasing M1Fab concentrations. The data were fitted to a pre-equilibrium model by using Equation 4.
Interestingly, a slower process was observed for the association reaction measured at 50 nm M1Fab concentration, where the time base was 5 s. This process could be accounted for by adding an exponential phase to the model (Fig. 4A). This indicated the presence of a slower process in the association reaction. In order to characterize this phase, we carried out association experiments in the fluorimeter by hand mixing. We added increasing concentrations of M1Fab to a cuvette containing 50 nm FITC-E7Ep and measured changes in fluorescence anisotropy. We observed a slow increase in the fluorescence anisotropy signal after adding M1Fab and therefore monitored reactions for about 30 min until an equilibrium value was reached (Fig. 4B). For all concentrations tested, the increase in anisotropy was well described by a single exponential function (supplemental Fig. S3). The average anisotropy value extrapolated to t = 0 from the fits of the data to a single exponential function was r = 0.039 (supplemental Table S1). Because the average anisotropy value of the free E7Ep fragment was r = 0.031 (supplemental Table S1), we assign the remaining change in anisotropy Δr = 0.008 (supplemental Table S1) to the bimolecular association reaction. The relative amplitude of the slow association phase increased with increasing M1Fab concentration, and for 1 μm M1Fab, 83% of the total anisotropy change corresponded to the slow association phase, and 17% corresponded to the fast bimolecular association phase (supplemental Table S1).
The presence of an exponential process in an association reaction can be due to either postbinding events or to pre-equilibrium conformational rearrangements. The concentration dependence of the slow association phase can discriminate between these two scenarios. In our case, the observed rate constant showed a marked decrease upon increasing M1Fab concentration (Fig. 4C and supplemental Table S1), which was indicative of the existence of a pre-equilibrium reaction (8, 47). The pre-equilibrium model depicted in Scheme 1 and Eq. 4 was used to analyze the data and to obtain the microscopic constants for each step. By using this model, we were able to obtain values for the microscopic rate constants corresponding to the pre-equilibrium reaction (k1 = 3.2 ± 0.3 × 10−3 s−1, k−1 = 2.0 ± 0.3 × 10−2 s−1, K1 = k1/k−1 = 1.64 ± 0.18 × 10−1; Scheme 1). Moreover, the fit of the data also yields a value for the equilibrium constant of the bimolecular association reaction (KD2 = 1.2 ± 0.3 × 10−7 m; Scheme 1). Finally, using the value of 6.4 ± 1.8 × 107 m−1 s−1 for the bimolecular association rate constant obtained by stopped-flow measurements (Fig. 4A) and the equilibrium constant KD2 for the binding reaction estimated from the fit to the pre-equilibrium model, we estimated the dissociation rate constant for the bimolecular association reaction (k−2 = 7.5 ± 2 s−1; Scheme 1). Using the fitted parameters, we calculated the global equilibrium constant for the two-step pre-equilibrium model, KD(global) = 8.3 × 10−7 m (see “Experimental Procedures” and Scheme 1). This value was 6-fold higher than the KD value determined independently in the equilibrium titration experiments, KD = 1.29 × 10−7 m (Fig. 2). This discrepancy suggested the presence of an additional reaction involving a rearrangement of the E7Ep·M1Fab complex, which we accounted for in the right-hand side of Scheme 1. From the equilibrium KD value, the pre-equilibrium association constant K1, and the bimolecular association equilibrium constant K2, we estimated an equilibrium constant for this reaction, K3, of 6 ± 1.5 (see “Experimental Procedures” and Scheme 1). The measured dissociation kinetics also lent support to the three-step model. On one hand, the slowest dissociation phase (0.026 s−1) was in excellent agreement with the k−1 value from Scheme 1 (0.020 s−1). On the other hand, the faster dissociation phase (0.63 s−1) agreed reasonably well with the model from Scheme 1, because it would be expected to correspond roughly to the product of k−2 times the fraction of E7Ep·M1Fab intermediate (k−2 × 1/K3) if the last equilibrium in the reaction is fast, which yields a value of 1.25 ± 0.46 s−1.
SCHEME 1.
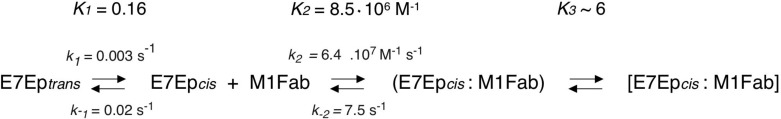
Minimal model for the E7-M1Fab interaction.
Temperature Dependence of the Pre-equilibrium Association Phase
The values of the pre-equilibrium kinetic rate constants were indicative of a slow process and could correspond to the isomerization of a Xaa-Pro peptide bond in either of the reactants. Inspection of the E7Ep sequence revealed the presence of two proline residues (Fig. 1A). Protein conformational changes limited by Xaa-Pro peptide bond isomerization often present characteristic temperature dependence, with an activation enthalpy close to −22 kcal/mol and a negligible activation change in heat capacity. We analyzed the temperature dependence of the slow phase by measuring the anisotropy change after mixing 600 nm M1Fab with 100 nm of E7Ep at different temperatures. All traces could be fitted to a single exponential function (supplemental Fig. S4), and as expected, the observed rate constant became faster as temperature was increased (supplemental Table S2). We estimated the activation parameters for the reaction from a Eyring plot of the data (ln(k(h/k*T)) versus 1/T), which showed a linear relationship, as expected for a reaction without a change in heat capacity (Fig. 5A). The activation enthalpy was ΔH‡ = −24.2 ± 0.68 kcal/mol. The linearity and the ΔH‡ value obtained was characteristic of prolyl isomerization reactions in peptides (48). This result suggested that the pre-equilibrium reaction could be limited by a prolyl isomerization event within E7Ep.
FIGURE 5.

Temperature dependence of the slow association phase. A, Eyring plot of the slow association phase, which was measured at different temperatures by mixing 100 nm FITC-E7Ep with 600 nm M1Fab. Traces were fit to a single exponential function to obtain the observed rate constants at each temperature (supplemental Fig. S4). Data were fit to a linear function from which the enthalpy of activation could be estimated (see “Experimental Procedures”). The value obtained was ΔH‡ = −24.2 ± 0.7 kcal/mol. B, sequence logo of the E7Ep region for all E7 proteins from the α9 papillomavirus species (type HPV16, -31, -33, -35, -35H, -52, -58, or -67). The numbering corresponds to the HPV16 E7 protein. Conserved proline residues are highlighted in black.
Prolyl Isomerization in E7Ep Determined from NMR
In order to analyze whether one or both of the Pro residues in E7Ep presented an isomerization equilibrium in the free form of the peptide, we conducted NMR experiments. Proton and carbon chemical shift resonances of the E7Ep peptide epitope were completely assigned using two-dimensional NMR experiments (supplemental Tables S3 and S4). The spectra revealed two sets of signals originating from Gly-40 to Gln-44 and from Glu-46 to Asp-48 in the sequence. The fact that residues showing the largest shift differences were either Pro or those adjacent to Pro indicated that the weaker set of signals arose from the cis isomers of Pro residues. Both Pro-41 and Pro-47 were involved in such exchange process. Cis and trans prolines were identified from the 13Cβ and 13Cγ chemical shifts, which differ significantly in both configurations. 13Cβ and 13Cγ chemical shifts for the cis isomer were found to be at 34.4 and 24.6 ppm, respectively, for Pro-41 and at 34.0 and 24.2 ppm, respectively, for Pro-47. For the trans isomer, the chemical shifts exhibited by the same nuclei were 31.9 and 26.9 ppm, respectively, for Pro-41 and 31.8 and 27.0 ppm, respectively, for Pro-47 (49) (Fig. 6A and supplemental Tables S3 and S4). Also, in the NOESY spectrum, typical strong Hδ-Hαi − 1 and Hδ-HNi − 1 NOEs were observed for the trans prolines and strong Hα-Hαi − 1 and Hδ-HNi − 1 NOEs for the cis ones (Fig. 6B). The mole fraction of the cis and trans isomers, xcis and xtrans, were obtained by integrating the volumes of the cross-peaks corresponding to cis and trans proline isomers at each temperature in the 1H-13C HMQC spectrum. For both proline residues in the peptide, the trans isomer was the most populated, with the cis isomer mole fraction, xcis, being 0.152 ± 0.020 and 0.049 ± 0.005 for Pro-41 and Pro-47, respectively, at 30 °C.
FIGURE 6.

E7Ep peptide proline chemical shift assignment. A, selected region of the 1H-13C HMQC spectrum showing the chemical shift assignment of CH2 groups of positions β (left) and γ (right) of prolines at 20 °C. For both Pro-41 (P41) and Pro-47 (P47), the trans isomer is much more abundant at equilibrium (85 and 95%, respectively). B, selected region of the NOESY spectrum at 20 °C showing the cis and trans chemical shift assignments. Characteristic Hδ-Hαi − 1 NOEs are observed for the trans prolines, whereas Hα-Hαi − 1 cross-peaks are detected in the cis isomers.
Conformational exchange rates were determined from the NOESY spectra at different mixing times (Fig. 7A). As at 20 °C, no exchange cross-peaks were observed, and the kinetic rate constants for the isomerization process were determined at 30 and 40 °C (supplemental Table S5). Selected regions of the NOESY spectra at different mixing times illustrating the build-up and decay of the Hδ exchange cross-peaks are shown in Fig. 7B. The mixing time dependences of the diagonal and cross-peak intensities show that the diagonal peaks decay exponentially, whereas the chemical exchange cross-peak has a rapid build-up phase and a competing decay arising from relaxation (Fig. 7B). Reasonably good fits were obtained by graphing the ratios as a function of τm) (Fig. 7B), from which the kinetic rate constants were estimated (supplemental Table S5). The rate constants for the trans to cis isomerization were 0.0023 s−1 (Pro-41) and 0.0015 s−1 (Pro-47), whereas the rate constants for the cis to trans isomerization were 0.029 s−1 (Pro-41) and 0.042 s−1 (Pro-47) (Table 1). In addition, equilibrium constants for the cis-trans proline interconversion obtained from the ratio between the kinetic forward and reverse rate constants agreed rather well with those obtained from integrating the volume of the signals of both isomers in the 1H-13C HMQC spectrum (supplemental Table S5). Finally, we calculated the enthalpy of activation for the isomerization reactions from the measurements at 30 and 40 °C. The ΔH‡ values for the trans to cis isomerization were 20.9 kcal/mol (Pro-41) and 20.8 kcal/mol (Pro-47), whereas the ΔH‡ values for the cis to trans isomerization were 16.6 kcal/mol (Pro-41) and 15.1 kcal/mol (Pro-47) (Table 1).
FIGURE 7.

Prolyl cis-trans isomerization kinetics estimated by NMR. A, selected region of the NOESY spectra at 30 °C recorded with different mixing times (τm) illustrating the variation of the chemical exchange cross-peak intensities for Hδ of Pro-47 (left panels) and Pro-41 (right panels). The exchange cross-peaks are boxed. B, examples of chemical exchange cross-peak intensity (left) and of diagonal cross-peak intensity (middle) as a function of mixing time. The right panel shows an example of the cross-peak to diagonal peak intensity ratio. The data were fit to Equation 9 to estimate the kinetic rates for proline isomerization (see “Experimental Procedures”).
TABLE 1.
Comparison between the slow association phase for E7Ep · M1Fab binding and prolyl isomerization in free E7Ep
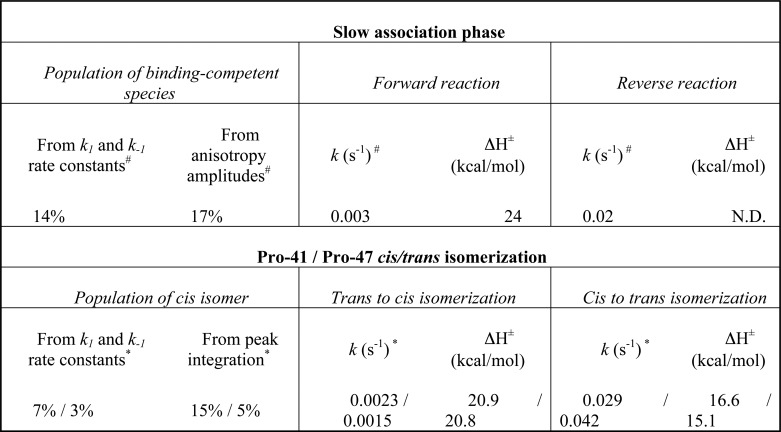
# Amplitudes and kinetic rate constants from association experiments were obtained from measurements performed at 20 °C (supplemental Tables S1 and S2).
* Isomer populations and kinetic rate constants from NMR experiments were extrapolated to 20 °C from the measured data at 30 and 40 °C (supplemental Table S5).
The Pre-equilibrium Association Phase Is Limited by trans to cis Prolyl Isomerization in E7Ep
The NMR experiments revealed that both proline residues within E7Ep presented a isomerization equilibrium between cis and trans configurations. We compared the equilibrium and kinetics of the pre-equilibrium association phase with the equilibrium and kinetics of prolyl isomerization. The equilibrium constants for the isomerizations in the peptide and for the pre-equilibrium reaction preceding complex formation were in agreement (Table 1). For both prolines, the trans conformer was populated to about 90%, compared with 10% of the cis conformer (Table 1). In the case of the pre-equilibrium, the value of K1 indicated that 86% of the molecules must isomerize prior to binding (Table 1). Additionally, the anisotropy amplitudes of the association phases indicated that the forward reaction of the pre-equilibrium was rate-limiting for 83% of the molecules (Fig. 4B, supplemental Table S1, and Table 1). We concluded that the pre-equilibrium reaction corresponded to the cis-trans isomerization of either Pro-41 or Pro-47.
The kinetic rate constants supported and further clarified this view. The forward rate constant for the pre-equilibrium association phase was 0.003 s−1, with a ΔH‡ of 24 kcal/mol. These values were in excellent agreement with the rate constants for trans to cis isomerization of both Pro-41 and Pro-47 at 0.002 s−1 and the activation enthalpies of 21 kcal/mol (Table 1). We propose that the forward reaction for the pre-equilibrium association is limited by trans to cis isomerization of Pro-41 or Pro-47. In turn, the reverse process of the pre-equilibrium association phase had a rate constant of 0.02 s−1, in agreement with the rate constants for cis to trans isomerization of both Pro-41 and Pro-47 at 0.03 and 0.04 s−1, respectively (Table 1). We thus assigned the reverse reaction for the pre-equilibrium association to the cis to trans isomerization of Pro-41 or Pro-47. Altogether, these results strongly suggested that the fast binding event corresponded to the association between M1Fab and the E7Ep cis configuration of Pro-41 or Pro-47, and the slower binding event corresponded to the association of the remaining E7Ep molecules, which was limited by trans to cis isomerization of Pro-41 or Pro-47.
E7Ep Mutants Identify the Proline Residue Involved in Binding and Isomerization
In order to confirm whether one or both E7Ep proline residues were involved in the M1Fab interaction, we first performed equilibrium binding experiments using FITC-labeled E7Ep mutants P41A, P47A, and the P41A/P47A double mutant. The P47A mutant bound to the M1Fab with an affinity (KD = 160 ± 8 nm) very similar to that of the wild type E7Ep peptide (129 ± 2 nm), whereas the FITC-labeled P41A E7Ep and double mutant E7Ep peptides presented no changes in anisotropy upon the addition of M1Fab up to the micromolar concentration range (Fig. 8A). Based on the concentrations tested, we estimated that if an interaction occurred, the KD values for the P41A and the double mutant peptides were in the millimolar range or higher. Therefore, these experiments allowed us to conclude that the Pro-41 residue was the sole proline residue determining M1Fab affinity. We further tested the association kinetics and found that the P47A mutant also presented a monoexponential slow association phase, as observed for the wild type E7Ep peptide (supplemental Fig. S5). This observed phase became slower at increasing M1Fab concentrations (Fig. 8B). The average anisotropy value extrapolated to t = 0 was r = 0.040 (supplemental Table S6), and considering the average anisotropy value of the free P47A E7Ep fragment (r = 0.032; supplemental Table S6), we obtained a change in anisotropy of Δr = 0.008 for the bimolecular association reaction, in remarkable agreement with the parameters of the wild type E7Ep peptide (supplemental Table S1). The relative amplitude of the slow and fast association phases at 1 μm M1Fab were 87 and 13% respectively, also in excellent agreement with the values obtained for wild type E7Ep (supplemental Tables S1 and S6). A fit of these data to the pre-equilibrium model yielded values similar to those of the wild type peptide (k1 = 3.2 ± 0.3 × 10−3 s−1, k−1 = 0.02 ± 0.2 10−2 s−1, and K = 117 ± 31 nm). These experiments confirmed that the slow association phase corresponded to the trans-cis isomerization of proline residue 41 in E7Ep and that this residue also was required for interaction with M1Fab.
FIGURE 8.

Equilibrium binding and association kinetics of the complex of E7Ep proline mutants with M1Fab. A, FITC fluorescence anisotropy change measured upon adding increasing amounts of M1Fab to a cuvette containing 100 nm FITC- P47A E7Ep (filled circles), 100 nm FITC- P41A E7Ep (open circles), or 100 nm FITC- P41A + P47A E7Ep (open triangles). The different M1Fab·antigen complexes were incubated in separate tubes for 30 min prior to measurements. The line is a fit of the data for the FITC-P47A E7Ep·M1Fab complex to a 1:1 binding model, from which the following dissociation constant was obtained (see “Experimental Procedures”): KD = 160 ± 8 nm. The P41A E7Ep and the P41A/P47A E7Ep mutants did not bind to the M1Fab at the concentrations tested, which prevented fitting of the data. B, concentration dependence of the slow association phase for the FITC-P47A E7Ep·M1Fab complex measured at 50 nm P47A E7Ep and at increasing M1Fab concentrations. The observed rate constants were obtained from monoexponential fitting of the kinetic traces (supplemental Fig. S6), and the line represents the fitting of the data to a pre-equilibrium model by using Equation 4.
Association Kinetics of the E7Ep·M1Fab Complex Followed by Circular Dichroism
In order to determine whether M1-E7 binding was accompanied by changes in secondary structure, we carried out circular dichroism (CD) experiments. As expected, the E7Ep fragment had a CD spectrum characteristic of a disordered polypeptide with a minimum at 200 nm (Fig. 9A, dotted line), whereas the M1Fab CD spectrum showed a high content of β-sheet structure, characteristic of Fabs (50) (Fig. 9A, solid line). A comparison of the spectrum of the E7Ep·M1Fab complex (Fig. 9B, dashed line) with that obtained by the sum of the individual spectra (Fig. 9B, solid line) showed significant differences at 230, 216, and 203 nm, indicating that structural changes had occurred upon binding.
FIGURE 9.

Conformational changes associated with formation of the E7Ep·M1Fab complex. A, far-UV CD spectrum of 5 μm M1Fab (continuous line) and of 5 μm E7Ep (dotted line). B, CD spectrum of a stoichiometric 5 μm E7Ep·M1Fab complex incubated for 30 min prior to measurement (dashed line) and CD spectrum resulting from the sum of the individual E7Ep and M1Fab spectra (solid line). C, pre-steady-state change in ellipticity upon formation of the E7Ep·M1Fab complex. The ellipticity change was measured at 203 nm after mixing 5 μm M1 with 5 μm E7Ep peptide. The time trace shows two phases with amplitudes of different sign, which were fitted independently to single exponential functions to give the observed rate constants for each phase (kobs1 = 6.5 ± 0.4 × 10−3 s−1 and kobs2 = 5.06 ± 0.8 × 10−3 s−1).
Next, we performed kinetic experiments to monitor structural changes upon E7Ep·M1Fab binding. In these experiments, the far-UV CD signal was recorded after the E7Ep fragment was added to a cuvette containing M1Fab by manual mixing. We chose to monitor structural changes at 203 nm, because it was the wavelength at which the largest changes in the CD signal took place. The bimolecular association phase is expected to be fast at the micromolar concentrations used in these experiments (5 μm) and completed within the dead time of the manual mixing (∼20 s). Interestingly, the time course of the reaction showed two phases with opposite amplitudes, with rates kobs1 = 6.5 ± 0.4 × 10−3 s−1 for the phase with positive amplitude and kobs2 = 5.1 ± 0.8 × 10−3 s−1 for the phase with negative amplitude (Fig. 9C). Both rate constants and their net rate (2.9 × 10−3 s−1) were in agreement with the limiting value of kobs = 3 × 10−3 s−1 expected for the pre-equilibrium isomerization phase at high protein concentration (the end point from the fit of the pre-equilibrium model; Fig. 4C). This result suggested that one of the phases in the circular dichroism experiments corresponded to the preassociation isomerization equilibrium of Pro-41 in E7Ep and that this reaction was accompanied by changes in secondary structure. The remaining circular dichroism phase could correspond to a trans-cis isomerization of Pro-47 in E7Ep that does not affect the interaction affinity or to an additional conformational rearrangement in E7Ep or M1Fab.
DISCUSSION
Some antibody·antigen recognition events involve additional species to the bound and unbound states, as is the case for the association between E7 and the M1 antibody studied here. We have determined rate constants for association, dissociation, and first-order conformational rearrangements occurring before and after the binding event, by means of kinetic and equilibrium studies followed by fluorescence spectroscopy, circular dichroism, and nuclear magnetic resonance. With these data, we propose a minimal kinetic model for the reaction (Scheme 1).
The dissociation constant for the complex between full-length E7 and M1Fab was in the nanomolar range, implying that M1 had underwent affinity maturation close to the affinity ceiling for antibodies (51) (Scheme 1 and Fig. 2). Kinetic analyses yielded a rate constant for association of E7Ep with M1Fab that approaches 6 × 107 s−1 m−1 (Scheme 1 and Fig. 4), among the fastest determined rates for antibody·antigen complexes (52). In immunological terms, fast binding of the antigen may be at least as important as high affinity, a phenomenon that has been referred to as kinetic maturation (46).
The mechanism of biomolecular recognition is often discussed in terms of conformational selection versus induced fit (53). In other words, either binding takes place only for a subpopulation of the unbound species or the two molecules form a complex that later rearranges. Our results suggest that both phenomena play a role in M1-E7 complex formation. On one hand, fluorescence anisotropy experiments revealed a slow association phase, with a rate constant that decreased with increasing M1Fab concentration (Fig. 4). This was indicative of a conformational change in the free reactants, prior to binding (8, 47) (Scheme 1). Thus, M1Ab-E7Ep recognition involves a conformational selection step. On the other hand, the comparison between kinetic and equilibrium constants indicated the presence of a third step in the binding reaction involving a first-order rearrangement of the complex that takes place after the initial association event and has an estimated equilibrium constant of about 6 (Scheme 1). This could be related to an induced fit step. Moreover, the values of the two phases observed in the dissociation reaction measured by both FITC fluorescence intensity and anisotropy (Fig. 3) were in agreement with this hypothesis. Additional interrupted binding (double-jump) experiments (54) are required to better define this induced fit step of the recognition mechanism.
The presence of two proline residues in the E7 epitope prompted us to assess whether the conformational selection event could be related to prolyl isomerization. NMR experiments revealed that both proline residues within E7Ep presented an isomerization equilibrium with a predominance of the trans over the cis isomer (Figs. 6 and 7), as expected for an intrinsically disordered peptide. The equilibrium and kinetic properties of the proline isomerization reactions in unbound E7Ep by NMR are in remarkable agreement with the corresponding parameters of the preassociation equilibrium (Figs. 5–7 and Table 1), which allowed us to assign the conformational selection event to a proline isomerization in E7Ep. The reaction parameters for isomerization of Pro-41 and Pro-47 are very similar and did not allow discrimination of the Pro residue involved. However, mutagenesis experiments clearly indicated that Pro-41 was necessary, whereas Pro-47 was dispensable for binding (Fig. 8). We determined that the M1 antibody binds to the cis isomer of the antigen, which is populated to about 10% (Scheme 1 and Table 1) and that the kinetics of the selection step is limited by the rate constant for trans to cis isomerization, which involves a conformational rearrangement in E7Ep.
We have narrowed down the E7 epitope to the intrinsically disordered E7Ep fragment (E7(36–48)) using ELISA and fluorescence anisotropy (Figs. 1 and 2). Interestingly, E7Ep contains the immunodominant B epitope, which was also targeted by antiproliferative single chain antibodies (55). Of the three main immunodominant epitopes of E7 (36, 37), two are located in the same region and contained within E7Ep. Peptide epitopes are often found in protein regions with residual conformations or conformational ensembles (14). The E7 epitope spans a region of E7 that indeed appears as disordered by NMR, crystallography, and circular dichroism spectroscopy (56, 57) (Fig. 9). It has been proposed that structural transitions within disordered regions could increase immunogenicity (58). Our data show that trans to cis isomerization of Pro-41 is a requirement for E7Ep·M1Fab complex formation (Scheme 1). It is plausible that presentation of a “locked” cis conformation by the antigen-presenting cells may enhance epitope immunogenicity.
The intrinsically disordered E7N domain can be described as a succession of conserved linear motifs that bind cellular targets, separated by linkers of variable sequence and length (59). The M1 E7Ep epitope is located in the last of the linkers in E7N, which joins the E7N CR2 region with the globular E7C domain and lacks known binding sites. The proline residues within E7Ep are conserved among HPV E7 proteins from the α9 species (Fig. 5B), but this region is highly divergent across E7 proteins from other HPV species (59). This may explain why M1 is able to discriminate the HPV16 E7 protein from the HPV18 E7 protein, which belongs to the closely related α7 species (supplemental Fig. S1). This ability of M1 to distinguish the E7 proteins from the two main high risk HPV types may prove useful for diagnostic applications.
We have recently described the kinetic interaction mechanism of the LXCXE linear motif within the E7N CR2 region with the retinoblastoma tumor suppressor RbAB domain (42). The LXCXE-RbAB interaction is a fast two-state process, indicating that sampling of the conformational ensemble in this region of the E7N domain is very fast. In contrast, the kinetic interaction mechanism of the E7Ep epitope with the M1 antibody displays a slow conformational selection step limited by prolyl isomerization. Because linear motifs are often rich in proline residues (60), this result suggests that linear motif recognition may be limited by proline isomerization in other cases. Proline isomerization within E7Ep may be coupled not only to antibody binding but also to rearrangement of the relative positions of the E7N and E7C domains. This could play an important role in the cases where the two E7 domains cooperate in the binding of cellular targets, such as binding of the retinoblastoma protein (42). The other linker regions in E7N are also rich in proline residues (59), suggesting that the isomerization of specific peptide bonds may govern other slow conformational transitions and recognition events in many E7 proteins. A picture emerges where different regions of the disordered E7N domain can present conformational equilibria on different time scales. This allows for a diversity of interaction mechanisms, which are a salient feature of intrinsically disordered domains and include both pre- and postbinding rearrangement events (61–63) as well as the existence of a variety of conformational states in the ligand-bound form (64).
We have investigated the recognition of an HPV E7 epitope, which was also found to be the main immunodominant B cell epitope in humans (65). In addition to its interest for diagnostic applications, the immune response toward this oncoprotein is the basis of therapeutic vaccines against HPV cancer under development (25, 66). We revealed a peptide epitope with two proline isomers that may constitute discrete structural states and interconvert in the minute time scale. Antibody isomerization extends the immune repertoire by allowing binding of more than one antigen by a single antibody primary sequence. In a similar way, proline isomerization may provide with alternative conformations that regulate binding to different antibodies or protein ligands. Further structural and thermodynamic studies will be required to completely dissect this interaction mechanism. It will be interesting to investigate whether there is a common theme in the rules governing the recognition of intrinsically disordered viral epitopes, such as those found in E7, by the immune system, because the IDP nature is abundantly represented among multifunctional viral proteins.
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica Grant PICT2010-1052 (to I. E. S.) and Instituto Nacional de Cancer, Argentina, Basic Research Grant 2012–2014 (to G. P. G.).

This article contains supplemental Tables S1–S6 and Figs. S1–S5.
- HPV
- human papillomavirus
- IDP
- intrinsically disordered protein.
REFERENCES
- 1. Poljak R. J., Amzel L. M., Avey H. P., Becka L. N. (1972) Structure of Fab′ New at 6 Å resolution. Nat. New Biol. 235, 137–140 [DOI] [PubMed] [Google Scholar]
- 2. Allcorn L. C., Martin A. C. (2002) SACS. Self-maintaining database of antibody crystal structure information. Bioinformatics 18, 175–181 [DOI] [PubMed] [Google Scholar]
- 3. Lund O., Nielsen M., Lundegaard C., Kesmir C., Brunak S. (2005) Immunological Bioinformatics, pp. 69–102, MIT Press, Cambridge, MA [Google Scholar]
- 4. Sundberg E. J., Mariuzza R. A. (2002) Molecular recognition in antibody-antigen complexes. Adv. Protein Chem. 61, 119–160 [DOI] [PubMed] [Google Scholar]
- 5. Wilson I. A., Stanfield R. L. (1994) Antibody-antigen interactions. New structures and new conformational changes. Curr. Opin. Struct. Biol. 4, 857–867 [DOI] [PubMed] [Google Scholar]
- 6. Pauling L. (1940) A theory of the structure and process formation of antibodies. J. Am. Chem. Soc. 62, 2643 [Google Scholar]
- 7. Foote J., Milstein C. (1994) Conformational isomerism and the diversity of antibodies. Proc. Natl. Acad. Sci. U.S.A. 91, 10370–10374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. James L. C., Roversi P., Tawfik D. S. (2003) Antibody multispecificity mediated by conformational diversity. Science 299, 1362–1367 [DOI] [PubMed] [Google Scholar]
- 9. Oldstone M. B. (1998) Molecular mimicry and immune-mediated diseases. FASEB J. 12, 1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. James L. C., Tawfik D. S. (2005) Structure and kinetics of a transient antibody binding intermediate reveal a kinetic discrimination mechanism in antigen recognition. Proc. Natl. Acad. Sci. U.S.A. 102, 12730–12735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leder L., Berger C., Bornhauser S., Wendt H., Ackermann F., Jelesarov I., Bosshard H. R. (1995) Spectroscopic, calorimetric, and kinetic demonstration of conformational adaptation in peptide-antibody recognition. Biochemistry 34, 16509–16518 [DOI] [PubMed] [Google Scholar]
- 12. Berger C., Weber-Bornhauser S., Eggenberger J., Hanes J., Plückthun A., Bosshard H. R. (1999) Antigen recognition by conformational selection. FEBS Lett. 450, 149–153 [DOI] [PubMed] [Google Scholar]
- 13. Cerutti M. L., Ferreiro D. U., Sanguineti S., Goldbaum F. A., de Prat-Gay G. (2006) Antibody recognition of a flexible epitope at the DNA binding site of the human papillomavirus transcriptional regulator E2. Biochemistry 45, 15520–15528 [DOI] [PubMed] [Google Scholar]
- 14. Dyson H. J., Wright P. E. (1995) Antigenic peptides. FASEB J. 9, 37–42 [DOI] [PubMed] [Google Scholar]
- 15. Eckert B., Martin A., Balbach J., Schmid F. X. (2005) Prolyl isomerization as a molecular timer in phage infection. Nat. Struct. Mol. Biol. 12, 619–623 [DOI] [PubMed] [Google Scholar]
- 16. Feige M. J., Groscurth S., Marcinowski M., Shimizu Y., Kessler H., Hendershot L. M., Buchner J. (2009) An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell 34, 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rajbhandari P., Finn G., Solodin N. M., Singarapu K. K., Sahu S. C., Markley J. L., Kadunc K. J., Ellison-Zelski S. J., Kariagina A., Haslam S. Z., Lu K. P., Alarid E. T. (2012) Regulation of estrogen receptor α N terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol. Cell Biol. 32, 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sarkar P., Reichman C., Saleh T., Birge R. B., Kalodimos C. G. (2007) Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol. Cell 25, 413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sibille N., Huvent I., Fauquant C., Verdegem D., Amniai L., Leroy A., Wieruszeski J. M., Lippens G., Landrieu I. (September 30, 2011) Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered Tau protein. Proteins 10.1002/prot.23210 [DOI] [PubMed] [Google Scholar]
- 20. Tugarinov V., Zvi A., Levy R., Anglister J. (1999) A cis proline turn linking two β-hairpin strands in the solution structure of an antibody-bound HIV-1IIIB V3 peptide. Nat. Struct. Biol. 6, 331–335 [DOI] [PubMed] [Google Scholar]
- 21. Tsikaris V., Vlachoyiannopoulos P. G., Panou-Pomonis E., Marraud M., Sakarellos C., Moutsopoulos H. M., Sakarellos-Daitsiotis M. (1996) Immunoreactivity and conformation of the P-P-G-M-R-P-P repetitive epitope of the Sm autoantigen. Int. J. Pept. Protein Res. 48, 319–327 [DOI] [PubMed] [Google Scholar]
- 22. McLaughlin-Drubin M. E., Münger K. (2009) The human papillomavirus E7 oncoprotein. Virology 384, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Münger K., Basile J. R., Duensing S., Eichten A., Gonzalez S. L., Grace M., Zacny V. L. (2001) Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 20, 7888–7898 [DOI] [PubMed] [Google Scholar]
- 24. Galloway D. A. (1992) Serological assays for the detection of HPV antibodies. IARC Sci. Publ., 147–161 [PubMed] [Google Scholar]
- 25. Cerutti M. L., Alonso L. G., Tatti S., de Prat-Gay G. Long-lasting immunoprotective and therapeutic effects of a hyperstable E7 oligomer based vaccine in a murine human papillomavirus tumor model. Int. J. Cancer 130, 1813–1820 [DOI] [PubMed] [Google Scholar]
- 26. Hung C. F., Ma B., Monie A., Tsen S. W., Wu T. C. (2008) Therapeutic human papillomavirus vaccines. Current clinical trials and future directions. Expert Opin. Biol. Ther. 8, 421–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uversky V. N., Roman A., Oldfield C. J., Dunker A. K. (2006) Protein intrinsic disorder and human papillomaviruses. Increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J. Proteome Res. 5, 1829–1842 [DOI] [PubMed] [Google Scholar]
- 28. Boehr D. D., Nussinov R., Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uversky V. N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43, 1090–1103 [DOI] [PubMed] [Google Scholar]
- 30. Tokuriki N., Oldfield C. J., Uversky V. N., Berezovsky I. N., Tawfik D. S. (2009) Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 34, 53–59 [DOI] [PubMed] [Google Scholar]
- 31. Alonso L. G., García-Alai M. M., Nadra A. D., Lapeña A. N., Almeida F. L., Gualfetti P., Prat-Gay G. D. (2002) High-risk (HPV16) human papillomavirus E7 oncoprotein is highly stable and extended, with conformational transitions that could explain its multiple cellular binding partners. Biochemistry 41, 10510–10518 [DOI] [PubMed] [Google Scholar]
- 32. García-Alai M. M., Alonso L. G., de Prat-Gay G. (2007) The N-terminal module of HPV16 E7 is an intrinsically disordered domain that confers conformational and recognition plasticity to the oncoprotein. Biochemistry 46, 10405–10412 [DOI] [PubMed] [Google Scholar]
- 33. Chemes L. B., Glavina J., Faivovich J., de Prat-Gay G., Sánchez I. E. (2012) Evolution of linear motifs within the papillomavirus E7 oncoprotein. J. Mol. Biol. 422, 336–346 [DOI] [PubMed] [Google Scholar]
- 34. Dantur K., Alonso L., Castaño E., Morelli L., Centeno-Crowley J. M., Vighi S., de Prat-Gay G. (2009) Cytosolic accumulation of HPV16 E7 oligomers supports different transformation routes for the prototypic viral oncoprotein. The amyloid-cancer connection. Int. J. Cancer 125, 1902–1911 [DOI] [PubMed] [Google Scholar]
- 35. Alonso L. G., Chemes L. B., Cerutti M. L., Dantur K. I., Prat-Gay G. (2012) Biochemical and structure-function analyses of the HPV E7 oncoprotein. in Small Tumour Viruses (Gaston K., ed) pp. 125–150, Caister Academic Press, Norfolk, UK [Google Scholar]
- 36. Tindle R. W., Smith J. A., Geysen H. M., Selvey L. A., Frazer I. H. (1990) Identification of B epitopes in human papillomavirus type 16 E7 open reading frame protein. J. Gen. Virol. 71, 1347–1354 [DOI] [PubMed] [Google Scholar]
- 37. Comerford S. A., McCance D. J., Dougan G., Tite J. P. (1991) Identification of T- and B-cell epitopes of the E7 protein of human papillomavirus type 16. J. Virol. 65, 4681–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chemes L. B., Sánchez I. E., Smal C., de Prat-Gay G. (2010) Targeting mechanism of the retinoblastoma tumor suppressor by a prototypical viral oncoprotein. Structural modularity, intrinsic disorder and phosphorylation of human papillomavirus E7. FEBS J. 277, 973–988 [DOI] [PubMed] [Google Scholar]
- 39. Smal C., Wetzler D. E., Dantur K. I., Chemes L. B., Garcia-Alai M. M., Dellarole M., Alonso L. G., Gaston K., de Prat-Gay G. (2009) The human papillomavirus E7-E2 interaction mechanism in vitro reveals a finely tuned system for modulating available E7 and E2 proteins. Biochemistry 48, 11939–11949 [DOI] [PubMed] [Google Scholar]
- 40. de Prat Gay G., Fersht A. R. (1994) Generation of a family of protein fragments for structure-folding studies. 1. Folding complementation of two fragments of chymotrypsin inhibitor-2 formed by cleavage at its unique methionine residue. Biochemistry 33, 7957–7963 [DOI] [PubMed] [Google Scholar]
- 41. de Prat Gay G., Ruiz-Sanz J., Fersht A. R. (1994) Generation of a family of protein fragments for structure-folding studies. 2. Kinetics of association of the two chymotrypsin inhibitor-2 fragments. Biochemistry 33, 7964–7970 [DOI] [PubMed] [Google Scholar]
- 42. Chemes L. B., Sánchez I. E., de Prat-Gay G. (2011) Kinetic recognition of the retinoblastoma tumor suppressor by a specific protein target. J. Mol. Biol. 412, 267–284 [DOI] [PubMed] [Google Scholar]
- 43. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 44. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 45. Hans-Ulrich B. (2006) Papillomavirus Research. From Natural History To Vaccines and Beyond, pp. 11–17, Caister Academic Press, Norfolk, UK [Google Scholar]
- 46. Foote J., Milstein C. (1991) Kinetic maturation of an immune response. Nature 352, 530–532 [DOI] [PubMed] [Google Scholar]
- 47. Vogt A. D., Di Cera E. (2012) Conformational selection or induced fit? A critical appraisal of kinetic mechanism. Biochemistry 51, 5894–5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harrison R. K., Stein R. L. (1990) Mechanistic studies of peptidyl prolyl cis-trans isomerase. Evidence for catalysis by distortion. Biochemistry 29, 1684–1689 [DOI] [PubMed] [Google Scholar]
- 49. Shen Y., Bax A. (2010) Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 46, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tetin S. Y., Prendergast F. G., Venyaminov S. Y. (2003) Accuracy of protein secondary structure determination from circular dichroism spectra based on immunoglobulin examples. Anal. Biochem. 321, 183–187 [DOI] [PubMed] [Google Scholar]
- 51. Batista F. D., Neuberger M. S. (1998) Affinity dependence of the B cell response to antigen. A threshold, a ceiling, and the importance of off-rate. Immunity 8, 751–759 [DOI] [PubMed] [Google Scholar]
- 52. Foote J., Eisen H. N. (1995) Kinetic and affinity limits on antibodies produced during immune responses. Proc. Natl. Acad. Sci. U.S.A. 92, 1254–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bosshard H. R. (2001) Molecular recognition by induced fit. How fit is the concept? News Physiol. Sci. 16, 171–173 [DOI] [PubMed] [Google Scholar]
- 54. Ferreiro D. U., de Prat-Gay G. (2003) A protein-DNA binding mechanism proceeds through multi-state or two-state parallel pathways. J. Mol. Biol. 331, 89–99 [DOI] [PubMed] [Google Scholar]
- 55. Accardi L., Donà M. G., Mileo A. M., Paggi M. G., Federico A., Torreri P., Petrucci T. C., Accardi R., Pim D., Tommasino M., Banks L., Chirullo B., Giorgi C. (2011) Retinoblastoma-independent antiproliferative activity of novel intracellular antibodies against the E7 oncoprotein in HPV 16-positive cells. BMC Cancer 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ohlenschläger O., Seiboth T., Zengerling H., Briese L., Marchanka A., Ramachandran R., Baum M., Korbas M., Meyer-Klaucke W., Dürst M., Görlach M. (2006) Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 25, 5953–5959 [DOI] [PubMed] [Google Scholar]
- 57. Liu X., Clements A., Zhao K., Marmorstein R. (2006) Structure of the human papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J. Biol. Chem. 281, 578–586 [DOI] [PubMed] [Google Scholar]
- 58. Waltho J. P., Feher V. A., Lerner R. A., Wright P. E. (1989) Conformation of a T cell stimulating peptide in aqueous solution. FEBS Lett. 250, 400–404 [DOI] [PubMed] [Google Scholar]
- 59. Chemes L. B., Glavina J., Alonso L. G., Marino-Buslje C., de Prat-Gay G., Sánchez I. E. (2012) Sequence evolution of the intrinsically disordered and globular domains of a model viral oncoprotein. PLoS One 7, e47661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Davey N. E., Van Roey K., Weatheritt R. J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., Gibson T. J. (2012) Attributes of short linear motifs. Mol. Biosyst. 8, 268–281 [DOI] [PubMed] [Google Scholar]
- 61. Choi U. B., McCann J. J., Weninger K. R., Bowen M. E. (2011) Beyond the random coil. Stochastic conformational switching in intrinsically disordered proteins. Structure 19, 566–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sugase K., Dyson H. J., Wright P. E. (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447, 1021–1025 [DOI] [PubMed] [Google Scholar]
- 63. Dogan J., Schmidt T., Mu X., Engström Å., Jemth P. (2012) Fast association and slow transitions in the interaction between two intrinsically disordered protein domains. J. Biol. Chem. 287, 34316–34324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tompa P., Fuxreiter M. (2008) Fuzzy complexes. Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8 [DOI] [PubMed] [Google Scholar]
- 65. Krchnák V., Vágner J., Suchánková A., Krcmár M., Ritterová L., Vonka V. (1990) Synthetic peptides derived from E7 region of human papillomavirus type 16 used as antigens in ELISA. J. Gen. Virol. 71, 2719–2724 [DOI] [PubMed] [Google Scholar]
- 66. Zeng Q., Peng S., Monie A., Yang M., Pang X., Hung C. F., Wu T. C. (2011) Control of cervicovaginal HPV-16 E7-expressing tumors by the combination of therapeutic HPV vaccination and vascular disrupting agents. Hum. Gene Ther. 22, 809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]