

Abstract
The ideal sedative–hypnotic drug would be a rapidly titratable intravenous agent with a high therapeutic index and minimal side effects. The current efforts to develop such agents are primarily focused on modifying the structures of existing drugs to improve their pharmacodynamic and pharmacokinetic properties. Drugs currently under development using this rational design approach include analogues of midazolam, propofol, and etomidate, such as remimazolam, PF0713, and cyclopropyl methoxycarbonyl-etomidate (MOC-etomidate), respectively. An alternative approach involves the rapid screening of large libraries of molecules for activity in structural or phenotypic assays that approximate anesthetic and target receptor interactions. Such high-throughput screening offers the potential for identifying completely novel classes of drugs. Anesthetic drug development is experiencing a resurgence of interest because there are new demands on our clinical practice that can be met, at least in part, with better agents. The goal of this review is to provide the reader with a glimpse of the novel anesthetic drugs and new developmental approaches that lie on the horizon.
Keywords: Anesthetic, etomidate, midazolam, propofol
INTRODUCTION
Over the last decade, the development of new sedative and anesthetic drugs has been driven by the changing demands of our clinical practice. Procedures once performed only in hospitals are now commonly conducted in outpatient settings on an increasingly older population with a greater number of significant comorbidities. In addition, efforts to constrain costs have increased the demand for intravenous agents that can be more easily (and safely) administered by nonspecialists and without the expensive equipment required for inhaled agents.[20] Ideally, such agents would provide anesthesia and/or sedation in a rapidly titratable manner (i.e., rapid onset of action and recovery) and side effects, such as respiratory and cardiovascular depression, nausea, vomiting, and pain on injection would be minimal. Finally, any such ideal drug would possess a high therapeutic index and be easy to formulate in an aqueous solution. Currently, there is no clinically available hypnotic agent that possesses all of these properties.
Midazolam and propofol are among the most commonly used sedative–hypnotics by anesthesiologists [Figure 1]. It is against these 2 agents that new ones will be judged. In general, midazolam is used to produce sedation and propofol to induce anesthesia. However, when used at appropriate doses, both agents may achieve either clinical endpoint. Midazolam and propofol enhance the inhibitory actions of GABAA receptors in the central nervous system by promoting channel opening and facilitating the diffusion of chloride ions into neuronal cells.[49] The resulting hyperpolarization reduces the ability of these cells to initiate an action potential, causing central nervous system depression. In spite of these mechanistic similarities, these 2 agents are thought to bind to distinct sites on the GABAA receptor and to modulate the receptor with different efficacies.[7,2,54,48,47] Unlike propofol, midazolam is highly water soluble and its action is pharmacologically reversible (with flumazenil), both of which are desirable properties. However, midazolam's onset of action is relatively slow and recovery may be prolonged by the presence of a pharmacologically active metabolite.[39,5] At doses sufficient to produce deep sedation or anesthesia, midazolam causes respiratory depression, a property that led to numerous fatalities when midazolam first entered the market. Compared with midazolam, propofol provides a more rapid onset of action and recovery. However, it may produce significant hypotension and respiratory depression, particularly in the elderly and critically ill.[55,10,17] It is poorly water soluble and is commonly formulated as an emulsion that supports bacterial growth and may cause a syndrome of metabolic acidosis with prolonged infusion.[23,41,3]
Figure 1.
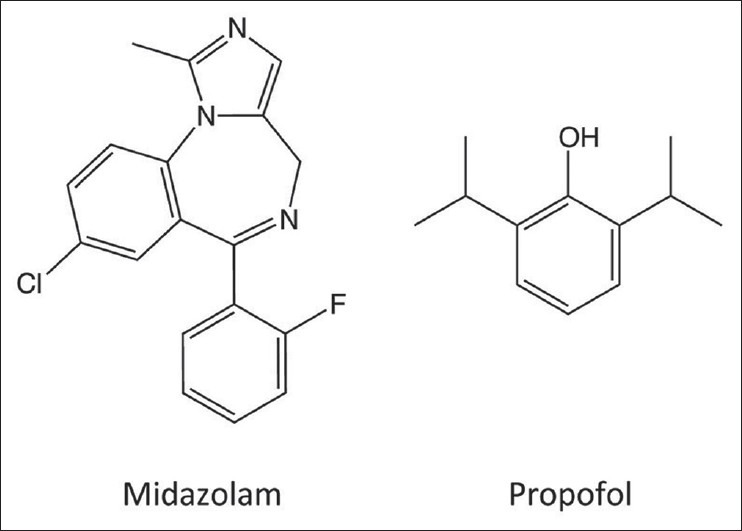
Structures of midazolam and propofol
Midazolam and propofol both possess significant advantages for sedation and hypnosis. However, their shortcomings present opportunities for targeting the development of novel anesthetic agents. Strategies for developing new drugs generally fall into 2 categories: rational design and high throughput screening. The remainder of this review will provide an overview of these 2 methods and discuss some of the anesthetic agents that are currently under development using these techniques.
RATIONAL DESIGN
Rational design uses information about the structure of a biologic target (e.g, an ion channel or enzyme) gleaned from X-ray crystallographic and computational modeling studies to design novel candidate drugs or to improve existing ones.[26] Most commonly, the goal is to maximize the affinity of the drug for the target responsible for its desirable pharmacologic action. In the case of anesthetic drugs, this approach has not been successfully applied largely because there is no high-resolution structural information that allows one to fully understand why anesthetics bind to the relevant protein targets (eg, the GABAA receptor) responsible for such anesthetic endpoints as hypnosis, amnesia, and immobility. An alternative goal is to minimize drug affinity for a target responsible for an undesirable side effect. This approach has been used to reduce etomidate binding to 11β-hydroxylase, thus eliminating the drug's ability to suppress steroid synthesis (see the section on carboetomidate below). Rational design may also involve the modification of a known drug to create analogues with improved pharmacokinetic properties or aqueous solubilities. In this case, little may be known about the structure of the drug's target. In one commonly applied approach, a metabolically labile ester moiety is incorporated into an existing drug to produce a new drug, which is readily susceptible to hydrolysis by plasma esterases.[4] This modification results in a drug that is metabolized more quickly, has a shorter duration of action, and can be titrated more easily during continuous intravenous infusion than the original compound. Because they are specifically designed to be rapidly metabolized, such agents are often referred to as “soft drugs.” Examples of soft drugs currently in common clinical practice include the opiate remifentanil and the beta-blocker esmolol.
Benzodiazepine and benzodiazepine-like drugs
Benzodiazepines are widely used in clinical anesthesiology as anxiolytics, amnestics, and sedative–hypnotics.[49] They are less commonly used as anesthetic induction agents because at the doses typically required to produce anesthesia, cardiovascular depression may be significant and recovery prolonged.[9] Among anesthesiologists, midazolam is the most commonly used benzodiazepine because it has a relatively short duration of action. However, there is still a demand for even shorter-acting agents that allow deep levels of sedation to be achieved during procedures while permitting predictable and rapid recovery afterward. Agents in this category that are currently being investigated include remimazolam (CNS 7056) and JM-1232(-).
Remimazolam (CNS 7056)
Remimazolam is an analogue of midazolam that utilizes the metabolically labile ester design approach to produce an ultra-short–acting benzodiazepine [Figure 2]. Remimazolam is rapidly hydrolyzed by nonspecific esterases to the carboxylic acid CNS 7054, which has an in vitro affinity for human GABAA receptors that is 400-fold lower than remimazolam.[31] Thus, the metabolite is not expected to produce important behavioral effects unless it accumulates with prolonged remimazolam administration and achieves high brain concentrations. Studies in animals have confirmed that remimazolam is a potent hypnotic, is rapidly metabolized, and has a significantly shorter duration of action than midazolam. In mice, for example, approximately equihypnotic doses of remimazolam and midazolam produce loss of righting reflexes for durations of several minutes and nearly an hour, respectively.[31] Remimazolam produces modest respiratory and cardiovascular depression at sedating doses, a side effect profile that it shares with midazolam and other benzodiazepines.[61,60]
Figure 2.
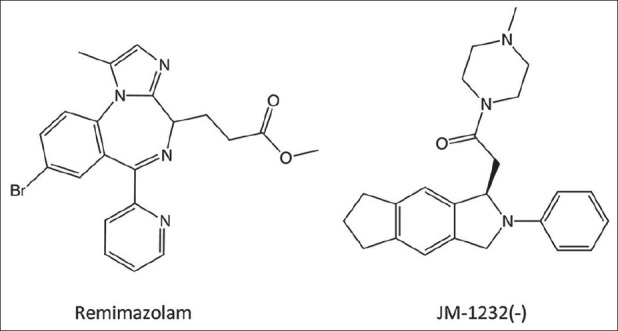
Remimazolam is a benzodiazepine that contains a metabolically labile ester moiety that renders it ultra-short acting. JM-1232(-) is a benzodiazepine-like sedative–hypnotic with a very high therapeutic index
Phase 1 clinical trials to assess the safety and efficacy of remimazolam have been completed and the results reported. These studies showed that remimazolam is more rapidly metabolized than midazolam and recovery is faster, consistent with prior studies in animals. Following 1-min intravenous infusions of remimazolam and midazolam (at equihypnotic doses), recovery times were 10 and 40 min, respectively.[1] This difference is less than that observed in mice, which may reflect differences between mice and humans in the rates with which they metabolize ester-containing soft drugs. Pharmacokinetic/pharmacodynamic modeling studies of remimazolam and midazolam suggest that even larger differences between the 2 drugs would be observed following prolonged infusion.[66] In the case of midazolam, the context-sensitive half-time (the time required for blood concentrations to decrease by half after infusion termination) is predicted to increase dramatically with midazolam infusion length, reaching 60 min after a simulated 8-h infusion.[66] In contrast, the predicted context-sensitive half-time of remimazolam reaches a maximum of only 7-8 min for infusions longer than 2 h. In these modeling studies, hypnotic recovery was also predicted to be highly dependent on infusion duration for midazolam, reaching 6 h after an 8-h infusion, whereas predicted recovery was essentially independent of infusion duration for remimazolam infusions greater than 2 h.
Based on the existing data, remimazolam shows great promise as a sedative agent for outpatient procedural sedation where predictable and rapid recovery is highly desirable. The ability to pharmacologically reverse its actions when inadvertent over-dosage leads to significant respiratory depression is also highly desirable, particularly when administered by practitioners who are not highly trained in airway management. Further human studies are needed to more completely characterize its action, to define optimal dosing regimens, and to determine whether metabolite accumulation with prolonged infusion slows recovery, particularly in patients with renal dysfunction.
JM-1232 (-) (MR04A3)
JM-1232 (-) is a nonbenzodiazepine sedative-hypnotic that was synthesized by Maruishi Pharmaceutical Co (Osaka, Japan) utilizing an isoindolin-1-one skeleton with the goal of increasing sedative potency, therapeutic index, and water solubility [Figure 2].[30] Although structurally not a benzodiazepine, JM-1232 (-) allosterically modulates GABAA receptor in a manner that seems to be identical to benzodiazepines and its activity can be inhibited by flumazenil, strongly suggesting that it binds to the same site on the GABAA receptor as classic benzodiazepines.[59,58,38] In addition to its sedative–hypnotic actions, JM-1232 (-) has been shown in animals to possess antinociceptive properties and have a therapeutic index of >38.5, which indicates a safety margin that is greater than propofol, midazolam, thiopental, and even etomidate.[30,38,8]
The results of human safety and efficacy trials of MR04A3, a 1% aqueous solution of JM-1232 (-), were published in early 2012. MR04A3 was found to have quick onset of action with a dose-dependent hypnotic effect and minimal hemodynamic depression at clinically relevant doses.[57] Based on this initial study, further study of MR04A3 seems warranted, as its pharmacokinetics could be faster than midazolam and its higher therapeutic index in rats may portend greater safety in humans.
Etomidate analogues
Etomidate is an imidazole-based anesthetic agent that was synthesized by Janssen Pharmaceuticals (Titusville, NJ, USA) in the early 1960s [Figure 3].[25] Originally developed as a potential antifungal agent, it was found to have potent hypnotic activity and a high therapeutic index when tested in rats. It was introduced into clinical practice in 1972 and gained in popularity as an anesthetic agent in the operating room and a sedative in the intensive care unit because it produced minimal cardiovascular depression.[16,37,21] Subsequently it was discovered that prolonged infusions of etomidate significantly increase mortality in critically ill patients by inhibiting 11β-hydroxylase activity and suppressing adrenocortical steroid synthesis.[35,65,14,22,64] Because of this side effect, etomidate is no longer administered as a prolonged infusion, but it is still used to induce anesthesia at the beginning of surgery particularly in the elderly and critically ill. However, after even a single etomidate bolus, adrenocortical function can be suppressed for days because it is so potent an inhibitor of 11β-hydroxylase.[15,63] The clinical importance of adrenocortical suppression produced by a single etomidate dose and its impact on mortality is hotly debated in the anesthesia and critical care fields with some recommending that etomidate use be abandoned completely.[29] This has led to efforts to develop etomidate analogues with reduced effects on adrenocortical function.
Figure 3.
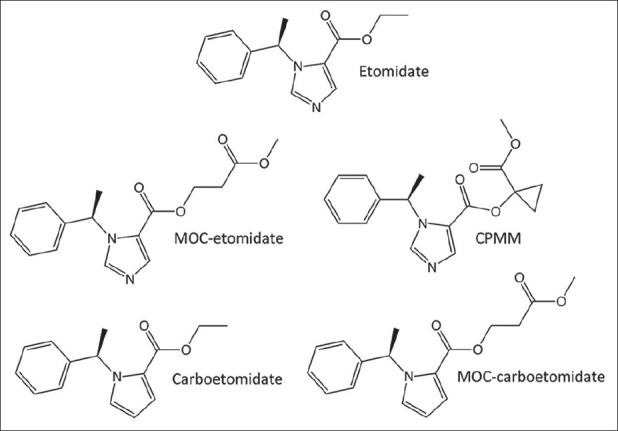
Methoxycarbonyl-etomidate (MOC-etomidate) is a rapidly metabolized etomidate analogue. Cyclopropyl MOC-etomidate is a more potent and longer-acting analogue of MOC-etomidate. Carboetomidate is a pyrrole etomidate analogue. MOC-carboetomidate has structural and pharmacologic properties present individually in MOC-etomidate and carboetomidate
Methoxycarbonyl etomidate and other spacer-linked etomidate esters
MOC-etomidate is a soft analogue of etomidate and the prototypical member of a new class of etomidate analogues termed “spacer-linked etomidate esters” [Figure 3].[12] It was designed with the goal of maintaining the desirable characteristics of etomidate (rapid onset, potent hypnosis, and hemodynamic stability) while providing rapid recovery from both adrenal suppression and hypnosis. Similar to remimazolam, remifentanil, and esmolol, MOC-etomidate contains a metabolically labile ester moiety that is rapidly hydrolyzed by nonspecific esterases.[12,13] The resulting carboxylic acid metabolite (MOC-ECA) has GABAA receptor, hypnotic, and adrenocortical inhibitory potencies that are 300- to 400-fold lower than those of MOC-etomidate.[43]
Animal experiments have confirmed that MOC-etomidate is metabolized extremely rapidly in vivo. They also showed that following single bolus administration or brief infusion, hypnotic, and adrenocortical recovery is significantly faster with MOC-etomidate than with etomidate.[12,43] However, subsequent studies revealed that when high doses of MOC-etomidate are given for prolonged periods of time, recovery times increase dramatically.[45] For example, rats recovered their righting reflexes 1.5 min after terminating a 5-min MOC-etomidate infusion, but 30 min after terminating a 30-min infusion.[45] Electroencephalographic recovery following prolonged MOC-etomidate infusion was similarly shown to be highly dependent on infusion duration. Analysis of the cerebrospinal fluid of rats following prolonged infusions of MOC-etomidate showed that MOC-ECA reached concentrations (>2 mM) sufficient to produce hypnosis. These metabolite concentrations declined with a time course of hours following infusion termination, explaining why recovery was so slow.[45]
Subsequent efforts in this area have focused on ameliorating the problem of metabolite accumulation by designing spacer-linked etomidate esters that are more potent than MOC-etomidate and metabolized more slowly.[27] More than a dozen spacer-linked etomidate esters have been synthesized and tested since MOC-etomidate was first introduced and several exhibit these favorable properties. One such compound, cyclopropyl MOC-etomidate (CPMM), is both more potent and longer acting than MOC-etomidate by nearly 10-fold [Figure 3]. Dosing requirements for continuous infusion (and consequently predicted metabolite concentrations achieved in the brain) are nearly 2 orders of magnitude lower with CPMM than with MOC-etomidate. Upon terminating CPMM infusions lasting either 5 min or 2 h, hypnotic and encephalographic recovery occurs in only 4 min.[24] Thus as observed with remimazolam (and remifentanil and esmolol), recovery times are independent of infusion duration. This compound is expected to reach clinical trials next year.
Carboetomidate
Homology modeling studies of 11β-hydroxylase-bound etomidate indicate that etomidate binds with high affinity primarily because the basic nitrogen in its imidazole ring forms a coordination bond with the heme iron at the enzyme's active site.[46] This conclusion is strengthened by X-ray crystallography studies showing that imidazole-containing ligands bind to other cytochrome P450 enzymes in a similar manner.[50,42,51] Carboetomidate is an analogue of etomidate in which this critical imidazole nitrogen has been replaced with a methylene group that cannot form a bond with iron [Figure 3].[11] In vitro studies using an adrenocortical carcinoma cell line have shown that this subtle molecular change reduces adrenocortical inhibitory potency by 2000-fold.[11] Unfortunately, this change also substantially reduces aqueous solubility. This low solubility may account for carboetomidate's relatively slow onset of action and implies that formulation will be more challenging than with etomidate. Carboetomidate retains etomidate's minimal effect on cardiovascular function, but unlike etomidate, carboetomidate neither inhibits steroid synthesis nor enhances proinflammatory cytokine production in a rat model of endotoxemia.[11,44] These findings suggest that it (or more water-soluble analogues) may be most suited for maintaining sedation or anesthesia in critically ill patients with sepsis.
Methoxycarbonyl carboetomidate
MOC-etomidate and carboetomidate each offer distinct advantages over etomidate. Through ultra-rapid metabolism, MOC-etomidate reduces the duration of adrenal suppression and allows rapid emergence from anesthesia.[12] Carboetomidate produces no significant adrenocortical suppression, but has a relatively slow onset of action and is difficult to formulate.[13,11] MOC-carboetomidate combines the chemical modifications present individually in MOC-etomidate and carboetomidate in an attempt to produce a single agent that possesses the advantages found in each of the 2 drugs [Figure 3].[43,44] MOC-carboetomidate enhances GABAA receptor function similar to etomidate, MOC-etomidate, and carboetomidate. As expected, it is metabolized more quickly than carboetomidate and unlike MOC-etomidate does not produce adrenocortical suppression.[43] However, similar to carboetomidate, it is poorly soluble in water and has a slow onset of action suggesting that that any advantage over carboetomidate, particularly for short-term use, may be modest.[43]
Other compounds
PF0713
PF0713 ((R, R)-2,6-di-sec-butylphenol) is a propofol analogue in which the two isopropyl groups have been replaced with sec-butyl groups [Figure 4].[56] It was originally synthesized in 1980 as a follow-up to propofol in attempt to improve on propofol's properties. Initial studies in mice indicated that it was similar in hypnotic potency to propofol, but had a slower onset of action. An abstract reporting the results of studies in rats also showed a similar potency to propofol, but a longer duration of hypnotic action.[53] Phase I clinical studies have shown PF0713 to be safe and effective as an intravenous induction agent.[52] Its principle advantage over propofol appears to be that it causes less pain on injection.[52] Whether this improvement is sufficient to justify continued development is unclear.
Figure 4.
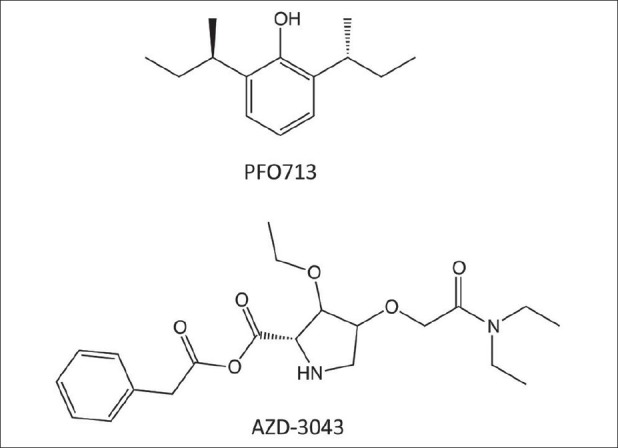
PFO713 is a propofol analogue that causes less pain on injection than propofol. AZD-3043 is a propanidid analogue, differing only by the presence of an additional methylene group and formulated as an emulsion
AZD-3043 (TD4756)
AZD-3043 is a close structural analogue of propanidid, a nonbarbiturate hypnotic that was introduced into clinical practice approximately 50 years ago [Figure 4].[37] The only structural difference between the two molecules is that AZD-3043 contains an additional methylene group, which increases hydrophobicity and hypnotic potency. AZD-3043 and propanidid also differ in their formulation as the former is formulated in an emulsion, whereas the latter was dissolved in cremophor, a vehicle that has been associated with anaphylactoid reactions. Animal studies have shown that AZD-3043 produces rapid-onset hypnosis and rapid recovery upon infusion termination, even after prolonged continuous infusion.[18] There have been no reports detailing the cardiovascular effects of AZD-3043. Given its structural similarity to propanidid, it seems likely that it will produce similar reductions in blood pressure. Clinicaltrials.gov records that three phase 1 clinical trials have been completed, but the results have not been reported. A planned fourth clinical trial focusing on elderly patients was withdrawn and it appears that AstraZeneca (Wilmington, DE, USA) is reconsidering further development of this compound.
HIGH-THROUGHPUT SCREENING
The above-described approach of altering existing, efficacious chemotypes to modulate activity, either on- or off-pathway, has a fairly high history of success. However, it will always be limited to some degree by the sterics or physicochemistry of the scaffold itself. More specific and efficacious drugs that might exist in compound space will always remain hidden. Thus, in order to broaden the search for these novel chemotypes, unbiased screening of large compound sets has become both possible and popular, and many examples of successful such approaches are available. However, this approach has not yet been reported as a pathway to new general anesthetics, thus the discussion here will use our recent work to illustrate the process itself, rather than any new compound or chemotype that has resulted.
The screen starts with an assay that is amenable to miniaturization and deployment in robotic, high-throughput mode. The key for a successful screen, however, is the mechanistic proximity of the assay used to the phenotypic activity desired. Herein lies the Achilles-heel of using this approach for general anesthetics—the targets and mechanisms are likely multiple and redundant. We describe a recent approach that used a surrogate target in the assay to illustrate the steps, problems, and potential solutions.
The first step in establishing the high-throughput assay is to select a drug target. In our case, a protein was selected that binds general anesthetics in a way that matches their in vivo EC50. Although this protein, apoferritin, is very unlikely to be involved in on-pathway effects of general anesthetics, this strong correlation between binding affinity and in vivo potency suggests physicochemical mimicry of the crystallographically proven apoferritin binding site to that of the actual physiologic targets.[36,62] Having selected a target, the next step is to develop a reporter system for binding in high-throughput mode. Typically, this is an optical readout—either fluorescence or absorbance. Fortunately, we had been developing a fluorescent anesthetic for imaging purposes[19] and repurposed it for a screening application. The compound, 1-aminoanthracene (1-AMA), is not only a reversible general anesthetic in both amphibia and mammals, but when bound in a hydrophobic protein cavity, its fluorescence yield is dramatically increased and shifted.[6] In addition, we had already demonstrated that it bound apoferritin with micromolar affinity. Thus, an assay that relied on competition with 1-AMA was developed and miniaturized to 384-well format for preliminary reagent testing, then to 1536-well format for quantitative high-throughput screening (qHTS).[28]
Performance of the assay is typically tested using small, validated compound sets consisting largely of existing pharmaceuticals with proven activities. The 1-AMA screen was tested against the Library Of Pharmacologically Active Compounds-Sigma (LOPAC) set, a collection of 1280 compounds, including at least one general anesthetic, propofol.[34] Each compound is tested at 7 concentrations, 77 μM to 25 nM, in order to prepare concentration–response curves and allow compound triage and determination of IC50 (fluorescence is inhibited with compound binding). Based on the character of the curves, compounds were classified as inactive or inconclusive (1074), or active (206). Of the active class, only 5 compounds had an inhibitory response greater than 60% at the highest concentration, and thus were considered “top actives”; this latter group included propofol. The others were a mixture of generally small hydrophobic molecules. These were further validated as binding to apoferritin using another approach (isothermal titration calorimetry), but were not characterized in in vivo studies.
Having validated the assay and its reliability in high-throughput mode, we moved on to conduct a larger, entirely automatic, robotic screen at the National Chemical Genomics Center (Rockville, MD, USA). In this case, the entire molecular libraries collection was screened, involving 351,367 compounds tested at 5 concentrations ranging from 7 to 150 μM. A total of 1509, 1536-well plates were used in this fully automated assay, a process that required only 4 days. Each plate contained positive [propofol, Figure 5] and negative (no compound) control wells, and the cross plate reproducibility was excellent. A total of ~7% of the entire library was found to consistently decrease the 1-AMA fluorescence signal, but less than a tenth of these decreased the signal more than 60% at the highest concentration [Figure 6]. Thus, 0.64% (2563) of the compounds were considered “top actives,” and these were pared down to less than 100 through a combination of chemotype clustering, cherry picking and re-assay [Figure 7] shows a group of concentration–effect curves from one cluster.
Figure 5.
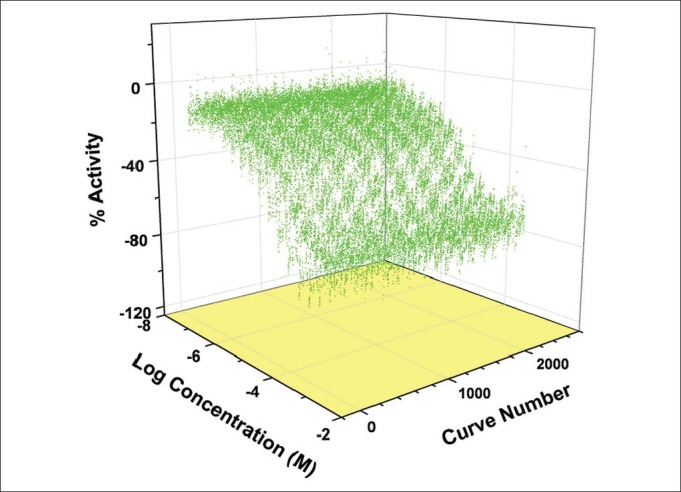
Positive control performance in 1536-well format in quantitative high-throughput screening mode. Each curve is a titration of the 1-AMA/apoferritin mixture with propofol from 7 to 150 μM from each of the 1509 separate plates. The average IC50 = 23 μM and MSR of 3.3
Figure 6.
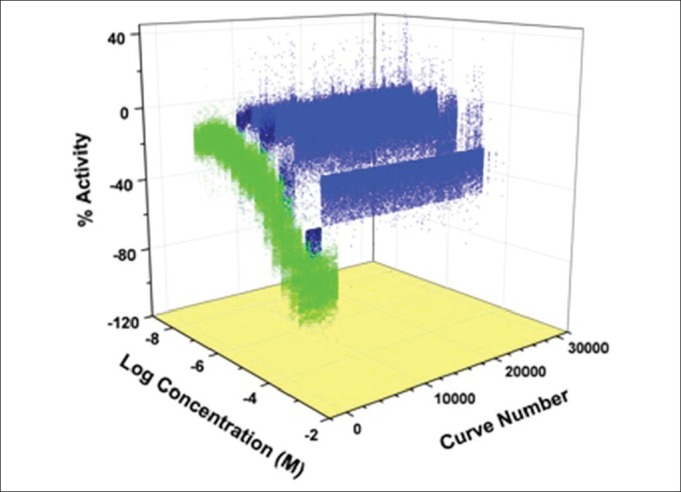
A 3D plot of the active group from the full screen. This set of curves represents about 7%, or almost 25,000 of compounds that showed inhibition of the 1-AMA fluorescence. The green curves are the positive controls (propofol), and the light blue are those that showed weak inhibition. The “top actives,” or those that inhibited greater than 60%, are the dark blue group, representing about 2500 compounds
Figure 7.
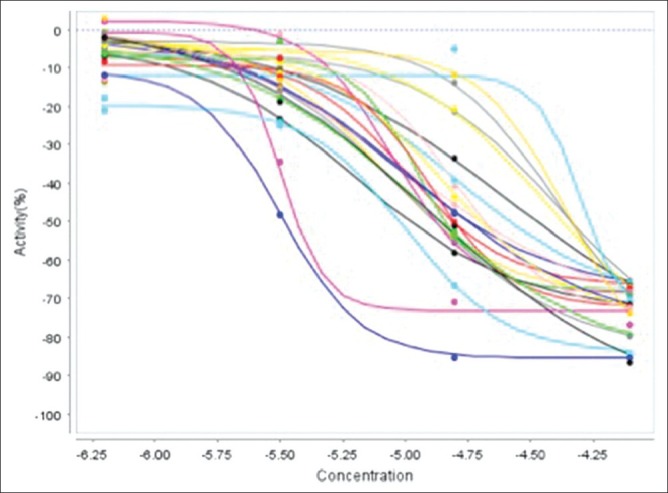
Concentration effect curves from the quantitative high-throughput screening run of a group of top-active compounds clustered on chemotype. Despite showing greater than 10-fold differences in potency, note compounds in this cluster inhibited greater than 60%, suggesting that further attention be devoted to this chemotype
At this point, we have a group of fairly diverse compounds selected only from a fluorescence, surrogate-based assay that has little to do with the activity sought. There are several confounders to a light-based assay that need to be considered before we even judge these as true binders of apoferritin, let alone having anesthetic activity. In other words, is the decrease in fluorescence intensity always caused by competitive binding with 1-AMA? For example, if the test compound absorbs light strongly at the 1-AMA excitation or emission wavelength, the fluorescence will be reduced independent of any effect on binding, producing false positives. This effect, known as "inner filter" can be calculated, if the absorption spectra are available for each compound, but appears to be small for most of the top actives. Fortunately, binding can be measured by an independent approach (such as isothermal titration calorimetry, or plasmon resonance spectroscopy) to verify that these compounds were indeed active in the surrogate assay. This step helps to clarify the validity of the assay used, which is especially important in this case as it relies on a surrogate target.
There also exists the possibility of false negatives, and these are much harder to detect due to the vast number of apparently inactive compounds. One prominent source of this error acknowledges that many of the compounds tested are quite hydrophobic, and may aggregate at the highest concentrations used in this project, which would effectively reduce the free concentration and thus the degree of competition. Also, although less likely, is the possibility that the compounds may themselves fluoresce, masking decreases in 1-AMA fluorescence, or even increasing it via FRET reactions.
After undertaking secondary validation experiments to address at least the false-positive problem, the most important issue remaining is whether the biologic activity sought, anesthesia, is enriched within the top active compounds. Because apoferritin appears to bind general anesthetics with GABAA co-agonist activity,[62,40] we considered an intermediate receptor-based assay, flunitrazepam binding to neuronal membranes. This notion was considered too restrictive in that the goal was not better GABAA co-agonists, but rather the broader goal of better general anesthetics. Thus, we jumped immediately to a phenotypic assay to evaluate anesthetic activity of our top active list.
Testing even 100 compounds in a rodent model would be difficult due to the mass of the often scarce compound required for testing, and the time and number of animals required. An intermediate organism was desired that could be evaluated with small amounts of compound and in high numbers quickly. The Xenopus tadpole was chosen as it is a complex vertebrate, and has been used as an anesthetic-testing organism for many years.[32] It exhibits reversible immobility to general anesthetic compounds at essentially the same concentrations as mammals. Thus, compounds were first screened in groups of 10 tadpoles at 100 μM compound in their pond water for an hour of exposure. The compound was eliminated if immobility did not occur in 100% of tadpoles. The remaining compounds were tested at 10 μM and then at 1 μM. Compounds were eliminated if they caused acute or delayed mortality at a concentration of 1 μM, or if mortality occurred in a delayed manner in the absence of 100% immobility acutely. Of the ~100 initial compounds, all have been screened in this manner, with a couple novel chemotypes producing reversible immobility in tadpoles to <10 μM. These compounds were then subjected to medicinal chemistry in attempts to further enhance activity. A probe compound representing a single novel chemotype has been selected for further optimization.
In summary, this process demonstrates that quantitative high-throughput screening is of very low yield, but the revealed compounds are of completely unique character to the field of anesthesia. The importance of the initial assay is demonstrated here in that only a small minority of the top active list turned out to be reversible immobilizers of tadpoles. This suggests that at least for activities not yet well associated with a target, or for those associated with multiple targets, phenotypic screening[33] might be a better, albeit lower, throughput approach than surrogate target qHTS screening.
CONCLUSION
The novel anesthetics on the horizon have been developed through the targeted modification of existing compounds in manners that improve their pharmacodynamic and/or pharmacokinetic properties. One common molecular paradigm for pharmacokinetic improvement is the transformation of the parent compound to a soft drug through the addition of an ester linkage, increasing its susceptibility to metabolism by nonspecific esterases in the bloodstream. Remifentanil and esmolol are prototypical soft drugs and remimazolam and MOC-etomidate are examples of novel anesthetic agents that make use of this approach. An alternative approach is to modify the structure of the parent compound to alter its pharmacodynamic effects, as in the case of carboetomidate.
qHTS is a newer and different approach to anesthetic development that involves the testing of hundreds of compounds for interaction with a surrogate target for anesthetic activity. Although of low yield without a true phenotypic screen, the technique offers the potential to identify completely novel compounds with anesthetic activity.
ACKNOWLEDGMENTS
Funded by R01-GM087316, R21-DA029253, R03-MH84836 and P01-GM55876. Thanks to Weiming Bu, David Liang, and Brian Weiser for their hard work on postscreening aspects of this work, and also to Ganesh Rai, Wendy Lea, Vince Setola, Chris Austin, Anton Simeonov, AjitJadhav, and David Maloney of the NCGC for all their work, tolerance, and support of this unique project.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2013/4/2/2/109179
Contributor Information
Hovig V. Chitilian, Email: hchitilian@partners.org.
Roderic G. Eckenhoff, Email: roderic.eckenhoff@uphs.upenn.edu.
Douglas E. Raines, Email: draines@partners.org.
REFERENCES
- 1.Antonik LJ, Goldwater DR, Kilpatrick GJ, Tilbrook GS, Borkett KM. A Placebo-and midazolam-controlled, phase I, single ascending-dose study evaluating the safety, pharmacokinetics, and pharmacodynamics of remimazolam (CNS 7056): Part I. Safety, efficacy, and basic pharmacokinetics. Anesth Analg. 2012;115:274–83. doi: 10.1213/ANE.0b013e31823f0c28. [DOI] [PubMed] [Google Scholar]
- 2.Bali M, Akabas MH. Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol. 2004;65:68–76. doi: 10.1124/mol.65.1.68. [DOI] [PubMed] [Google Scholar]
- 3.Bennett SN, McNeil MM, Bland LA, Arduino MJ, Villarino ME, Perrotta DM, et al. Postoperative infections traced to contamination of an intravenous anesthetic, propofol. N Engl J Med. 1995;333:147–54. doi: 10.1056/NEJM199507203330303. [DOI] [PubMed] [Google Scholar]
- 4.Bodor N, Buchwald P. Soft drug design: General principles and recent applications. Med Res Rev. 2000;20:58–101. doi: 10.1002/(sici)1098-1128(200001)20:1<58::aid-med3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 5.Boulieu R, Lehmann B, Salord F, Fisher C, Morlet D. Pharmacokinetics of midazolam and its main metabolite 1-hydroxymidazolam in intensive care patients. Eur J Drug Metab Pharmacokinet. 1998;23:255–8. doi: 10.1007/BF03189348. [DOI] [PubMed] [Google Scholar]
- 6.Butts C, Xi J, Brannigan G, Klein ML, Saad AA, Venkatachalan SP, et al. Identification of a fluorescent general anesthetic,1-aminoanthracene. Proc Natl Acad Sci U S A. 2009;106:6501–6. doi: 10.1073/pnas.0810590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campagna-Slater V, Weaver DF. Anaesthetic binding sites for etomidate and propofol on a GABAA receptor model. Neurosci Lett. 2007;418:28–33. doi: 10.1016/j.neulet.2007.02.091. [DOI] [PubMed] [Google Scholar]
- 8.Chiba S, Nishiyama T, Yamada Y. The antinociceptive effects and pharmacological properties of JM-1232(-): A novel isoindoline derivative. Anesth Analg. 2009;108:1008–14. doi: 10.1213/ane.0b013e318193678f. [DOI] [PubMed] [Google Scholar]
- 9.Choi YF, Wong TW, Lau CC. Midazolam is more likely to cause hypotension than etomidate in emergency department rapid sequence intubation. Emerg Med J. 2004;21:700–2. doi: 10.1136/emj.2002.004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Claeys MA, Gepts E, Camu F. Haemodynamic changes during anaesthesia induced and maintained with propofol. Br J Anaesth. 1988;60:3–9. doi: 10.1093/bja/60.1.3. [DOI] [PubMed] [Google Scholar]
- 11.Cotten JF, Forman SA, Laha JK, Cuny GD, Husain SS, Miller KW, et al. Carboetomidate: A pyrrole analog of etomidate designed not to suppress adrenocortical function. Anesthesiology. 2010;112:637–44. doi: 10.1097/ALN.0b013e3181cf40ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cotten JF, Husain SS, Forman SA, Miller KW, Kelly EW, Nguyen HH, et al. Methoxycarbonyl-etomidate: A novel rapidly metabolized and ultra-short-acting etomidate analogue that does not produce prolonged adrenocortical suppression. Anesthesiology. 2009;111:240–9. doi: 10.1097/ALN.0b013e3181ae63d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cotten JF, Le Ge R, Banacos N, Pejo E, Husain SS, Williams JH, et al. Closed-loop continuous infusions of etomidate and etomidate analogs in rats: A comparative study of dosing and the impact on adrenocortical function. Anesthesiology. 2011;115:764–73. doi: 10.1097/ALN.0b013e31821950de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Jong FH, Mallios C, Jansen C, Scheck PA, Lamberts SW. Etomidate suppresses adrenocortical function by inhibition of 11 beta-hydroxylation. J Clin Endocrinol Metab. 1984;59:1143–7. doi: 10.1210/jcem-59-6-1143. [DOI] [PubMed] [Google Scholar]
- 15.den Brinker M, Hokken-Koelega AC, Hazelzet JA, de Jong FH, Hop WC, Joosten KF. One single dose of etomidate negatively influences adrenocortical performance for at least 24h in children with meningococcal sepsis. Intensive Care Med. 2008;34:163–8. doi: 10.1007/s00134-007-0836-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doenicke A, Wagner E, Beetz KH. Arterial blood gas analyses following administration of three short-acting i.v. hypnotics (propanidid, etomidate, methohexitone) Anaesthesist. 1973;22:353–6. [PubMed] [Google Scholar]
- 17.Dundee JW, Robinson FP, McCollum JS, Patterson CC. Sensitivity to propofol in the elderly. Anaesthesia. 1986;41:482–5. doi: 10.1111/j.1365-2044.1986.tb13271.x. [DOI] [PubMed] [Google Scholar]
- 18.Egan TD, Obara S, Jenkins TE, Jaw-Tsai SS, Amagasu S, Cook DR, et al. AZD-3043, A novel, metabolically labile sedative-hypnotic agent with rapid and predictable emergence from hypnosis. Anesthesiology. 2012;116:1267–77. doi: 10.1097/ALN.0b013e31825685a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emerson D, Liao Z, Eckenhoff RG, Dmochowski IJ. A novel fluorescent general anesthetic enables imaging of the in vivo sites of action. Anesthesiology. 2012;116:1363. doi: 10.1097/ALN.0b013e31824cb4b1. [DOI] [PubMed] [Google Scholar]
- 20.Forman SA. Molecular approaches to improving general anesthetics. Anesthesiol Clin. 2010;28:761–71. doi: 10.1016/j.anclin.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forman SA. Clinical and molecular pharmacology of etomidate. Anesthesiology. 2011;114:695–707. doi: 10.1097/ALN.0b013e3181ff72b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fragen RJ, Shanks CA, Molteni A, Avram MJ. Effects of etomidate on hormonal responses to surgical stress. Anesthesiology. 1984;61:652–6. doi: 10.1097/00000542-198412000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Fudickar A, Bein B, Tonner PH. Propofol infusion syndrome in anaesthesia and intensive care medicine. Curr Opin Anaesthesiol. 2006;19:404–10. doi: 10.1097/01.aco.0000236140.08228.f1. [DOI] [PubMed] [Google Scholar]
- 24.Ge R, Pejo E, Husain SS, Cotten JF, Raines DE. Encephalographic and hypnotic recoveries following brief and prolonged infusions of etomidate and optimized soft etomidate analogs. Anesthesiology. 2012;117:1037–43. doi: 10.1097/ALN.0b013e31826d3de2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godefroi EF, Janssen PA, Vandereycken CA, Vanheertum AH, Niemegeers CJ. Dl-1-(1-Arylalkyl) Imidazole-5-Carboxylate Esters. A novel type of hypnotic agents. J Med Chem. 1965;8:220–3. doi: 10.1021/jm00326a017. [DOI] [PubMed] [Google Scholar]
- 26.Huggins DJ, Sherman W, Tidor B. Rational approaches to improving selectivity in drug design. J Med Chem. 2012;55:1424–44. doi: 10.1021/jm2010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Husain SS, Pejo E, Ge R, Raines DE. Modification of methoxycarbonyl etomidate's interester spacer optimizes in vitro metabolic stability and in vivo hypnotic potency and duration of hypnotic action. Anesthesiology. 2012;117:1027–36. doi: 10.1097/ALN.0b013e31826d3bef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, et al. Quantitative high throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103:11473–8. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson WL., Jr Should we use etomidate as an induction agent for endotracheal intubation in patients with septic shock.: A critical appraisal? Chest. 2005;127:1031–8. doi: 10.1378/chest.127.3.1031. [DOI] [PubMed] [Google Scholar]
- 30.Kanamitsu N, Osaki T, Itsuji Y, Yoshimura M, Tsujimoto H, Soga M. Novel water-soluble sedative-hypnotic agents: Isoindolin-1-one derivatives. Chem Pharm Bull (Tokyo) 2007;55:1682–8. doi: 10.1248/cpb.55.1682. [DOI] [PubMed] [Google Scholar]
- 31.Kilpatrick GJ, McIntyre MS, Cox RF, Stafford JA, Pacofsky GJ, Lovell GG, et al. CNS 7056: A novel ultra-short-acting Benzodiazepine. Anesthesiology. 2007;107:60–6. doi: 10.1097/01.anes.0000267503.85085.c0. [DOI] [PubMed] [Google Scholar]
- 32.Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA (A) receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338–51. [PubMed] [Google Scholar]
- 33.Laggner C, Kokel D, Setola V, Tolia A, Lin H, Irwin JJ, et al. Chemical informatics and target identification in a zebrafish phenotypic screen. Nat Chem Biol. 2011;8:144–6. doi: 10.1038/nchembio.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lea WA, Jadhav A, Xi J, Lu L, Austin CP, Simeonov A, et al. A high throughput approach for identification of novel general anesthetics. PLoS One. 2009;4:e7150. doi: 10.1371/journal.pone.0007150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ledingham IM, Watt I. Influence of sedation on mortality in critically ill multiple trauma patients. Lancet. 1983;1:1270. doi: 10.1016/s0140-6736(83)92712-5. [DOI] [PubMed] [Google Scholar]
- 36.Liu R, Loll PJ, Eckenhoff RG. Structural basis for high affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005;19:567–76. doi: 10.1096/fj.04-3171com. [DOI] [PubMed] [Google Scholar]
- 37.Morgan M, Lumley J, Whitwam JG. Etomidate, a new water-soluble non-barbiturate intravenous induction agent. Lancet. 1975;1:955–6. doi: 10.1016/s0140-6736(75)92011-5. [DOI] [PubMed] [Google Scholar]
- 38.Nishiyama T, Chiba S, Yamada Y. Antinociceptive property of intrathecal and intraperitoneal administration of a novel water-soluble isoindolin-1-one derivative, JM 1232 (-) in rats. Eur J Pharmacol. 2008;596:56–61. doi: 10.1016/j.ejphar.2008.07.054. [DOI] [PubMed] [Google Scholar]
- 39.Nordt SP, Clark RF. Midazolam: A review of therapeutic uses and toxicity. J Emerg Med. 1997;15:357–65. doi: 10.1016/s0736-4679(97)00022-x. [DOI] [PubMed] [Google Scholar]
- 40.Oakley S, Vedula S, Xi J, Bu W, Liu R, Eckenhoff RG, et al. Recognition of anesthetic barbituates by a protein binding site: A high resolution structural analysis. PLoS One. 2012;7:e32070. doi: 10.1371/journal.pone.0032070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otterspoor LC, Kalkman CJ, Cremer OL. Update on the propofol infusion syndrome in ICU management of patients with head injury. Curr Opin Anaesthesiol. 2008;21:544–51. doi: 10.1097/ACO.0b013e32830f44fb. [DOI] [PubMed] [Google Scholar]
- 42.Ouellet H, Podust LM, de Montellano PR. Mycobacterium tuberculosis CYP130: Crystal structure, biophysical characterization, and interactions with antifungal azole drugs. J Biol Chem. 2008;283:5069–80. doi: 10.1074/jbc.M708734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pejo E, Cotten JF, Kelly EW, Le Ge R, Cuny GD, Laha JK, et al. In vivo and in vitro pharmacological studies of methoxycarbonyl-carboetomidate. Anesth Analg. 2012;115:297–304. doi: 10.1213/ANE.0b013e3182320559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pejo E, Feng Y, Chao W, Cotten JF, Le Ge R, Raines DE. Differential effects of etomidate and its pyrrole analogue carboetomidate on the adrenocortical and cytokine responses to endotoxemia. Crit Care Med. 2012;40:187–92. doi: 10.1097/CCM.0b013e31822d7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pejo E, Ge R, Banacos N, Cotten JF, Husain SS, Raines DE. Electroencephalographic recovery, hypnotic emergence, and the effects of metabolite after continuous infusions of a rapidly metabolized eomidate analog in rats. Anesthesiology. 2012;116:1057–65. doi: 10.1097/ALN.0b013e3182515403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roumen L, Sanders MP, Pieterse K, Hilbers PA, Plate R, Custers E, et al. Construction of 3D models of the CYP11B family as a tool to predict ligand binding characteristics. J Comput Aided Mol Des. 2007;21:455–71. doi: 10.1007/s10822-007-9128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruesch D, Neumann E, Wulf H, Forman SA. An allosteric coagonist model for propofol effects on alpha1beta2 gamma 2L gamma-aminobutyric acid type A receptors. Anesthesiology. 2012;116:47–55. doi: 10.1097/ALN.0b013e31823d0c36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rusch D, Forman SA. Classic benzodiazepines modulate the open-close equilibrium in alpha1beta2 gamma 2L gamma-aminobutyric acid type A receptors. Anesthesiology. 2005;102:783–92. doi: 10.1097/00000542-200504000-00014. [DOI] [PubMed] [Google Scholar]
- 49.Saari TI, Uusi-Oukari M, Ahonen J, Olkkola KT. Enhancement of GABAergic activity: Neuropharmacological effects of benzodiazepines and therapeutic use in anesthesiology. Pharmacol Rev. 2011;63:243–67. doi: 10.1124/pr.110.002717. [DOI] [PubMed] [Google Scholar]
- 50.Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR. Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl) imidazole at 1.9-A resolution: Insight into the range of P450 conformations and the coordination of redox partner binding. J Biol Chem. 2004;279:27294–301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 51.Seward HE, Roujeinikova A, McLean KJ, Munro AW, Leys D. Crystal structure of the Mycobacterium tuberculosis P450 CYP121-fluconazole complex reveals new azole drug-P450 binding mode. J Biol Chem. 2006;281:39437–43. doi: 10.1074/jbc.M607665200. [DOI] [PubMed] [Google Scholar]
- 52.Siegel LC, Konstantatos A. PF0713 produced rapid induction of general anesthesia without injection pain in a phase 1 study. Anesthesiology. 2009;A463 Available from: http://www.asaabstracts.com/strands/asaabstracts/abstract.htm;jsessionid=DA93833E41FF06A7178351B6D992EC52?year=2008&index=2&absnum=1096 . [Google Scholar]
- 53.Siegel LC, Pelc LR, Shaff K. Dose response of PF0713, a novel investigational intravenous anesthetic agent. Anesthesiology. 2008;A463 Available from: http://www.asaabstracts.com/strands/asaabstracts/abstract.htm;jsessionid=DA93833E41FF06A7178351B6D992EC52?year=2008&index=2&absnum=1096 . [Google Scholar]
- 54.Sigel E. Mapping of the benzodiazepine recognition site on GABA (A) receptors. Curr Top Med Chem. 2002;2:833–9. doi: 10.2174/1568026023393444. [DOI] [PubMed] [Google Scholar]
- 55.Skues MA, Prys-Roberts C. The pharmacology of propofol. J Clin Anesth. 1989;1:387–400. doi: 10.1016/0952-8180(89)90080-9. [DOI] [PubMed] [Google Scholar]
- 56.Sneyd JR, Rigby-Jones AE. New drugs and technologies, intravenous anaesthesia is on the move (again) Br J Anaesth. 2010;105:246–54. doi: 10.1093/bja/aeq190. [DOI] [PubMed] [Google Scholar]
- 57.Sneyd JR, Rigby-Jones AE, Cross M, Tominaga H, Shimizu S, Ohkura T, et al. First human administration of MR04A3: A novel water-soluble nonbenzodiazepine sedative. Anesthesiology. 2012;116:385–95. doi: 10.1097/ALN.0b013e318242b2af. [DOI] [PubMed] [Google Scholar]
- 58.Takamatsu I, Sekiguchi M, Yonamine R, Wada K, Kazama T. The effect of a new water-soluble sedative-hypnotic drug, JM-1232(-), on long-term potentiation in the CA1 region of the mouse hippocampus. Anesth Analg. 2011;113:1043–9. doi: 10.1213/ANE.0b013e3182291782. [DOI] [PubMed] [Google Scholar]
- 59.Uemura S, Fujita T, Sakaguchi Y, Kumamoto E. Actions of a novel water-soluble benzodiazepine-receptor agonist JM-1232(-) on synaptic transmission in adult rat spinal substantia gelatinosa neurons. Biochem Biophys Res Commun. 2012;418:695–700. doi: 10.1016/j.bbrc.2012.01.080. [DOI] [PubMed] [Google Scholar]
- 60.Upton R, Martinez A, Grant C. A dose escalation study in sheep of the effects of the benzodiazepine CNS 7056 on sedation, the EEG and the respiratory and cardiovascular systems. Br J Pharmacol. 2008;155:52–61. doi: 10.1038/bjp.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Upton RN, Martinez AM, Grant C. Comparison of the sedative properties of CNS 7056, midazolam, and propofol in sheep. Br J Anaesth. 2009;103:848–57. doi: 10.1093/bja/aep269. [DOI] [PubMed] [Google Scholar]
- 62.Vedula S, Brannigan G, Economou NJ, Xi J, Hall MA, Dailey WP, et al. A unitary anesthetic protein binding site at high resolution. J Biol Chem. 2009;284:24176–84. doi: 10.1074/jbc.M109.017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vinclair M, Broux C, Faure P, Brun J, Genty C, Jacquot C, et al. Duration of adrenal inhibition following a single dose of etomidate in critically ill patients. Intensive Care Med. 2008;34:714–9. doi: 10.1007/s00134-007-0970-y. [DOI] [PubMed] [Google Scholar]
- 64.Wagner RL, White PF, Kan PB, Rosenthal MH, Feldman D. Inhibition of adrenal steroidogenesis by the anesthetic etomidate. N Engl J Med. 1984;310:1415–21. doi: 10.1056/NEJM198405313102202. [DOI] [PubMed] [Google Scholar]
- 65.Watt I, Ledingham IM. Mortality amongst multiple trauma patients admitted to an intensive therapy unit. Anaesthesia. 1984;39:973–81. doi: 10.1111/j.1365-2044.1984.tb08885.x. [DOI] [PubMed] [Google Scholar]
- 66.Wiltshire HR, Kilpatrick GJ, Tilbrook GS, Borkett KM. A Placebo and Midazolam-Controlled Phase I Single Ascending Dose Study Evaluating the Safety, Pharmacokinetics, and Pharmacodynamics of Remimazolam (CNS 7056). Part II: Population Pharmacokinetic and Pharmacodynamic Modeling and Simulation. Anesth Analg. 2012;115:284–96. doi: 10.1213/ANE.0b013e318241f68a. [DOI] [PubMed] [Google Scholar]