

Summary
Epigenetics is defined as heritable changes to the transcriptome that are independent on changes in the genome. The biochemical principles that govern epigenetic modifications are DNA methylation and posttranslational modifications of histones, amongst which acetylation/deacetylation and methylation/demethylation of lysine residues are the most widely studied. Such epigenetic changes are involved in many physiological and pathological processes. This essay will focus on the biology of histone methylation and demethylation reactions, highlighting how these reactions depend on “metabolic” cofactors like S-Adenosylmethionine, Flavin Adenine Dinucleotide and α-ketoglutarate. Furthermore, we will illustrate that the absence of lysine methyltranferases and demethylases can lead to altered metabolic homeostasis. Altogether, this provides preliminary evidence that lysine methyltranferases and demethylases could act as molecular transducers of metabolic signals to chromatin, and sets the stage for further research into this area to consolidate these enticing initial observations.
Introduction
Increased life expectancy in industrialized countries goes hand-in-hand with a steady progression of chronic multifactorial diseases, such as type 2 diabetes mellitus (T2D), obesity, and hyperlipidemia, which result from the combined action of many genes and environmental factors, such as diet or exercise. Metabolic homeostasis is maintained by an intricate regulatory circuitry, controlled to a large extent by transcriptional mechanisms (Chawla et al., 2001; Francis et al., 2003; Feige and Auwerx, 2007). Metabolism therefore represents a sensitive indicator of the efficiency of these transcriptional mechanisms. Transcriptional control is achieved through a complex molecular circuitry that involves individual transcription factors, the basal transcriptional machinery, and multiprotein coregulatory complexes. These coregulators fine-tune transcriptional processes and are proposed to act as metabolic sensors, which translate changes in metabolism into alterations in gene expression by affecting the activity of transcription factors, as well as changing the structure of the epigenome (reviewed by Smith and O’Malley, 2004; Spiegelman and Heinrich, 2004; Rosenfeld et al., 2006; Feige and Auwerx, 2007 and Haberland et al. 2009).
Within the eukaryotic cell nucleus, genetic information in the DNA is organized in a highly conserved structural polymer, termed chromatin, which supports and controls the functions of the genome. The fundamental repeating unit of chromatin is the nucleosome, which consists of 146 base pairs of DNA wrapped around an octamer of core histone proteins (an H3-H4 tetramer and two H2A-H2b dimers) (recently reviewed by Laskowski and Thornton, 2008). Linker histones of the H1 class associate with DNA between single nucleosomes establishing a higher level of organization, the so-called ‘solenoid’ helical fibers (30 nm fibers) (Laskowski and Thornton, 2008). Core histone proteins are evolutionarily conserved and consist of a globular domain and a flexible charged N-terminal histone tail, which is covalently modified by different enzymes mainly at specific lysine and/or arginine residues and constitutes, together with DNA methylation pattern, the “histone code” which is then properly read and translated into transcriptional changes (reviewed by Jenuwein and Allis, 2001; Berger, 2007; Kouzarides, 2007 and Ruthenburg et al., 2007). The best-known and most widely studied covalent modifications of the histone tail include acetylation, phosphorylation, methylation (Fig.1A) and ubiquitination. Depending on nucleosome condensation, chromatin is more or less accessible to different enzymes and proteins, most of which regulate gene expression. Euchromatin is generally referred to transcriptionally active relaxed chromatin, while the silent inaccessible chromatin is called heterochromatin. Chromatin-modifying enzymes regulate nucleosome condensation and protein accessibility to chromatin. For the purposes of transcription, histone modifications can be divided into those that correlate with activation and those that correlate with repression even though the truth is likely to be that any given modification can activate or repress genes under different conditions, underscoring the context dependence of these histone modifications (Vakoc et al., 2005; Berger, 2007; Bernstein et al., 2007; Kouzarides, 2007; Ruthenburg et al., 2007).
Figure 1.
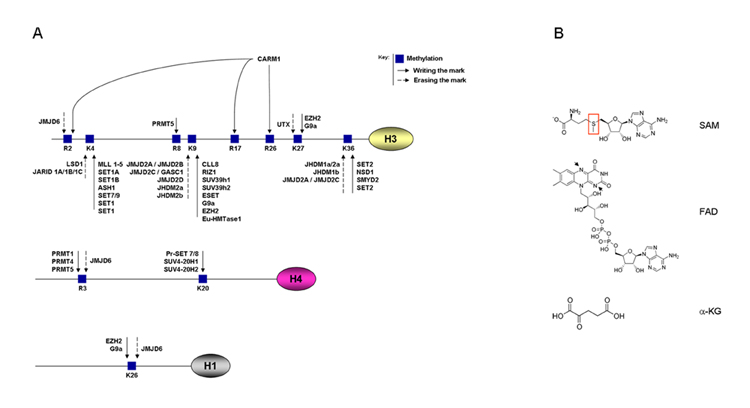
(A) Schematic representation of post-translational modifications of histone proteins with a particular focus on histone methylation. (B) Chemical structures of SAM, FAD and α-KG. Red ring on SAM highlights the reactive sulfur atom. Arrows on FAD highlight the nitrogen atoms, which are protonized in FADH2
Since the well established role of DNA methylation (reviewed by Suzuki and Bird, 2008 and Ling and Groop, 2009) and the emerging function of histone acetyltransferases and deacetylases (reviewed by Smith and O’Malley, 2004; Spiegelman and Heinrich, 2004; Rosenfeld et al., 2006; Feige and Auwerx, 2007 and Haberland et al., 2009) in the control of gene expression and metabolism has been already reviewed, we will focus here on the existing evidence for a potential role of histone methylation in metabolic adaptation.
Histone methylation and demethylation in the control of gene expression
Histone methylation increases the basicity and hydrophobicity of the histone tail and the affinity of certain proteins, such as transcription factors, toward DNA. There are three classes of histone methyltransferase (HMTs) enzymes: SET domain lysine methyltransferases, non-SET domain lysine methyltransferases, and arginine methyltransferases. All three classes use S-adenosylmethionine (SAM) as a coenzyme to transfer methyl groups (Smith and Denu, 2009). Lysine methyltransferases have striking target specificity and they usually modify one single lysine on a single histone and their output can be either the activation or repression of transcription.
The SET domain-containing class of methyltransferases is the best characterized and has been associated with metabolic diseases (Lee J. et al., 2008; El-Osta et al., 2008 and Brasacchio et al., 2009). The SET domain is an evolutionary conserved domain, initially identified in Drosophila PEV (positive effect variegation) suppressor SU(VAR)39 (Tschiersch et al., 1994), the polycomb group protein Enhancer of zeste (Jones and Gelbart, 1993) and the trithorax group protein Trithorax (Stassen et al., 1995). The major role of these proteins is thought to be modulation of gene activity via histone methylation and alteration of chromatin structure (Rea et al., 2000). Several non-histone proteins have also been identified as target of methyltransferases. In particular, the tumor suppressors p53 (Chuikov et al., 2004; Huang et al., 2010) and pRb (Munro et al., 2010) and the estrogen receptor alpha (ERα) (Subramanian et al., 2008) are substrates of SET-domain containing methyltransferases, while the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is a substrate of the arginine methyltransferase CARM1 (Teyssier et al., 2005).
For a number of years following the discovery of HMTs, the existence of histone demethylases (HDMs) was contentious. The discovery of the first HMD, Lysine Specific Demethylase 1 (LSD1) (Shi et al., 2004), has led to the identification of additional demethylases. These enzymes are distinguished by the nature of their demethylase domains, with distinct reactions being catalyzed by the LSD1 domain and the JmjC (Jumonji C) domain demethylases. LSD1 is a highly conserved protein, homologous to other FAD-dependent oxidases (like the monoamine oxidases involved in serotonin metabolism), that consists of two subdomains: a FAD-binding and a substrate-binding domain (Anand and Marmorstein, 2007). LSD1 demethylates monomethylated H3K4 and represses transcription (Shi et al., 2004). However, when LSD1 is present in a complex with the androgen receptor, it demethylates monomethylated H3K9 and activates transcription (Metzger et al., 2005). It is important to point out that H3K9, as well as H3K36, can also be demethylated by the JmjC-containing demethylases, which are generally known as transcriptional activators (Tsukada et al., 2006).
The jumonji gene was identified by a mouse gene trap approach and is essential for the development of multiple tissues (Takeuchi, 1997). Over 100 proteins from many species, including bacteria, fungi, plants and animals contain a JmjC domain, a predicted metalloenzyme catalytic motif. More recently the JmjC-domain containing proteins were identified as transcriptional coactivators involved in chromatin-dependent transcriptional regulation by demethylating H3K9 (Tsukada et al., 2006). Five JmjC domain subfamilies maintain histones demethylated, i.e. JHDM1, JHDM2, JMJD2, JMJD3 and JARID1 (Tsukada et al., 2006). Recently, another JMJC-domain containing protein, JMJD6, was identified as a histone demethylase with specificity to H3R2me2 and H4R3me2, demonstrating that also arginine methylation marks are reversible changes (Chang et al., 2007). JmjC-domain-containing enzymes convert their substrates in a manner different from that of LSD1 and are able to act on trimethylated lysines as well (Smith and Denu, 2009). Their demethylation activity depends on two coenzymes, α-ketoglutarate, which stabilizes the enzyme/substrate complex, and Fe2+ ions (Tsukada et al., 2006). Furthermore, it needs to be stressed that although the demetylation of H3 starts to be understood, the proteins that demethylate other histone proteins are still largely unknown, providing a fertile terrain of future research (Fig.1A).
The production of cofactors for HMTs and HDMs are dependent on the cellular energy status
Both HMTs and HDMs depend, for their activity, on the presence of specific metabolic coenzymes (Tsukada et al., 2006; Anand and Marmorstein, 2007; Smith and Denu, 2009), whose biosynthesis is dependent on intracellular ATP levels (Fig.1B and 2A).
Figure 2.
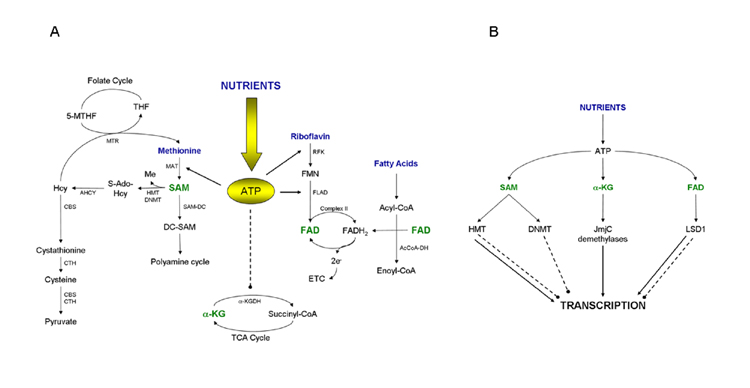
(A) Schematic representation of how cofactor biosynthesis is dependent on intracellular energy status. The ATP produced by the intracellular metabolism of nutrients is the key molecule for the biosynthesis of cofactors. MAT converts methionine to SAM by using ATP. RFK and FLAD use also ATP to synthesize FAD from riboflavin. High intracellular ATP levels in contrast inhibit α-KGDH, a key enzyme in the TCA (tri-carboxylic-acid cycle) that converts α-ketoglutarate to succinyl-CoA (Smith et al., 1974). (B) Schematic representation of how intracellular nutrient signals are integrated to reprogram transcription. Solid arrows indicate activation of transcription, dotted lines indicate inhibition of transcription.
Words depicted in blue indicate precursors for coenzymes biosynthesis; words in green indicate coenzymes directly involved in methylation (SAM) and/or demethylation (FAD and α-KG) reactions.
S-Adenosyl-methionine (SAM) is the common coenzyme involved in reactions with methyl group transfer (Smith and Denu, 2009). The methyl group attached to the sulfur atom in SAM is chemically reactive (Fig.1B). This allows donation of this group to an acceptor substrate in transmethylation reactions, catalyzed by methyltransferases and involving various acceptors like nucleic acids, proteins and lipids. SAM is produced in the cytosol by the reaction between methionine and ATP catalyzed by S-adenosyl methionine transferase (MAT) and links energy homeostasis to the methylation of lysines and arginines in proteins and cytosines in DNA (Fig.2A). Interestingly, recently MAT was shown to be present in the nucleus, where its presence correlated with histone H3K27 trimethylation, suggesting that it controlled the local supply of SAM (Reytor et al. 2009). By taking part in methylation reactions, SAM is demethylated to S-Adenosyl-Homocysteine (S-AdoHCy), which is then converted to homocysteine (Hcy) by S-adenosyl-homocysteinase (AHCY). Hcy can then be either recycled back to methionine by 5-methyltetrahydrofolate-homocysteine methyl transferase (MTR) through transfer of a methyl group from 5-methyltetrahydrofolate (5-MTHF) (Finkelstein and Martin, 2000), or fully metabolized to cysteine and pyruvate by two rounds of transsulfuration reactions catalyzed by cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CTH) (Finkelstein and Martin, 2000). High intracellular energy levels increase cellular SAM concentrations and both DNA methyl transferase (DNMT) and HMT activity, with a potential epigenetic impact on gene regulation (Chiang et al., 2009).
Besides its role as methyl donor in methylation reactions, methylation independent SAM metabolism can also influence transcription. SAM can be decarboxylated by the SAM decarboxylase (SAM-DC) to decarboxy-SAM (DC-SAM) and enter the polyamine cycle (Fig.2A). Polyamines are abundant multivalent organic cations, largely bound to RNA and DNA and known to regulate many cellular functions like transcription and cell growth. Interestingly, mice transgenic for the rate-limiting enzyme in the polyamine pathway, spermine/spermidine acetyltransferase (SSAT), have an improved metabolic profile (Pirinen et al., 2007). Polyamine flux is accelerated in these mice resulting in enhanced ATP consumption and highlighting a role for polyamine catabolism in the regulation of energy metabolism (Pirinen et al., 2007).
To catalyze demethylation reactions, the LSD1 or the Jumonji domain classes of HDMs require different metabolic cofactors, flavin adenine dinucleotide (FAD) (Anand and Marmorstein, 2007) and α-ketoglutarate (α-KG) (Tsukada et al., 2006), respectively. FAD is a redox coenzyme existing in two different redox states, which is involved in several important reactions in metabolism. FAD consists of a riboflavin moiety (vitamin B2) bound to the phosphate group of an ADP molecule (Fig.1B), thus clearly requiring ATP to be synthesized. Starting from riboflavin, flavin mononucleotide (FMN) is first generated by riboflavin kinase (RFK)-dependent phosphorylation. FMN is then converted to FAD by FAD-synthase (FLAD), which transfers an AMP moiety from an ATP molecule to the FMN (Fig.2A). The reduced form of FAD, FADH2, is an energy carrier. When oxidized back to FAD, FADH2 sends its two high energy electrons through the electron transport chain (ETC) to produce 1,5 equivalents of ATP by oxidative phosphorylation. Any oxidoreductase enzyme that, like the histone demethylase LSD1, uses FAD as an electron carrier is called a flavoprotein. Flavoproteins are essential in many metabolic reactions, as illustrated by the enzyme complex succinate dehydrogenase (complex II) that oxidizes succinate to fumarate in the citric acid cycle, thereby reducing FAD to FADH2. Other well-studied flavoproteins include acyl CoA dehydrogenase, α-ketoglutarate dehydrogenase (α-KGDH) and a component of the pyruvate dehydrogenase complex, illustrating the tight link between LSD1 and intermediary metabolism (Fig.2A).
α-Ketoglutarate (Fig.1B) (α-KG), the coenzyme of the JmjC class of HDMs (Tsukada et al., 2006), is a key intermediate in the Krebs cycle, coming after isocitrate and before succinyl CoA. Anaplerotic reactions can replenish the cycle at this juncture by synthesizing α-KG through the action of glutamate dehydrogenase on glutamate. α-Ketoglutarate is the substrate of the α-KGDH reaction by which it is converted in succinyl-CoA in the presence of FAD and NAD (Fig.2A). This reaction is a critical and highly regulated step of the Krebs cycle mainly controlled by feedback regulation. Succinyl-CoA, FADH2 and NADH, have all been shown to inhibit the reaction (Smith et al., 1974). Interestingly, α-KGDH is also inhibited by high intracellular ATP content (Smith et al., 1974) (Fig.2A).
Based on the dependence of the HMT and HDMs on metabolic cofactors, whose availability on their turn is governed by the intracellular energy content, these effectors of reversible methylation are well positioned to reprogram gene expression in function of the metabolic milieu.
SET7 – LSD1: methylation / demethylation in the pathogenesis of metabolic disorders
The first SET-domain HMT to be studied for its role in metabolism, was MLL3, a member of the mixed-lineage leukemia (MLL) family (Ansari and Mandal, 2010). MLL3 is a component of the ASC-2/NCOA6 (ASCOM) complex, which possesses histone methylation activity toward H3K4 and is involved in transcriptional coactivation (Lee J. et al., 2008). MLL3−/− mice have significantly less white adipose tissue, associated with a favorable overall metabolic profile, likely achieved through the inhibition of adipogenesis. The MLL3−/− mice are also resistant to high-fat diet-induced hepatosteatosis because of the role of ASCOM in coactivating the Liver X Receptor (LXR) (Lee S. et al., 2008). These studies on MLL3 were amongst the first to link histone methylation with certain features of metabolic diseases.
The SET7 HMT has been recently shown to be involved in the pathogenesis of vascular complications of type1 and type 2 diabetes (El-Osta et al., 2008; Brasacchio et al., 2009). Both in vitro and in vivo, transient hyperglycemia causes a sustained expression and activation of the transcription factor NFkB-p65. This is associated on the one hand with SET7 recruitment and H3K4 monomethylation (H3K4me1) (El-Osta et al., 2008) and on the other hand with persistent H3K9 demethylation and recruitment of LSD1 to the promoter of the NFkB-p65 gene (Brasacchio et al., 2009). Although not analyzed, it is tempting to speculate that the transient hyperglycemia, by increasing the intracellular ATP content, boosts SAM levels, which then could activate the SET7 methyltransferase. Along a similar line, the oxidative phosphorylation-induced production of FAD could also activate LSD1.
LSD1 has also been reported to be involved in the pathogenesis of vascular complications of diabetes (Reddy et al., 2008). In mouse Vascular Smooth Muscle Cells (mVSMC) from db/db mice, a mouse model of diabesity, both the basal and Tumor Necrosis Factor-α (TNFα)-induced expression of the inflammatory genes Monocyte Chemoattractant Protein 1 (MCP-1) and Interleukin 6 (IL-6) are increased. This is associated with an increase in the activating H3K4 dimethylation mark and reduced LSD1 recruitment to the promoters of the genes. Interestingly, high-glucose treatment recapitulates this in vivo observation in cultured human VSMC, suggesting that glucose can by itself increase the expression of inflammatory genes, like MCP-1 and IL-6, through alterations in histone methylation and affecting the metabolic memory of VSMCs.
Although this interesting set of studies suggests a metabolic function of HMTs and HDMs, they are far from nailing that point and they raise many additional questions. In fact, none of these studies provided hard biochemical evidence that the levels of the metabolic cofactors, SAM (for the SET-domain HMTs) and FAD (for LSD1) were affected by the metabolic changes - a conditio sine qua non to establish the link between methylation/demethylation and metabolism. Furthermore, it is unclear how these HMTs and HDMs are selectively targeted to certain promoters (e.g. NFkB-p65 gene) to achieve highly specific effects on certain histone methylation sites but not others. From the apparent contradictory results, showing on the one hand LSD1 recruitment and persistent demethylation of H3K9 (Brasacchio et al., 2009) or on the other hand LSD1 depletion and hypermethylation of H3K4 (Reddy et al., 2008), it also emerges that the role of high glucose levels in the control of LSD1 activity and methylation is complex and contentious.
JHDM2a/KDM3A (JmjC-domain-containing histone demethylase 2A/Lysine Demethylase 3A): a novel potential key regulator of energy homeostasis
One of the best characterized JmjC-domain-containing histone demethylases is JHDM2a/KDM3A, which has been recently demonstrated to play a role in the regulation of metabolic gene expression. Two coinciding studies, show that JHDM2a−/− mice develop adult-onset obesity associated with reduced energy expenditure and fat oxidation (Tateishi et al., 2009; Inagaki et al., 2009). The mechanisms proposed in these two studies are different. In the first study, the authors attribute the obesity observed in JHDM2a−/− mice to impaired brown adipose tissue function, likely due to a reduced expression of genes involved in the control of energy metabolism, as UCP1 and PPARα and an impaired β-adrenergic signaling (Tateishi et al., 2009). Interestingly, the JHDM2a−/− mice developed by Inagaki, showed also a full blown metabolic syndrome, but differently from Tateishi et al., these investigators did not find alterations in brown adipose tissue (Inagaki et al., 2009), In contrast, they showed that a set of 30 genes was specifically downregulated in white adipose tissue, including the anti-adipogenic transcription factor COUP-TFII (Xu et al., 2008), an inhibitor of fat storage like ApoC1 (Jong et al., 2001), the insulin-dependent glucose transporter GLUT4 (Stenbit et al., 1997) and ADAMTS9, a gene associated to type 2 diabetes by meta-analysis of genome-wide association studies (Zeggini et al., 2008). It is tempting to speculate that in situation of energy affluence, the accumulation of the metabolic cofactor α-KG within the cell, which activates the JmjC-domain-containing HDMs (Tsukada et al., 2006) that function as transcriptional coactivators, contributes to the induction of ATP consuming proteins (like UCP1) or anti-adipogenic factors (like COUP-TFII). When this regulatory process is impaired in vivo in JHDM2a−/− mice, one can imagine that an imbalance of intracellular energy homeostasis will ensue, which translates into metabolic phenotypes like obesity. Taken together these studies constitute preliminary evidence that JmjC-domain-containing HDMs could integrate metabolic signals with transcription, but unfortunately they did not include the level of metabolic information required to establish this link beyond reasonable doubt.
Conclusions and future perspectives
The epigenome has been hypothesized to provide an important part of the interface between the environment and the regulation of gene expression (reviewed by Ladurner, 2006 and Feinberg, 2007). Clearly, one of the most important factors in the environment is the availability of calories (energy). Energy-rich substrates, such as carbohydrates and fats, are converted into ATP, with concomitant increases in the levels of metabolites such as acetyl-CoA, NAD+/NADH, SAM, α-KG, and FAD by processes like glycolysis, fatty acid oxidation and oxidative phosphorylation. Mitochondria are crucial in many aspects of the transformation of these calories into usable forms of energy. The above mentioned metabolites, in turn, are the high-energy substrates used as coenzymes by chromatin and DNA modifying enzymes to drive epigenetic modifications and alter gene expression (Ladurner, 2006; Tsukada et al., 2006; Anand and Marmorstein, 2007; Feige and Auwerx, 2007; Chang et al., 2009; Wellen et al., 2009). Sensing these intermediary factors is hence the principal way, how the cell is informed about the availability of calories.
For the lysine acetyl transferases (KATs) and deacetylases (KDACs), solid evidence is emerging that they act as real energy sensors, that are respectively activated by high and low intracellular energy levels, and translate these metabolic signals into alterations in chromatin structure and transcription (reviewed by Feige and Auwerx, 2007, Dominy et al., 2009; Jeninga et al., 2010 and Zhao et al., 2010). Furthermore, the characterization of these KATs and KDACs, has unequivocally demonstrated a role for some of these metabolic sensors in the control metabolic flexibility. This suggest also that these KAT and KDACs could contribute to the pathogenesis of common complex disorders of the metabolic, cardiovascular and nervous systems, which all show an increased incidence in “westernized” societies.
In analogy to the KATs and KDACs, and supported by the preliminary evidence reviewed here, it is reasonable to speculate that enzymes, like the HMTs, HDMs, could fulfil similar functions (Fig.2B). From the phenotypic characterization of various genetically engineered mouse models for the HMTs and HDMs, it became clear that their absence translates into striking metabolic abnormalities, underscoring the fact that they control metabolic processes. Furthermore, the dependence of HMTs and HDMs on metabolic coenzymes, such as SAM, FAD, and α-KG, is well-established biochemically. What is, however, missing for the HMTs and HDMs is evidence how the rapidly changing physiological and developmental homeostatic context affects the dynamic levels of their metabolic coenzymes, and how this then is integrated by the HMTs and HDMs to alter “transcriptional homeostasis” in a highly specific manner. Within this context, particular attention should be given to solve the apparent contradiction that the cellular energy levels positively regulate the synthesis of both coenzymes for HMTs and HDMs. Furthermore, clarification is required about how these nuclear enzymes capture these metabolic coenzymes, which are often produced in different cellular compartments (mitochondria and cytoplasm). We predict that future studies aimed at altering the levels and compartmentalization of these intermediary factors through, physiological, pharmacological and genetic approaches will shed light on their relevance to synchronize histone methylation/demethylation control transcription with the metabolic milieu and to maintain cellular homeostasis across generations. A better understanding of the biology and activity of these HMT and HDM enzymes may help not only to understand their role in metabolism, but also to exploit them perhaps as new targets for prevention and treatment of metabolic diseases.
Acknowledgements
This work was supported by grants of the Ecole Polytechnique Fédérale de Lausanne, Swiss National Science Foundation, NIH (DK59820), and the European Research Council Ideas programme (Sirtuins; ERC-2008-AdG23118).
References
- Anand R, Marmorstein R. Structure and mechanism of lysine-specific demethylase enzymes. J. Biol. Chem. 2007;282:35425–35429. doi: 10.1074/jbc.R700027200. [DOI] [PubMed] [Google Scholar]
- Ansari KI, Mandal SS. Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J. 2010;277:1790–1804. doi: 10.1111/j.1742-4658.2010.07606.x. [DOI] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, Calkin AC, Brownlee M, Cooper ME, El-Osta A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58:1229–1236. doi: 10.2337/db08-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- Chiang EP, Wang YC, Chen WW, Tang FY. Effects of insulin and glucose on cellular metabolic fluxes in homocysteine transsulfuration, remethylation, S-adenosylmethionine synthesis, and global deoxyribonucleic acid methylation. J. Clin. Endocrinol. Metab. 2009;94:1017–1025. doi: 10.1210/jc.2008-2038. [DOI] [PubMed] [Google Scholar]
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- Dominy JE, Jr, Lee Y, et al. Nutrient-dependent regulation of PGC-1[alpha]’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim. Biophys. Acta - Proteins & Proteomics. doi: 10.1016/j.bbapap.2009.11.023. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008;205:2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17:292–301. doi: 10.1016/j.tcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Finkelstein J, Martin J. Homocysteine. Int J Biochem Cell Biol. 2000;32:385–389. doi: 10.1016/s1357-2725(99)00138-7. [DOI] [PubMed] [Google Scholar]
- Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annual Review in Physiology. 2003;65:261–311. doi: 10.1146/annurev.physiol.65.092101.142528. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Dorsey J, Chuikov S, Pérez-Burgos L, Zhang X, Jenuwein T, Reinberg D, Berger SL. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285:9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T, Tachibana M, Magoori K, Kudo H, Tanaka T, Okamura M, Naito M, Kodama T, Shinkai Y, Sakai J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes to Cells. 2009;14:991–1001. doi: 10.1111/j.1365-2443.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- Jeninga EH, Schoonjans K, Auwerx J. Reversible acetylation of PGC-1 – Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene. 2010 doi: 10.1038/onc.2010.206. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jones RS, Gelbart WM. The Drosophila Polycomb-group gene Enhancer of zeste contains a region with sequence similarity to trithorax. Mol. Cell. Biol. 1993;13:6357–6366. doi: 10.1128/mcb.13.10.6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong MC, Voshol PJ, Muurling M, Dahlmans VE, Romijn JA, Pijl H, Havekes LM. Protection from obesity and insulin resistance in mice overexpressing human apolipoprotein C1. Diabetes. 2001;50:2779–2785. doi: 10.2337/diabetes.50.12.2779. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Ladurner AG. Rheostat control of gene expression by metabolites. Mol. Cell. 2006;24:1–11. doi: 10.1016/j.molcel.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Thornton JM. Understanding the molecular machinery of genetics through 3D structures. Nat. Rev. Genet. 2008;9:141–151. doi: 10.1038/nrg2273. [DOI] [PubMed] [Google Scholar]
- Lee J, Saha PK, Yang QH, Lee S, Park JY, Suh Y, Lee SK, Chan L, Roeder RG, Lee JW. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc. Natl. Acad. Sci. USA. 2008;105:19229–19234. doi: 10.1073/pnas.0810100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee J, Lee SK, Lee JW. Activating signal cointegrator-2 is an essential adaptor to recruit histone H3 lysine 4 methyltransferases MLL3 and MLL4 to the liver X receptors. Mol. Endocrinol. 2008;22:1312–1319. doi: 10.1210/me.2008-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58:2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Munro S, Khaire N, Inche A, Carr S, La Thangue NB. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene. 2010;29:2357–2367. doi: 10.1038/onc.2009.511. [DOI] [PubMed] [Google Scholar]
- Pirinen E, Kuulasmaa T, Pietilä M, Heikkinen S, Tusa M, Itkonen P, Boman S, Skommer J, Virkamäki A, Hohtola E, Kettunen M, Fatrai S, Kansanen E, Koota S, Niiranen K, Parkkinen J, Levonen AL, Ylä-Herttuala S, Hiltunen JK, Alhonen L, Smith U, Jänne J, Laakso M. Enhanced polyamine catabolism alters homeostatic control of white adipose tissue mass, energy expenditure, and glucose metabolism. Mol. Cell. Biol. 2007;27:4953–4967. doi: 10.1128/MCB.02034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Reddy M, Villeneuve LM, Wang M, Lanting L, Natarajan R. Role of the lysine-specific demethylase 1 in the proinflammatory phenotype of vascular smooth muscle cells of diabetic mice. Circ. Res. 2008;103:615–623. doi: 10.1161/CIRCRESAHA.108.175190. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Reytor E, Pérez-Miguelsanz J, Alvarez L, Pérez-Sala D, Pajares MA. Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J. 2009;23:3347–3360. doi: 10.1096/fj.09-130187. [DOI] [PubMed] [Google Scholar]
- Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell. Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Smith C, Bryla J, Williamson JR. Regulation of mitochondrial alpha-ketoglutarate metabolism by product inhibition at alpha-ketoglutarate dehydrogenase. J Biol Chem. 1974;249:1497–1505. [PubMed] [Google Scholar]
- Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Stassen M, Bailey D, Nelson S, Chinwalla V, Harte PJ. The Drosophila trithorax proteins contain a novel variant of the nuclear receptor type DNA binding domain and an ancient conserved motif found in other chromosomal proteins. Mech. Dev. 1995;52:209–223. doi: 10.1016/0925-4773(95)00402-m. [DOI] [PubMed] [Google Scholar]
- Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, Houseknecht K, Katz EB, Charron MJ. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat. Med. 1997;3:1096–1101. doi: 10.1038/nm1097-1096. [DOI] [PubMed] [Google Scholar]
- Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D, Peng J, Cheng X, Vertino PM. Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol. Cell. 2008;30:336–347. doi: 10.1016/j.molcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Takeuchi T. A gene trap approach to identify genes that control development. Dev. Growth Differ. 1997;39:127–134. doi: 10.1046/j.1440-169x.1997.t01-1-00001.x. [DOI] [PubMed] [Google Scholar]
- Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teyssier C, Ma H, Emter R, Kralli A, Stallcup MR. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev. 2005;19:466–473. doi: 10.1101/gad.1295005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschiersch B, Hofmann A, Krauss V, Dorn R, Korge G, Reuter G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 1994;13:3822–3831. doi: 10.1002/j.1460-2075.1994.tb06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Wellen K, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Yu S, Hsu CH, Eguchi J, Rosen ED. The orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II is a critical regulator of adipogenesis. PNAS. 2008;105:2421–2426. doi: 10.1073/pnas.0707082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Boström KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jørgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CN, Payne F, Perry JR, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjögren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Wellcome Trust Case Control Consortium. Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]