

Abstract
Determining mechanistic details about how drugs kill cancer cells is critical for predicting which cancers will respond to given therapeutic regimens and for identifying effective combinations of drugs that more potently kill cancer cells while sparing normal cells. The BCL2-family of proteins and bioactive sphingolipids are intricately linked during apoptotic cell death. In fact, many chemotherapeutic drugs are known to cause accumulation of the pro-apoptotic sphingolipid ceramide, however the mechanism by which this occurs is not completely understood. Herein, we demonstrate that direct inhibition of anti-apoptotic BCL2 proteins with ABT-263 is sufficient to induce C16-ceramide synthesis in multiple cell lines, including human leukemia and myeloma cells. ABT-263 activates ceramide synthase (CerS) activity only in cells expressing BAK or in cells capable of activating BAK. Importantly, recombinant BAK is sufficient to increase in vitro CerS activity in microsomes purified from Bak knock-out (KO) cells and activated BAK more potently activates CerS than inactive BAK. Likewise, ABT-263 addition to wild-type, but not Bak deficient microsomes, increases CerS in vitro activity. Furthermore. we present a feed-forward model by which BAK activation of CerS, by chemotherapeutic drugs, leads to elevated ceramides that result in synergistic channel formation by ceramide (or one of its metabolites) and BAX/BAK.
Keywords: ABT-263, ABT-737, sphingolipid, hematology, ceramide, BCL2, apoptosis, leukemia
INTRODUCTION
One family of proteins that is a critical regulator of programmed cell death, or apoptosis, is the B-cell lymphoma 2 (BCL2) family [1, 2]. Since the initial discovery of BCL2, as the genetic driver mutation in follicular B-cell lymphoma, more than 20 additional family members have been identified based on homology to a small amino acid motif, known as the BCL2 homology 3 (BH3) domain [3]. Most members of the family promote apoptosis, but six members have been shown to perform primarily anti-apoptotic functions (BCL2, BCLxL/BCL2L1, BCLw/BCL2L2, BFL1/BCL2A1, MCL1, and BCLb/BCL2L10) [4]. The main function of each of the anti-apoptotic BCL2-like proteins is to antagonize the pore forming functions of the pro-apoptotic BCL2-like proteins BAK and BAX in the mitochondrial outer membrane during the induction phase of apoptosis. However, there is evidence that these proteins can also perform additional functions some of which may impinge indirectly on, or be completely unrelated to, the regulation of mitochondrial outer membrane permeabilization (MOMP) [5].
Mounting evidence indicates that inhibiting the action of the anti-apoptotic BCL2 proteins in multiple tumor types has significant efficacy in cell culture systems, animal models and in patients [6–13]. In fact, several small molecule BH3 mimetics have been identified and are being tested in various clinical and pre-clinical trials. One of the most promising molecules, ABT-737, has a very high specificity for BCL2, BCLxL and BCLw (note: the orally bioavailable version of ABT-737 is ABT-263, which is used herein). ABT-263/737 has shown promise against many tumor types, including leukemias and solid tumors, as a single agent and in combination therapeutic strategies, respectively [6, 14–17]. Pre-clinical studies have demonstrated that drug resistance to ABT-263/ABT-737, in lung and leukemia samples, is commonly associated with increased expression of MCL1, which is not inhibited by either of these compounds [18–20]. A role for anti-apoptotic BCL2 family members in drug resistance is not limited to treatments that specifically target the BCL2 genes. Increased expression of BCL2, BCLxL and BCLb has been observed in AML cell lines resistant to high levels of busulfan treatment [21]. Increased copy numbers of the genomic loci encoding BCLw, MCL1 or BCLb were identified in various cell lines that were treated with, and became resistant to, etoposide, campothecin, or Ara-C, respectively [22]. Taken together, there is promise in targeting the BCL2 family in both primary and drug resistant tumors, but as with all therapeutic strategies we must try to find combinatorial strategies to limit tumor recurrence. Whether these combinatorial strategies include targeting individual BCL2 members or factors that regulate the BCL2 members is still to be determined.
Ceramides are known to regulate apoptosis at least in part through induction of MOMP. Ceramides have been shown to form channels in membranes capable of allowing the passage of proteins [23, 24]. In addition, ceramide generation is upstream of, and has been reported to be important for, BAX activation in cells [25–27]. Indeed, in vitro data suggest that ceramide metabolites regulate MOMP induction via activation of BAX and BAK at mitochondrial membranes [28]. Recent reports indicate that ceramide and BAX are capable of forming a novel channel with characteristics consistent with those required for MOMP induction [29]. Although several mechanisms have been proposed for ceramide-induction of MOMP during apoptosis, most, if not all, of the literature suggests that it is via an interaction between ceramides (or one of its metabolites) and BCL2 proteins [30]. Thus, due to the intimate interplay between BCL2-like family members and bioactive sphingohpids in apoptotic regulation, drugs that target both ceramide metabolism and anti-apoptotic BCL2 proteins may be good candidates for synergistic cancer therapies.
Many apoptotic stimuli are known to have pleiotropic effects on cells, making it difficult to deconvolute signaling pathways and apoptotic effectors. ABT-263 is a drug that specifically inhibits BCL2/BCLxL/BCLw and has been demonstrated to have a therapeutic window against cancer, compared to normal cells [6, 14–17]. ABT-263 specifically interacts with the hydrophobic groove of BCL2/ BCLxL/BCLw, resulting in the release of bound pro-apoptotic BCL2-like proteins and activation of the pro-apoptotic multi-domain BCL2 proteins BAK and BAX. Herein, we utilize a variety of engineered human leukemia and myeloma cell lines to demonstrate that ABT-263 is sufficient to induce C16-ceramide generation and increased in situ CerS activity. ABT-263 is incapable of elevating CerS activity or C16-ceramide levels in human leukemia and myeloma cells stably overexpressing MCL1. In vitro assays demonstrate that purified BAK is sufficient to activate CerS activity. In addition, activated BAK (via addition of tBid) is a more potent activator of CerS than inactive BAK. Importantly, ABT-263 activates in vitro CerS activity in membranes isolated from wild type baby mouse kidney cells, but not in those isolated from Bak knockout cells. Thus, data support a model in which activation of BAK induces elevated CerS activity and C16-ceramide generation. Furthermore, by combining data presented herein with data from the literature, we present a new inclusive model by which BAK-induced ceramide accumulation potentiates a feed-forward mechanism to execute apoptosis.
EXPERIMENTAL PROCEDURES
Cell culture
Human leukemia and myeloma cell lines, U937, K562, RPMI8226, and MV411 were obtained from ATCC (Manassas, VA, USA) and cultured in RPMI media containing 10% FBS, 1% L-glutamine and 1% Penicillin/Streptomycin. Cells were maintained according to the manufacturer’s protocol and were not cultured for more than thirty passages. Cells were routinely assessed for mycoplasma using ‘MycoSensor PCR assay kit’ cat #302108 from Agilent Technologies (Santa Clara. CA, USA) according to manufacturer’s protocol. Both cell culture supernatant and cell lysates were tested for mycoplasma. In addition, cells were routinely examined for morphological characteristics and were tested for consistent IC50 to ABT-263, since each line has a distinguishable level of sensitivity to this drug.
Stably infected U937 or RPMI8226 cells were obtained by infecting parental cells with MIGRX replication incompetent viral supernatant. MIGRX is a murine stem cell based retroviral vector that contains a multiple cloning site (MCS) followed by an internal ribosomal entry sequence (IRES) and GFP. MCL1, BCLxL, or a fragment of the estrogen receptor (ER; negative control) were inserted into the MCS. Retrovirus-contaning supernatant was produced by transient transfection of HEK-293t cells with equal amounts of a packaging construct, amphotropic SVpsi-, and the respective MIGRX plasmid. Tissue culture supernatant was collected starting 72 hours after transfection and used to infect U937 or RPMI8226 cells in the presence of 4 μg/ml polybrene during centrifugation; 2200 rpm for 1.5 hour at 30C. At 48 hours post-infection, GFP positive cells were sorted using FACS. Stable cells lines were maintained as described for the parental cells.
Baby mouse kidney epithelial (BMK) cells, wt and Bak KO, (kind gift from Dr. E. White. Rutgers University) were maintained in DMEM, supplemented with 2 mM L-glutamine, 5% fetal bovine serum. Cells were routinely assessed for mycoplasma using ‘MycoSensor PCR assay kit’ cat #302108 from Agilent Technologies (Santa Clara, CA, USA) according to manufacturer’s protocol. Both cell culture supernatant and cell lysates were tested for mycoplasma. In addition, cells were routinely examined for morphological characteristics. Cells were utilized for no more than ten passages. 1 x 106 cells were seeded into 10 cm2 dishes. 24 hours following plating, the media was changed to that containing either vehicle or the indicated concentration of ABT-263. At the indicated time points cells were harvested for quantification of ceramides or alternatively in vitro CerS activity measured as described below.
In situ C17-sphingosine Labeling
Cells were labeled with CI7-sphingosine (1 μM, 15 min, Avanti Polar Lipids), washed three times with cold phosphate-buffered saline, and collected via scraping and centrifugation. The reaction was stopped by the addition of 2 ml of extraction solvent containing ethyl acetate/2-propanol/water (60/30/10, v/v) supplemented with internal standard for EI/LC/MS analysis. Lipids were extracted twice, dried under a stream of nitrogen, resuspended in 150 μl of 1 mM NH4COOH in 0.2% HCOOH in methanol, and analyzed at the MUSC Lipidomics core facility by EI/LC/MS.
Relative cell viability assays
Exponentially growing human leukemia or myeloma cells were seeded in 96-well dishes (5,000 RPMI8226 cells, 5,000 U937 cells, 7,500 K562 or 7,500 MV411 cells per well) and immediately treated with the indicated drug concentrations in a total volume of 100 μl per well. All treatments were done in triplicate. ABT-263 (cat# CT-A263) was obtained from Chemietek (Indianapolis, IN, USA). Cells were incubated for 48 hours and then 10 μl of Alamar Blue reagent (Invitrogen, Grand Island, NY, USA, cat# DAL1100) was added to each well. Plates were then incubated and the reduction in Alamar Blue fluorescence was determined on a SPECTRAmax Gemini EM plate reader every hour until untreated wells were mid-linear, approximately 4,000 arbitrary units. Wells containing only 100 μl of complete RPMI media plus 10 μl of Alamar Blue were averaged and subtracted from all experimental readings. Drug treatment regimens were then normalized to vehicle treated cells. Each graph shown is a representative experiment of at least three biological replicates.
In Vitro Ceramide Synthase Activity
Baby mouse kidney epithelial (BMK) cells were grown to 70% confluence, washed twice with ice cold phosphate-buffered saline, harvested by scraping in lysis buffer (20 mM Hepes (pH 7.4), 2 mM KCl, 2 mM MgCl2 250 mM sucrose, protease inhibitors), and lysed via 10 passages through a 28-gauge insulin syringe. Intact cells and nuclei were removed via centrifugation at 1,000 ×g for 10 min, and the mitochondrial enriched fraction obtained via centrifugation at 12,000 ×g for 10 min. Microsomes were obtained from the supernatant of the 12,000 x g spin via a 100,000 ×g centrifugation for 1 h. Ceramide synthase activity was measured in the microsomes or where indicated the mitochondrial enriched fraction (10–50 μg as indicated) as previously described [31]. Briefly, a reaction mix (100 μl of final volume) containing 15 μM C17-sphingosine and 50 μM C16 fatty acyl-CoA in 25 mM potassium phosphate buffer (pH 7.4) was pre-warmed at 37 °C for 5 min. The enzyme reaction was initiated via addition of the enzyme source (microsomes or mitochondria), and after 15 mm at 37 °C was terminated via the addition of 2 ml of extraction solvent containing ethyl acetate/2-propanol/water (60/30/10, v/v) supplemented with internal standard for EI/LC/MS analysis. Lipids are extracted twice, dried under a stream of nitrogen, and resuspended into 150 μl of 1 mM NH4COOH in 0.2% HCOOH in methanol and analyzed by EI/LC/MS.
Where indicated, CerS activity was measured in the presence of added recombinant proteins. Recombinant BAX or BAK were obtained as previously described [32, 33]. Where indicated, BAK activation was achieved via the addition of a 5 ng of BAK and 10 ng recombinant tBid (purchased from AnaSpec) at 37C. Where indicated, recombinant proteins were added to microsomes 15 min prior to the initiation of the enzymatic reaction (achieved via addition of substrates).
Sphingolipidomic mass spectrometry
5 X 106 leukemia or myeloma cells were seeded in each well of a 6-well dish in 4 ml total RPMI (containing 10% FBS, 1% pen/strep, 1% L-glutamine) and treated with the indicated amount of ABT-263 or DMSO, in biological triplicates. At indicated times, cells were harvested, washed two times in 5 mL of ice cold 1 x Phosphate buffered saline and then snap frozen in liquid nitrogen. In BMK cells, 1 x 106 cells were seeded in 10 cm2 dishes and after 36 h were treated with ABT-263. At 2 and 8 hrs, cells were washed twice with ice cold 1 x Phosphate buffered saline and then scraped into 5 mL ice cold 1 x Phosphate buffered saline. Cells were pelleted, the supernatant removed, and the cell pellet snap frozen in liquid nitrogen. For leukemia, myeloma, and BMK cells, quantification of sphingolipids species was performed by the Lipidomics Core Facility at the Medical University of South Carolina (MUSC) on a Thermo Finnigan TSQ 7000, triple-stage quadrupole mass spectrometer operating in a Multiple Reaction Monitoring (MRM) positive ionization mode as described [34]. Data were normalized to total lipid phosphate.
Plasmid construction
Total human Universal RNA (Clontech, cat. #636538, Mountain View, CA, USA) was used as a template to amplify BCLxl and MCL1 using the following oligs: 5 hu BCLxl atg RI, 5′-GCGCGAATTCACCATGTCTCAGAGCAACCGGGAG-3′; 3 hu BCLxl stop xhol, 5′-GCGCCTCGAGTTATTTCCGACTGAAGAGTGAGCCCAG-3′; 5 hu MCL1 atg Bam, 5′-GCGCGGATCCACCATGTTTGGCCTCAAAAGAAACGC-3′; 3 hu MCL1 stop sail, 5′-GCGCGTCGACTTATCTTATTAGATATGCCAAACC-3′. PCR amplified fragments were ligated into TOPO blunt cloning kit from Invitrogen (Carlsbad, CA, USA). Full-length BCLxL and full-length MCL1 were then digested with EcoRI and Xhol or BamHI and Sail, respectively. Fragments were then ligated into the MIGRX retroviral vector digested with EcoRI and Xhol (for BCLxl) or BglII and XhoI (for MCL1). All clones were sequenced for accuracy. MIG-ER was developed by digesting pBABE-ER (described previously [35]) with BamHI and Sail and ligating the fragment into MIGRX digested with BglII and XhoI.
Establishment of U937 and RPMI-8226 stable cell lines
Parental U937 and RPMI-8226 cell lines were maintained as described above. GFP expressing infectious virus was produced by transiently transfecting 293T cells with the described MIGRX-based plasmids and the packaging plasmid SV(psi)- and polyethyleneamine at a ratio of 1:2.5, DNA to PEL 72 hours post transaction viral supernatant was harvested and incubated with U937 or RPMI-8226 cells in the presence of 4 micrograms per milliliter of polybrene. This was centnfuged at 30C for 1 hour at 2000 rpm in a clinical tabletop centrifuge. 72 hours post infection the cells were sorted at the James Graham Brown Cancer Center Flow Cytometry Core Facility using standard methods.
RESULTS
ABT-263 induces generation of C16-ceramide in leukemia cells
Our previously published results demonstrate synergism between ABT-263 and drugs that modulate sphingolipid metabolism [36]. Specifically, data indicate that the combination of ABT-263 with drugs that inhibit metabolism of ceramide to pro-proliferative sphingolipids (e.g. sphingosine-1-phosphate or glucosylceramide) synergistically kill leukemia cells [36]. Sensitivity to ABT-263 correlates with basal ceramide levels and the synergistic killing is not universal, but rather dependent on the accumulation of total ceramide [36]. Thus, we hypothesized that ABT-263 regulates ceramide metabolism. To test this, we utilized three different human leukemia cell lines with varying sensitivities to ABT-263, namely histiocytic lymphoma U937 cells, chronic myelogenous K562 leukemia cells, and myelocytic MV411 leukemia cells. First, a dose response curve of ABT-263 for 48 h was performed and viable cells were determined via Alamar blue assay to determine the appropriate concentration at which to administer ABT-263 for ceramide analysis (Figure 1A). The IC50 values for ABT-263 were 7.5 μM, 0.4 μM, and 0.03 μM for U937, K562, and MV411 cells, respectively. Cells were treated for 8 or 24 h at their corresponding IC50 for ABT-263 or vehicle control (DMSO) and individual ceramides quantified via HPLC-MS/MS as described in the experimental procedures. In all cell lines, the only ceramide species that was significantly altered following ABT-263 treatment was C16-ceramide (Figure 1B, C, D). There were slight elevations in C22- and C22:1-ceramides (Figure 1B, C, D). However, these were not statistically significant elevations and are very minor species in these leukemia cells. In contrast, C16-ceramide is a major species that is elevated in a statistically significant manner (Figure 1B, C, D). These data indicate that inhibition of anti-apoptotic BCL2-like proteins, with ABT-263, causes accumulation of C16-ceramide.
Figure 1. Treatment of human leukemia cell lines with the BCL2/BCLxL /BCLw inhibitor, ABT-263, causes accumulation of C16-ceramide.

A, U937, K562 and MV411 cells were incubated with increasing doses of ABT-263 (0.7 nM to 45 μM) for 48 hours and the relative numbers of viable cells were determined by reduction of alamar blue. All data points represent the compilation of at least biological triplicates. B, U937 cells were treated with 1.6 μM of ABT-263, or vehicle (DMSO), for 8 and 24 hours and the total amount of cellular ceramides were determined by HPLC-MS/MS. C, K562 cells were treated with 150 nM of ABT-263, or vehicle, for 8 and 24 hours and the total amount of cellular ceramides were determined by HPLC-MS/MS. D, MV411 cells were treated with 50 nM of ABT-263, or vehicle (DMSO), for 8 and 24 hours and the total amount of cellular ceramides were determined by HPLC-MS/MS. D, ***, P=<0.0005; **, P=<0.005; *,p=<0.05.
ABT-263-induced C16-ceramide generation is inhibited by overexpression of MCL-1
ABT-263 induces apoptosis via its ability to bind to and inhibit the activity of anti-apoptotic BCL2 proteins BCL2, BCLxL, and BCLw. Thus, we hypothesized that ABT-263-induced C16-ceramide generation is via its inhibition of one or more of these anti-apoptotic BCL2 proteins. To test this hypothesis, U937 cells were infected with a retrovirus expressing green fluorescent protein (GFP) and either an irrelevant control protein fragment (ER), BCLxL, or MCL1, separated by an IRES [37]. Infected cells were sorted using fluorescent activated cell sorting and the established cell lines were validated for GFP and the respective BCL2-like protein expression (Supplemental Figure 1). The anti-apoptotic BCLxL protein is inhibited by ABT-263, whereas MCL1 is not inhibited by ABT-263 [38]. Interestingly, over-expression of either BCLxL or MCL1 in U937 cells does not dramatically alter their sensitivity to ABt-263 after 48 hours of treatment. Ceramides were quantified following treatment (8 or 24 h) of U937-ER, U937-BCLxL or U937-MCL1 cells with ABT-263 or vehicle (DMSO), as previously described. Treatment of control U937-ER and U937-BCLxL with ABT-263 lead to significant increase in the level of the major ceramide species C16-ceramide at both time points examined (Figure 2A, B). In stark contrast, C16-ceramide was not elevated following treatment of U937-MCL1 cells with ABT-263 (Figure 2C). In addition to C16-ceramide, ABT-263 also induced elevations in the minor species C22- and C22:1-ceramide in the U937-ER cells and to a lesser extent in the U937-BCL-xL cells that were not observed in the U937-MCL1 cells (Figure 2A and B). However, these are very minor species (Figure 1B) and are elevated mainly at 24 hours, a time point after cleavage of caspase 3 (previously published to be as early as 2 hours [36]). It is not known which enzymes are responsible for the generation of C22- and C22:1-ceramides, nor the location in the cell in which they are generated. It is possible that they are important to the apoptotic process and thus their generation should not be completely ignored. In summary, these data suggest that ABT-263-induced ceramide generation is via its inhibition of known target anti-apoptotic BCL2-like proteins.
Figure 2. ABT-263 induced ceramide accumulation and CerS activity can be inhibited by MCL1 in U937 cells.

A, U937 cells stably expressing GFP and a portion of an irrelevant protein, a portion of ER (U937-ER) were treated with 1.6 μM of ABT-263, or vehicle (DMSO), for 8 and 24 hours and the total amount of cellular ceramides were determined by HPLC-MS/MS. B, U937 cells stably expressing GFP and BCLxL (U937-BCLxL) were treated with 1.6 μM of ABT-263, or vehicle (DMSO), for 8 and 24 hours and the total amount of cellular ceramides were determined by HPLC-MS/MS. C, U937 cells stably expressing GFP and MCL1 (U937-MCL1) were treated with 1.6 μM of ABT-263, or vehicle (DMSO), for 8 and 24 hours and the total amount of cellular ceramides were determined by HPLC-MS/MS. D. U937-MIG ER cells (U937 cells stably infected with MIG-ER virus) were incubated with 1.6 μM of ABT-263, or vehicle (DMSO), for 1 hour and 45 minutes. 1μM C17-sphingosine was then added to the media for an additional 15 minutes. Cells were immediately lysed and the amount of C17-ceramide in the cells were quantitated by HPLC-MS/MS. E, U937-BCLxL cells were incubated with 1.6 μM of ABT-263, or vehicle (DMSO), for 1 hour and 45 minutes. 1μM of C17-sphingosine was then added to the media for an additional 15 minutes. Cells were immediately lysed and the amount of C17-ceramide in the cells were quantitated by HPLC-MS/MS. F, U937-MCL1 cells were incubated with 1.6 μM of ABT-263, or vehicle (DMSO), for 1 hour and 45 minutes. 1μM of C17-sphingosine was then added to the media for an additional 15 minutes. Cells were immediately lysed and the amount of C17-ceramide in the cells were quantitated by HPLC-MS/MS. Lipids were normalized to total lipid phosphate and data expressed as a fold-change of the untreated control. All data points represent the compilation of at least biological triplicates. ***, p=<0.0005; **, p=<0.005; *, p=<0.05.
ABT-263-induced ceramide generation is via activation of long-chain ceramide synthase activity
BCL2-like anti-apoptotic proteins are localized to mitochondria and ER membranes. Likewise, ceramide synthase (CerS) activity has been detected in ER, mitochondrial associated membranes, and mitochondria. Furthermore, previous work has demonstrated that CerS co-localize to membrane rafts that are enriched with BAX and CerS are required for irradiation-induced apoptosis in some cells [27, 40]. Therefore, we hypothesized that inhibition of BCL2-like proteins ultimately leads to activation of CerS activity. There are six CerS isoforms that catalyze the synthesis of ceramide from sphinganine or sphingosine utilizing fatty acyl-CoAs of varying chain lengths. Each CerS isoform preferentially utilizes particular fatty acyl CoAs as substrates and thus is responsible for the synthesis of particular ceramide species. ABT-263 induces accumulation specifically of C16-ceramide. Thus we hypothesized that ABT-263-induced C16-ceramide generation is via activation of CerS. To test this hypothesis, we performed in situ CerS activity assays. The three stable U937 cell lines were treated with ABT-263 for two hours. C17-sphingosine was added to the cells for 15 minutes and then lipids were extracted. Metabolism of C17-sphingosine into ceramides with 17 carbon sphingoid base backbones was quantitated by HPLC-MS/MS. Consistent with the increase in C16-ceramide observed in previous experiments, in situ CerS activity was significantly elevated in both U937-ER and U937-BCLxL cells following ABT-263 treatment (Figure 2D, E). However, there was no increase in in situ CerS activity in U937-MCL1 cells treated with ABT-263 (Figure 2F). To determine if the lack of ceramide generation in MCL1 overexpressing cells was specific to U937 cells, we utilized a human myeloma cell line, RPMI8226, also engineered to stably overexpress MCL1 (Supplemental Figure 2). RPMI-MCL1 cells are resistant to ABT-263 as evidenced by an elevated IC50 (Supplemental Figure 2). RPMI-ER and RPMI-MCL1 cells were treated with ABT-263 for 2 or 8 h and ceramides measured. Consistent with the data obtained in U937 cells, C16-ceramide is generated following treatment of RPMI-ER cells with the IC50 of ABT-263; RPMI-MCL1 cells when treated with the IC50 of ABT-263 for 2 or 8 h do not generate C16-ceramide (Figure 3 A, B). In situ CerS activity was measured in cells treated with vehicle and following treatment with ABT-263 for 2 h. In situ CerS activity was increased following treatment with ABT-263 in RPMI-ER cells, but not in RPMI-MCL1 cells (Figure 3C, D). These data suggest that ABT-263 induces C16-ceramide generation in a CerS dependent manner.
Figure 3. MCL1 inhibits ABT-263 induced C16-ceramide generation and increased in situ CerS activation in RPMI8226 human plasma cell leukemia.

A, RPMI8226 cells stably expressing GFP and a portion of an irrelevant protein, ER. (RPMI-ER) were treated with 0.5 μM of ABT-263, or vehicle (DMSO), for 2 and 8 hours and C16-ceramide quantified by HPLC-MS/MS. B, RPMI8226 stably expressing GFP and MCL1 (RPMI-MCL1) were treated with 5 μM of ABT-263, or vehicle (DMSO), for 2 and 8 hours and C16-ceramide quantified by HPLC-MS/MS. C. RPMI-ER cells were incubated with 0.5 μM of ABT-263, or vehicle (DMSO), for 1 hour and 45 minutes. 1μM C17-sphingosine was then added to the media for an additional 15 minutes. Cells were immediately lysed and the amount of C17-ceramide in the cells were quantitated by HPLC-MS/MS. D, RPMI-MCL1 cells were incubated with 5 μM of ABT-263, or vehicle (DMSO), for 1 hour and 45 minutes. 1μM of C17-sphingosine was then added to the media for an additional 15 minutes. Cells were immediately lysed and the amount of C17-ceramide in the cells were quantitated by HPLC-MS/MS. Lipids were normalized to total lipid phosphate and data expressed as a fold-change of the untreated control. All data points represent the compilation of at least biological triplicates. ***, p=<0.0005; **, p=<0.005; *, p=<0.05.
BAK is sufficient to induce activation of CerS
ABT-263 inhibits anti-apoptotic BCL2-like proteins (BCL2, BCLxL, and BCLw) via binding to their hydrophobic groove and displacing bound pro-apoptotic BCL2 proteins, leading to the activation of BAK and/ or BAX. MCL1 only binds BAK and ABT-263 is incapable of displacing BAK from MCL1 [41]. We previously reported that BAK is required for CerS-mediated long-chain ceramide generation during apoptosis [31]. Thus, we hypothesized that ABT-263 activates CerS in a BAK dependent manner. To test this hypothesis, we isolated mitochondria from either wild-type (wt) or Bak deficient (BakKO) baby mouse kidney (BMK) cells and performed in vitro CerS enzyme activity assays. Increasing amounts of purified mitochondria, isolated from either wt or Bak KO BMK cells, showed increasing CerS activity, however this activity was significantly lower in the Bak KO mitochondria, as demonstrated by decreased to conversion of C17-sphingosine into C16-C17-ceramide. suggesting that CerS activity is greatly diminished (Figure 4A). To determine whether the presence of Bak is sufficient to activate CerS activity, we isolated microsomes from Bak KO BMK cells and added increasing amounts of soluble recombinant Bax or Bak (Figure 4B). In vitro CerS enzymatic activity was determined by monitoring the conversion of C17-sphingosine into C16-C17-ceramide Soluble Bak was sufficient to activate CerS enzymatic activation on Bak KO microsomes in a dose dependent manner. whereas soluble Bax was not capable of increasing CerS activity.
Figure 4. Bak is necessary and sufficient for ceramide synthase activity, in vitro.
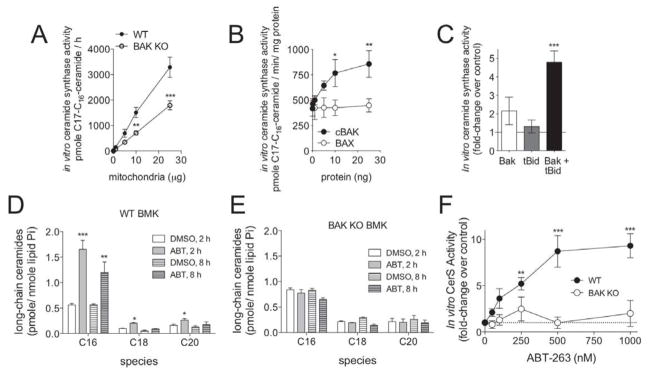
A, Bak deficient mitochondria are severely deficient in CerS activity. Increasing amounts of mitochondria isolated from wild-type (wt) or Bak deficient (Bak KO) baby mouse kidney (BMK) cells were incubated with C17-sphingosine for 15 minutes. Lipids were extracted and the conversion of C17-sphingosine into C17-ceramide was quantitated by HPLC-MS/MS. Data shown are the combination of biological triplicates done in duplicate. B, Soluble Bak, but not Bax, activates CerS on Bak KO microsomes. Microsomes isolated from Bak KO BMK cells (10 μg) were incubated with increasing concentrations of cleaved. soluble Bak (cBak, closed circles) or soluble Bax (open circles) for 15 mm and the activity of CerS was determined. C, Activated Bak is a more potent activator of CerS, in vitro. Microsomes from Bak KO BMK cells (10 μg) were incubated with recombinant Bak (5 ng), cleaved, active Bid (tBid) (10 ng), or Bak and tBid (5 ng Bak; 10 ng tBid). Reactions were incubated with C17-sphingosine for 15 minutes and the conversion to C17-ceramide was quantitated by HPLC-MS/MS. D. Baby mouse kidney cells established from wild-type mice (WT BMK) were treated with 2.67 μM of ABT-263 (ABT) or vehicle (DMSO) for 2 and 8 hours. Lipids were extracted and the total cellular levels of individual ceramide species were quantitated by HPLC-MS/MS. E, Baby mouse kidney cells established from Bak KO mice (BAK KO BMK) were treated with 2.67 μM of ABT-263 (ABT) or vehicle (DMSO) for 2 and 8 hours. Lipids were extracted and the total cellular levels of individual ceramide species were quantitated by HPLC-MS/MS. C, Heavy membranes were isolated from WT (closed circles) or Bak KO (open circles) BMK cells and incubated with increasing concentrations of ABT-263. The relative increase in the conversion of C17-sphingosine to C17-ceramide is indicated, as compared to vehicle treated heavy membranes from the respective genotype of BMK cell. Dashed line indicates no fold change. Each point represents biological triplicates done in duplicate. ***, p=<0.0005; **, p=<0.005; *, p=<0.05.
Bak is known to undergo activating conformational changes during apoptosis. ABT-263 treatment of cells induces activation of Bak. Thus, we hypothesized that activated Bak is a more potent activator of CerS activity than “non-activated” Bak. Certain BH3-only pro-apoptotic proteins, such as cleaved Bid (tBid), are known to cause activation of Bak. Therefore, we tested whether addition of purified tBid could influence the ability of Bak to activate CerS activity on purified Bak deficient microsomes (Figure 4C). When a minimal amount of soluble Bak (5 ng) was added to microsomes isolated from Bak KO BMK cells, we observed a slight increase in CerS activity. Purified tBid alone (10 ng) was not sufficient to alter CerS enzymatic activity in the absence of Bak. In contrast, when recombinant tBid was added with soluble Bak to microsomes there was a dramatic increase in CerS enzymatic activity, indicating that activated Bak is a more potent inducer of CerS enzymatic activity.
BAK is necessary for ABT-263-induced activation of CerS
Data indicate that ABT-263 activates in situ CerS activity, leading to an increased accumulation of C16-ceramide. The ability of ABT-263 to induce C16-ceramide can be inhibited by the expression of MCL1. Thus, we hypothesized that Bak is necessary for ABT-263 induced CerS activation and C16-ceramide generation. To formally determine the requirement of Bak on ABT-263-induced ceramide accumulation we treated Bak KO or wt BMK cells with 2.67 μM of ABT-263 and quantified ceramides at 2 and 8 h. As in the human leukemia cells, treatment of wt BMK cells with ABT-263 causes a significant increase in the level of C16-ceramide (Figure 4D). In stark contrast, treatment of Bak KO BMK cells with ABT-263 did not elevate C16-ceramide (Figure 4E). In vitro CerS activity assays were performed to determine if ABT-263 can activate CerS activity in a cell free system. ABT-263 induced a dose dependent increase in conversion of C17-sphingosine to C16-C17-ceramide in heavy membranes isolated from wt BMK cells. However, ABT-263 did not elevate in vitro CerS activity in heavy membranes isolated from Bak KO cells at any dose tested (Figure 4F). These data demonstrate that the ability of ABT-263 to cause C16-ceramide accumulation is dependent on CerS activation by Bak.
DISCUSSION
One key hallmark of cancer is the ability to evade apoptosis and one primary way in which cancers accomplish this is via upregulation of anti-apoptotic BCL2-like proteins [18, 19,41]. Cancer cells rely on anti-apoptotic BCL2-like proteins for survival and should in theory be highly sensitive to cytotoxic therapies that are aimed at their inhibition. Thus, much effort has been devoted to designing small molecule inhibitors that interfere with the pro-survival function of these oncoproteins [15].
ABT-263 is a potent inhibitor of three anti-apoptotic BCL2-like proteins, namely BCL2, BCLxL and BCLw [41]. Our data indicate that ABT-263 treatment is sufficient to induce generation of C16-ceramide. Importantly, generation of ceramide was not specific to a particular cell type as ABT-263 induced generation of C16-ceramide in multiple human leukemia cell lines as well as in kidney epithelial cells. Data indicate that ABT-263-induced C16-ceramide generation is dependent on its ability to inhibit anti-apoptotic BCL2-like proteins. ABT-263 inhibits BCL2, BCLxL, and BCLw by binding to their hydrophobic pocket and displacing bound pro-apoptotic BCL2-like proteins, including BAX and BAK [6]. Indeed, ABT-263 treatment of cells is sufficient to induce activation of BAX and/ or BAK and induction of MOMP. ABT-263 has a low affinity for the hydrophobic pocket of MCL1 and thus is unable to inhibit the anti-apoptotic functions of MCL1 MCL1 binds to BAK but not BAX. As ABT-263 has a low affinity for the hydrophobic pocket of MCL1, it is unable to induce the release of BAK. Thus, cells overexpressing MCL1 should have lower levels of free BAK available for activation of CerS. Indeed, data indicate that this is in fact the case. Overexpression of MCL1 in either U937 or RPMI8226 cell lines prevented the ability of ABT-263 to induce ceramide generation. If ABT-263 were indeed generating ceramide via a BAK activation of CerS, then following ABT-263 treatment of the parental cells CerS activity should be elevated. ABT-263 treatment of the control cells increased in situ CerS activity; this increase in activity was not observed in cells overexpressing MCL1. Data are consistent with the model that overexpression of MCL1 binds to BAK and prevents it from activating CerS. Consistent with this model, purified recombinant BAK was sufficient to induce activation of CerS in an in vitro assay. This was specific to BAK as the seemingly functional redundant protein BAX was incapable of activating in vitro CerS activity. Importantly, ABT-263 alone was also sufficient to induce activation of CerS in vitro. Again ABT-263 activation of CerS only occurred in the presence of BAK as no activation was observed when heavy membranes purified from BAK KO cells were utilized as the enzyme source.
Cells that become resistant to ABT-263 often overexpress MCL1 and our data suggest that this resistance may be due, in part, to an inability of MCL1 expressing cells to generate long-chain ceramides. Thus, one potential avenue of therapeutic exploration should be to identify drugs that can specifically inhibit the MCL1/BAK interaction, thereby leading to activation of CerS activity. Indeed, there are potential inhibitors that have recently been developed that do inhibit MCL1 and could be utilized in this capacity. However, many of these lack specificity and have off-target effects as evidenced from their ability to kill both wild-type and Bax/Bak double deficient cells. Thus, more specific inhibitors are needed to increase the specificity of cancer treatments.
In addition, ABT-263 treatment is known to have negative side-affects in patients, such as thrombocytopenia [42]. We propose that combining ABT-263 with other clinically relevant drugs will be beneficial by limiting the possibility of drug resistance and by lowering the effective dose of ABT-263 required, which would in turn, decrease the chances of patients developing detrimental side-effects. To this end, we have recently reported that drugs that inhibit metabolism of ceramide to either of the pro-proliferative metabolites sphingosine-1-phosphate or glucosylceramide can be combined with ABT-263 to synergistically kill a variety of human leukemia cell lines [36]. The combination of these drugs may allow it to be utilized at lower doses. Alternatively, these drug combinations may also prevent recurrence of cancers.
Taken together, data presented here indicate that ABT-263 induced generation of C16-ceramide is dependent on BAK activation of CerS. Future work will aim to determine the specific CerS that are regulated by BAK. CerS are key enzymes in the de novo and salvage pathways of ceramide generation. Six CerS isoforms are present in mammalian cells, each with its own fatty acyl CoA preference [43], Thus, specific CerS isoforms generate ceramides of specific chain lengths, which have been shown to contribute differently to cellular responses to stress stimuli [44, 45]. BAK-dependent CerS activity is restricted to particular long-chain fatty acyl-CoA substrates that are consistent with those preferred by CerS 4, 5 and 6 (see above and [31]). Once we identify the CerS isoforms that are regulated by BAK, we can define means for influencing specific CerS isoforms responsible for ceramide generation. This is not a trivial task as CerS activity is in part regulated by heterodimerization with other CerS (for example Cer2 with either Cer5 or CerS6) and knockdown of the expression of one CerS impacts the expression of the other CerS [46,47].
Data presented in this manuscript are consistent with previous reports of cross-talk between sphingolipids and BCL2-like proteins in the induction of MOMP. Data suggest a feed-forward model by which ABT-263, and likely other chemotherapeutic drugs, lead to BAK-dependent activation of CerS (Figure 5-1). CerS activation, in turn, leads to ceramide-induced inhibition of anti-apoptotic BCL2-like proteins (Figure 5-2) and synergistic channel formation by ceramide (or one of its metabolites) and BAX/BAK (Figure 5-3). Indeed, in vivo data from the Kolesnick laboratory demonstrate that ceramide production is required for radiation-induced apoptosis [40]. Furthermore, they also show that CerS activation of ceramide production and co-localization of ceramide with BAX in platforms in the outer mitochondrial membrane to induce apoptosis [27]. Data from Colombini and co-workers indicate that ceramide alone is capable of forming large and stable channels in planar phospholipid membranes in the absence of proteins as well as in mitochondrial outer membranes of isolated mitochondria [48]. Thus. BAK activation could lead to the activation of CerS and generation of sufficient ceramide to form channels large enough to induce MOMP (Figure 5-4). Data from the Colombini laboratory also indicate that these ceramide channels are regulated by BCL2-like proteins [29, 30, 49, 50]. Anti-apoptotic proteins like BCLxL and the BCL2 homologue Ced-9 inhibit ceramide channels [49]. Indeed, BCLxL binds ceramide in the same hydrophobic pocket that it binds to pro-apoptotic BCL2-like proteins and ABT-263 is capable of displacing bound ceramide [30]. Thus, ABT-263 could effectively prevent BCLxL or BCL2 inhibition of ceramide channels. Alternatively, ceramide binding to the hydrophobic pocket of BCL2-like anti-apoptotic proteins may prevent their interaction with pro-apoptotic BCL2-like proteins (Figure 5-2 and Figure 5-5). Thus, ceramide could, in effect, behave as a pleotropic version of ABT-263 that is capable of binding more promiscuously than ABT-263 to anti-apoptotic BCL2-like family members, such as MCL1, to displace BAK and further potentiate the production of ceramide (Figure 5-5). Thus, ABT-263 functions at least in part by increasing the concentration of BAK available to activate CerS and induce ceramide generation that would function in a positive feedback loop to (1) form ceramide channels in mitochondrial outer membranes, (2) activate BAX and BAK to form channels, (3) induce the formation of ceramide-BAX hybrid channels, (4) bind to the hydrophobic pocket of BCLxL and release bound pro-apoptotic BCL2-like proteins. Interesting, we also published recently that ceramide metabolites are capable of inducing activation of BAK and BAX [28]. While, this data might seem contradictory, it actually supports the notion of a positive feed-forward process whereby the BAK-induced ceramide is further metabolized to species that activate BAX and BAK. The activated BAK would lead to further activation of CerS and increased ceramides (and its metabolites) that would induce MOMP through the multiple potential pathways outlined above. The sphingolipid species that initially leads to the activation of BAK and synergizes with BAK and BAX to induce MOMP most likely depends on both the cell type as well as the apoptotic stimulus.
Figure 5.
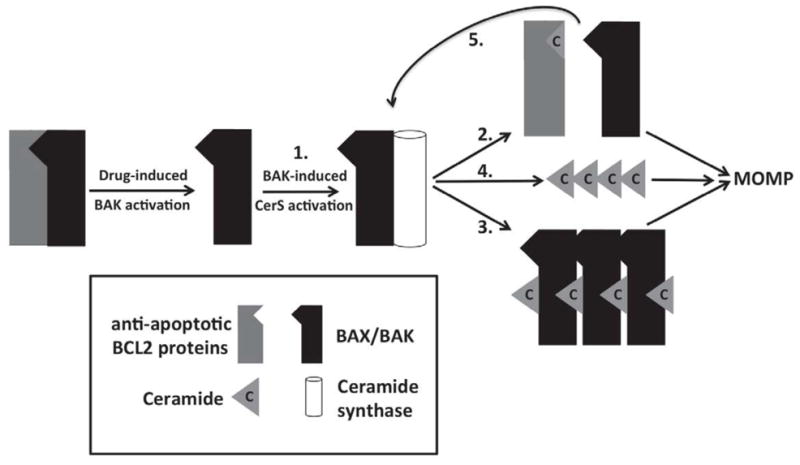
Feed-forward model for how the generation of ceramide, through a BAK-dependent mechanism, can contribute to MOMP-induced apoptosis. See text for discussion for detailed description.
Supplementary Material
Acknowledgments
We thank all members of the Beverly and Siskind laboratories for technical assistance and helpful discussion. In addition, we thank all member of the laboratories of Drs. Obeid and Hannun for their helpful discussions and support and in particular Dr. Lina M. Obeid. Research reported in this publication was supported by: National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health, [R01DK093462] (to L.J.S.), the Lipidomics Shared Resource of the Hollings Cancer Center at the Medical University of South Carolina supported by a Cancer Center Support Grant [P30 CA138313], pilot project from the [NIH/NCRR P20 RR17677] COBRE in Lipidomics and Pathobiology (to L.J.S); NIH, Molecular Targets COBRE, [8P20GM103482-10] (to L.J.B.), Wendy Will Case Cancer Fund, [GB220413] (to L.J.B.), Kosair Pediatric Cancer Research Program award (to L.J.B.) and the James Graham Brown Cancer Center, NIH R01 [CA157740] (to J.E.C.), March of Dimes Foundation [5-FY11-74] (to J.E.C.]
Abbreviations
- BCL2
B-cell lymphoma
- CerS
ceramide synthase
- MCL1
myeloid cell leukemia gene 1
- BAK
BCL2 antagonist/killer
- BH3
BCL2 homology domain 3
- MOMP
mitochondria outer membranes permeabilization
- AML
acute myeloid leukemia
- Ara-C
Arabinofuranosyl Cytidine
- MIG-RX
murine stem cell virus vector containing a MCS, IRES, and GFP
- MCS
multiple cloning site
- IRES
internal ribosomal entry sequence
- HEK-293t
human embryonic kidney cell line 293 with large T antigen
- BMK
baby mouse kidney cells
- GFP
green fluorescent protein
- FACS
fluorescent activated cell sorting
- EI/LC/MS
electrospray ionization/liquid chromatography/mass spectrometry
- PEI
polyethyleneimine
- DMSO
dimethylsulfoxide
- HPLC-MS/MS
high performance liquid chromatography-tandem mass spectrometry
Footnotes
Author Contributions
Levi J. Beverly and Leah J. Siskind conceived the experiments and wrote the manuscript. Lauren A. Howell, Maria Hernandez-Corbacho, Lavona Casson, Levi J. Beverly and Leah J. Siskind performed the experiments. Jerry E. Chipuk provided critical reagents and contributed to writing the manuscript.
References
- 1.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 2.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 3.Lanave C, Santamaria M, Saccone C. Comparative genomics: the evolutionary history of the Bcl-2 family. Gene. 2004;333:71–79. doi: 10.1016/j.gene.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 4.Sorenson CM. Bcl-2 family members and disease. Biochim Biophys Acta. 2004;1644:169–177. doi: 10.1016/j.bbamcr.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Beverly LJ. Regulation of anti-apoptotic BCL2-proteins by non-canonical interactions: The next step forward or two steps back? J Cell Biochem. 2012;113:3–12. doi: 10.1002/jcb.23335. [DOI] [PubMed] [Google Scholar]
- 6.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 7.Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, Oleksijew A, O’Connor JM, Wang B, Frost DJ, Bauch J, Marsh K, Tahir SK, Yang X, Tse C, Fesik SW, Rosenberg SH, Elmore SW. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14:3268–3277. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- 8.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 9.Cosulich SC, Worrall V, Hedge PJ, Green S, Clarke PR. Regulation of apoptosis by BH3 domains in a cell-free system. Curr Biol. 1997;7:913–920. doi: 10.1016/s0960-9822(06)00410-6. [DOI] [PubMed] [Google Scholar]
- 10.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nature chemical biology. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 13.Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nature clinical practice Oncology. 2006;3:388–398. doi: 10.1038/ncponc0538. [DOI] [PubMed] [Google Scholar]
- 14.Kohl TM, Hellinger C, Ahmed F, Buske C, Hiddemann W, Bohlander SK, Spiekermann K. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763–1772. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- 15.Wesarg E, Hoffarth S, Wiewrodt R, Kröll M, Biesterfeld S, Huber C, Schuler M. Targeting BCL-2 family proteins to overcome drug resistance in non-small cell lung cancer. Int J Cancer. 2007;121:2387–2394. doi: 10.1002/ijc.22977. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Bélec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–2309. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 19.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valdez BC, Murray D, Ramdas L, de Lima M, Jones R, Kornblau S, Betancourt D, Li Y, Champlin RE, Andersson BS. Altered gene expression in busulfan-resistant human myeloid leukemia. Leuk Res. 2008;32:1684–1697. doi: 10.1016/j.leukres.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yasui K, Mihara S, Zhao C, Okamoto H, Saito-Ohara F, Tomida A, Funato T, Yokomizo A, Naito S, Imoto I, Tsuruo T, Inazawa J. Alteration in copy numbers of genes as a mechanism for acquired drug resistance. Cancer Res. 2004;64:1403–1410. doi: 10.1158/0008-5472.can-3263-2. [DOI] [PubMed] [Google Scholar]
- 23.Colombini M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim Biophys Acta. 2010;1797:1239–1244. doi: 10.1016/j.bbabio.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 24.Siskind LJ, Kolesnick RN, Colombini M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion. 2006;6:118–125. doi: 10.1016/j.mito.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HJ, Mun JY, Chun YJ, Choi KH, Kim MY. Bax-dependent apoptosis induced by ceramide in HL-60 cells. FEBS Lett. 2001;505:264–268. doi: 10.1016/s0014-5793(01)02836-8. [DOI] [PubMed] [Google Scholar]
- 26.von Haefen C, Wieder T, Gillissen B, Starck L, Graupner V, Dorken B, Daniel PT. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–4019. doi: 10.1038/sj.onc.1205497. [DOI] [PubMed] [Google Scholar]
- 27.Lee H, Rotolo JA, Mesicek J, Penate-Medina T, Rimner A, Liao WC, Yin X, Ragupathi G, Ehleiter D, Gulbins E, Zhai D, Reed JC, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS One. 2011;6:e19783. doi: 10.1371/journal.pone.0019783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000. doi: 10.1016/j.cell.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganesan V, Perera MN, Colombini D, Datskovskiy D, Chadha K, Colombini M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis. 2010;15:553–562. doi: 10.1007/s10495-009-0449-0. [DOI] [PubMed] [Google Scholar]
- 30.Perera MN, Lin SH, Peterson YK, Bielawska A, Szulc ZM, Bittman R, Colombini M. Bax and Bcl-xL exert their regulation on different sites of the ceramide channel. Biochem J. 2012;445:81–91. doi: 10.1042/BJ20112103. [DOI] [PubMed] [Google Scholar]
- 31.Siskind LJ, Mullen TD, Romero Rosales K, Clarke CJ, Hernandez-Corbacho MJ, Edinger AL, Obeid LM. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J Biol Chem. 2010;285:11818–11826. doi: 10.1074/jbc.M109.078121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 33.Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24:677–688. doi: 10.1016/j.molcel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 34.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Ronchini C, Capobianco AJ. Notch(ic)-ER chimeras display hormone-dependent transformation, nuclear accumulation, phosphorylation and CBF1 activation. Oncogene. 2000;19:3914–3924. doi: 10.1038/sj.onc.1203719. [DOI] [PubMed] [Google Scholar]
- 36.Casson LKHL, Mathews LA, Ferrer M, Southall N, Guha R, Keller JM, Thomas C, Siskind LJ, Beverly LJ. Inhibition of ceramide metabolism sensitizes human leukemia cells to inhibition of BCL2-like proteins. PLoS One. 2013;8:e54525. doi: 10.1371/journal.pone.0054525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beverly LJ, Lockwood WW, Shah PP, Erdjument-Bromage H, Varmus H. Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilinl. Proc Natl Acad Sci USA. 2012;109:E119–126. doi: 10.1073/pnas.1119167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 39.Beverly LJ. Oncogenic driver supersedes BM preparation as the critical determinant of leukemic outcome: a one day protocol for BM harvest, infection and transplantation. Bone Marrow Transplant. 2013 doi: 10.1038/bmt.2012.273. ePub ahead of print. [DOI] [PubMed] [Google Scholar]
- 40.Deng X, Yin X, Allan R, Lu DD, Maurer CW, Haimovitz-Friedman A, Fuks Z, Shaham S, Kolesnick R. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science. 2008;322:110–115. doi: 10.1126/science.1158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, Yue P, Robati M, Phipson B, Fairlie WD, Lee EF, Campbell KJ, Vandenberg CJ, Cory S, Roberts AW, Ludlam MJ, Huang DC, Bouillet P. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012;119:5807–5816. doi: 10.1182/blood-2011-12-400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, Josefsson EC, Alwis I, Ono A, Willcox A, Andrews RK, Mason KD, Salem HH, Huang DC, Kile BT, Roberts AW, Jackson SP. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118:1663–1674. doi: 10.1182/blood-2011-04-347849. [DOI] [PubMed] [Google Scholar]
- 43.Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 44.Kroesen BJ, Jacobs S, Pettus BJ, Sietsma H, Kok JW, Hannun YA, de Leij LF. BcR-induced apoptosis involves differential regulation of C16 and C24-ceramide formation and sphingolipid-dependent activation of the proteasome. J Biol Chem. 2003;278:14723–14731. doi: 10.1074/jbc.M210756200. [DOI] [PubMed] [Google Scholar]
- 45.Senkal CE, Ponnusamy S, Rossi MJ, Bialewski J, Sinha D, Jiang JC, Jazwinski SM, Hannun YA, Ogretmen B. Role of human longevity assurance gene 1 and C18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Mol Cancer Ther. 2007;6:712–722. doi: 10.1158/1535-7163.MCT-06-0558. [DOI] [PubMed] [Google Scholar]
- 46.Laviad EL, Kelly S, Merrill AH, Jr, Futerman AH. Modulation of ceramide synthase activity via dimerization. J Biol chem. 2012;287:21025–21033. doi: 10.1074/jbc.M112.363580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullen TD, Spassieva S, Jenkins RW, Kitatani K, Bielawski J, Hannun YA, Obeid LM. Selective knockdown of ceramide synthases reveals complex interregulation of sphingolipid metabolism. J Lipid Res. 52:68–77. doi: 10.1194/jlr.M009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem. 2002;277:26796–26803. doi: 10.1074/jbc.M200754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siskind LJ, Feinstein L, Yu T, Davis JS, Jones D, Choi J, Zuckerman JE, Tan W, Hill RB, Hardwick JM, Colombini M. Anti-apoptotic Bcl-2 family proteins disassemble ceramide channels. J Biol Chem. 2008;283:6622–30. doi: 10.1074/jbc.M706115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ganesan V, Colombini M. Regulation of ceramide channels by Bcl-2 family proteins. FEBS Lett. 2010;584:2128–2134. doi: 10.1016/j.febslet.2010.02.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.