

Abstract
Cells can sense and migrate towards higher concentrations of adhesive cues such as the glycoproteins of the extracellular matrix and soluble cues such as growth factors. Here, we outline a method to create opposing gradients of adhesive and soluble cues in a microfluidic chamber, which is compatible with live cell imaging. A copolymer of poly-L-lysine and polyethylene glycol (PLL-PEG) is employed to passivate glass coverslips and prevent non-specific adsorption of biomolecules and cells. Next, microcontact printing or dip pen lithography are used to create tracks of streptavidin on the passivated surfaces to serve as anchoring points for the biotinylated peptide arginine-glycine-aspartic acid (RGD) as the adhesive cue. A microfluidic device is placed onto the modified surface and used to create the gradient of adhesive cues (100% RGD to 0% RGD) on the streptavidin tracks. Finally, the same microfluidic device is used to create a gradient of a chemoattractant such as fetal bovine serum (FBS), as the soluble cue in the opposite direction of the gradient of adhesive cues.
Keywords: Bioengineering, Issue 74, Microbiology, Cellular Biology, Biochemistry, Molecular Biology, Biophysics, Cell migration, live cell imaging, soluble and adherent gradients, microcontact printing, dip pen lithography, microfluidics, RGD, PEG, biotin, streptavidin, chemotaxis, chemoattractant, imaging
Introduction
Directed cell migration is a fundamental property of many cells and is a key aspect of many normal physiological processes, including embryonic development, defense against infection and wound healing. In addition, cell migration also plays a prominent role in many diseases such as vascular disease, tumour cell metastasis and chronic inflammation1,2. While the classical states of cell migration - polarization, protrusion extension, formation of adhesion, force generation and rear retraction - are generally accepted3,4 , the elucidation of spatiotemporal mechanisms by which signal integration is coordinated has been more challenging.
The extracellular matrix (ECM) serves as a substrate for cell adhesion, and through inherent chemical5 and physical6 diversity, provides directional cues for cell navigation7,8. In addition to these adhesive cues9, soluble factors10-12 such as chemokines and growth factors can induce directed cell migration through chemoattractant receptors and their downstream motogenic signalling. Currently, it is not known whether engagement and signalling through adhesion receptors (i.e. integrins) or chemoattractant receptors, such as receptor tyrosine kinase (RTK), are dominant. Nor is it known whether hierarchies of receptor systems are cell type specific.
Live cell microscopy gives a wealth of information that is not accessible in bulk assays and can be combined with microfluidic devices to generate gradients of immobilized 13,14 and soluble cues15 16. The method described here utilizes a series of simple and established steps for surface modification in conjunction with a commercially available microfluidic chamber to create an imaging assay for cell migration that can easily be implemented in a cell biology laboratory (Figure 1). The assembled microfluidic device has optical qualities that are comparable to glass17 and the gradients of diffusible molecules are stable for at least 16 hr. The system can be used for epifluorescence and advanced microscopy techniques. Unlike other chemotaxis setups18, this system is suitable for recording slowly migrating, adherent cells. Importantly, the system is modular and easily allows the introduction of alternative adhesive or soluble migration cues and examination of various cell types.
Protocol
1. Passivation of Glass Coverslips with PLL-PEG-biotin
This step is designed to passivate the surface so that cells adhere and migrate onto specific regions on the glass coverslip that are created with microcontract printing (Step 2-3a) or dip pen lithography (Step 3b). For the passivation, a co-polymer of poly-L-lysine (PLL) and polyethylene glycol (PEG) is used in which 20% of PEG molecules are grafted to biotin (PLL-PEG-biotin).
To clean glass coverslips (18 mm x 18 mm), submerge them in a rack in 100% ethanol and place the rack in a sonicating bath for 30 min. Dry the glass coverslips in air or under a stream of nitrogen.
Next, sonicate the glass coverslips for 30 min in 1 M NaOH. Afterwards rinse the glass surfaces by carefully dipping the rack in a beaker filled with 1.5 L Milli-Q H2O. Repeat the rinsing process three times using fresh Milli-Q H2O each time. Dry the glass coverslips in an oven at 60 °C.
Place half of the clean glass coverslips individually in a humid chamber, which is made of a 24-well plate partially filled with H2O. Drop 15 μl of PLL-PEG-biotin (1 mg/ml) in PBS on each glass coverslip. Take the other half of the remaining clean glass coverslips and carefully sandwich the PEG solution. Leave the coverslips for 1 hr in the humid chamber. Slide the sandwiched coverslips gently away from each other without scraping the PEG-coated surface and rinse them with Milli-Q H2O. The water should slide off easily on the PEG-treated side. If necessary, air-dry the glass coverslips on paper towel with the treated surface facing upward. Store the glass coverslips in a vacuum desiccator until further use.
2. Fabrication of Stamps for Microcontact Printing
We describe two processes to pattern streptavidin tracks onto the passivated glass coverslips. The first one, microcontact printing, typically requires the fabrication of a silicon master from which a PDMS stamp is cast. In our case, the pattern on the PDMS stamp are 100 μm-wide lines that are spaced 290 μm center-to-center with a feature height of 25 μm. Below, we describe briefly how to fabricate a suitable silicon master (Step 2.1-2.3) and PDMS stamp (Step 2.4). However, there are multiple protocols for microcontact printing described in the literature19-23, which are suitable for printing protein patterns onto glass coverslips. Masters can also be custom made and purchased from commercial sources.
First, clean the silicon wafer (4" in diameter) by incubating it in piranha solution (3:1 v:v of sulphuric acid and 30% hydrogen peroxide) for 20 min. Rinse the wafer thoroughly 10 times with MilliQ H2O. Dry the wafer in an oven at 150 °C O/N.
Rinse the clean and dried silicon wafer with 100% acetone, followed by 100% isopropyl alcohol. Next spin-coat 2 ml of GM1070 SU-8 photoresist (Gersteltec Sarl) onto the wafer at 3,100 rpm for 40 sec to create a 25 μm thick layer. The sample is then baked on a hot plate at 65 °C for 5 min followed by a further 5 min at 95 °C.
Expose the SU-8 photoresist to UV light for 60 sec under a photomask with the desired pattern (for example using a Quintel Q6000 mask aligner) and repeat the baking process describe in 2.2. Then the sample is developed by immersing the wafer in SU-8 developer (Gersteltec Sarl) for 4 min, rinsed in 100% isopropyl alcohol and dried under nitrogen flow. The area of the SU-8 photoresist that was not exposed to UV light is thus removed, creating the inverse pattern of the PDMS stamp.
The next step is to cast a PDMS stamp from the silicon master. First, thoroughly mix 1 part (w/w) of the curing agent with 10 parts of elastomer prepolymer (Sylgard 184). To remove any air bubbles, degas the mixture in a desiccator attached to a vacuum line for 1 hr. Next, pour the PDMS elastomer onto the silicon master inside a Petri dish to about 1 cm in height and cure the master and PDMS mixture in an oven at 70 °C for 1 hr. Finally, the PDMS stamp is removed from the silicon master and cut to size.
3. Patterning Streptavidin or Adhesive Cues onto Passivated Glass Coverslips
After passivating glass coverslips, protein patterns are printed with microcontact printing (Step 3a) or dip-pen lithography (Step 3b). In our case, streptavidin is printed as lines, which form the basis for RGD gradients (Step 4). The same procedure can also be used for printing matrix proteins to create adhesive patterns.
An alternative method to microcontact printing for the patterning of biomolecules onto surfaces is dip pen lithography. In dip pen lithography, a cantilever is used to transfer a small volume of solution onto the surface. The size of the cantilevers, viscosity of the solution, contact time of the cantilever on the surface, hydrophobicity of the surface and humidity in the printing chamber determine the size of the printed features. The advantage of this method is that different patterns can be printed for each experiment, as there is no need to fabricate masters and stamps. The disadvantage is that it takes longer to print a given pattern and that dip pen lithography requires a specialist instrument that may not be available in many laboratories. The dip pen lithography instrument that we used was the NanoeNabler from Bioforce Nanoscience. Here, we printed small dots that were <10 μm in diameter, for which we used SPT10 cantilevers.
3a. Microcontact Printing (μCP) Technique
To clean and make the PDMS stamp more hydrophilic, treat the patterned side of the PDMS stamps in an UV and ozone (for example, UV Ozone Procleaner from Bioforce Nanoscience) for 1.5 hr. Alternatively, use a plasma cleaner for a shorter time for the same purpose. This process creates an oxidized PDMS surface24, which improves the printing process.
Immediately after the ozone or plasma treatment, place and spread 10 μl streptavidin (1 mg/ml in PBS) onto the PDMS stamps and leave for 1 hr in a humid chamber (such as a partially filled 6-well plate, see Step 1.3). If tracks should be visible under the fluorescent microscope, use streptavidin that is conjugated to a fluorophore such as streptavidin-AlexaFluor350.
Remove excess streptavidin from the stamp with tissue paper and air dry the stamp for approximately 1 min. Lightly press the stamp onto the PLL-PEG-biotin-coated glass coverslip.
If fluorescent streptavidin was used in Step 3.a2, examine the pattern using under an epifluorescence microscope. Store printed surfaces in a desiccator.
3b. Dip Pen Lithography
First, mix 1 part of 1 mg/ml streptavidin or streptavidin-AlexaFluor350 with 10 parts of glycerol (v/v) and load 5 μl of the mixture onto the cantilever. Then begin the printing process as described in the operation manual of the dip pen lithography instrument. To reduce the spot size to approximately 5 μm, adjust printing speed, contact time of the cantilever on the surface or vertical distance of the cantilever to the surface. In the example shown in Figure 2c, dots were printed in a line with row and column separation set to 10-15 μm. This distance is sufficient to prevent dots from merging into each other but allows the viewing of multiple dots in a single field of view with most microscope settings.
Store printed surfaces in a desiccator.
4. Creating Adhesive Gradients onto Streptavidin-pattern Surfaces
We create adhesive gradients of biotin-RGD onto glass coverslips that are coated with streptavidin (Figure 3) or contain streptavidin patterns on passivated glass coverslips (Figure 5). To create a surface gradient, we used a commercially available microfluidic device (called sticky-Slide Chemotaxis 3D from Ibidi) that creates a stable solution gradient by passive diffusion. To compete for streptavidin binding sites, we employed two opposing gradients of biotin-RGD and biotin that was not conjugated to RGD (see Figure 1).
Adhere the streptavidin-coated or -patterned glass coverslips onto the sticky side of the sticky-Slide Chemotaxis 3D device. To ensure that there is no leakage for several hours, the edges of the device are sealed with a thin layer of warmed Vaseline and with a second layer of a mixture of 1 part of Vaseline to 1 part of paraffin wax (w/w).
Fill the channel of the device with 6 μl PBS in the channel. Then fill the two reservoirs with 70 μl of 0.03 μg/μl biotin-4-fluorescein in PBS and 70 μl of 0.03 μg/μl biotin-RGD in PBS, respectively. Incubate the samples at RT in the dark for 1 hr.
Remove the biotin solutions and carefully rinse the surface while still attached to the microfluidic device twice with PBS. Mark the direction of the adhesive gradient and if needed, store the device filled with PBS at 4 °C for but ensure that the device does not dry out. If required, analyze the adhesive gradient with a fluorescent microscope.
5. Labelling of Cells with Fluorophores for Live Cell Imaging
Cells are labelled with fluorescent dyes that assist cell tracking under the fluorescent microscope. Common probes are fluorescent organelle markers, such Syto 64 Red, which labels the cell nucleus. To do so, cells are first washed three times with PBS, then incubated with 1 μM Syto 64 Red in culture media for 45 min and finally washed a further three times with PBS. Please note that cells should not be kept in PBS for long periods of time.
Alternative dyes for cell tracking are CellTracker series that stain the cytoplasm and are retained during cell migration and division. Transfection of cells with fluorescent fusion protein is particularly useful to record the subcellular distributions of a protein of interest during migration. However, fluorescent proteins typically photo-bleach faster than organic dyes so that long-term observations may not be possible.
6. Loading of Cells with a Soluble Gradient into the Microfluidics Device
Count fluorescently labelled cells and split into two tubes of equal cell numbers. Wash and resuspend one tube of cells with media containing the chemoattractant, such as low glucose Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS). Wash and resuspend the other tube of cells in media without the chemoattractant, i.e. DMEM with 0% FBS. The final concentration of cells in each tube should be 5 x 105 cells/ml.
Remove all the solutions from the microfluidic device (from Step 4.3) and place it on the microscope stage (see Step 7). Load 70 μl of cells (5 x 105 cells/ml in 0% FBS DMEM) into one reservoir and 70 μl of HeLa cells (5 x 105 cells/ml in 10% FBS DMEM) into another. Loosely place a tape over the reservoirs to avoid evaporation. If cells on the surface are pre-incubated prior to imaging, it is advisable that the cells are loaded into the device in growth media without the gradient. The device is then placed on the microscope stage and the media replaced with gradient media i.e. DMEM without FBS in one reservoir and DMEM with 10% FBS in the other.
7. Live Cell Imaging and Data Analysis
Switch on the microscope (Nikon Ti-E) and warm up the stage at least 1 hr before starting the experiment.
Ensure that the channel of the microfluidic device is in the field of view and cells are in focus. Select image acquisition parameter such as exposure times and fluorescent filters, image sequence of bright field and fluorescence that captures cells and tracks as well as time points and recording length (e.g. every 15 min for 16 hr).
Analyze data with appropriate softwares such as ImageJ Manual Tracking or Ibidi Chemotaxis and Migration Tool.
Representative Results
To understand how cell integrate migratory signals25, we have developed a method to image cells with fluorescence microscopy that migrate in an environment with competing adhesive and soluble gradients (Figure 1). Adhesive tracks that contained fluorescent streptavidin and biotinylated RGD were created with microcontact printed tracks and dip pen lithography (Figure 2). Successful microcontact printing is indicated by the line profile of the fluorescence intensity across the tracks where adhesive tracks and anti-fouling regions can clearly be distinguished (Figure 2b). Dots printed with dip pen lithography appear rounded, not tear shaped, indicating a successful printing process. Surface chemistry and humidity influences the outcome of the printing process. Generally speaking, the more hydrophobic the surface is, the better the printing outcomes.
A gradient of adhesive cues on the printed track were created with a simple microfluidics device by loading biotin-4-fluorescein and biotin-RGD into the opposite side of a microfluidic chamber (Figure 3). Since both molecules bind competitively to the streptavidin track on the glass surface, an immobilized gradient of RGD ligands is generated. After removing the biotin/biotin-RGD solution, the same chamber can be used to introduce a soluble gradient, which is stable within the microfluidic channel for at least 16 hr (Figure 4).
We introduced HeLa cells into the chamber with a chemoattractant gradient, for which FBS was used. At low densities, HeLa cells preferentially adhered to and migrated on the streptavidin/RGD tracks (Ncells = 50) and avoided the anti-fouling PEG regions (Ncells = 19). The position of individual cells was recorded over 16 hr and analyzed with a cell tracking software. In an experiment where adhesive and soluble gradients are opposed to each other (Figure 5), the trajectories of individual cells showed that more HeLa cells migrated towards the source of the chemoattractant (red traces in Figure 5c, Ntraces = 36) than towards higher concentrations of adherent RGD (black traces in Figure 5c, Ntraces = 14). The average distance that cells migrated against (black traces) or in (red traces) the direction of the FBS gradient (x-component of average displacement of each trajectory) was 0.1700 ± 0.1172 and 0.2278 ± 0.1280, respectively (P < 0.0001, Student t-test) although there was no statistically significant difference in average displacement between the black traces (56.18 ± 32.27 μm) and the red traces (72.86 ± 45.05 μm; P > 0.05, Student t-test). HeLa cells migrate with an average speed of 17.47 ± 4.72 μm/hr in the presence of both gradient of adhesive cue (i.e. RGD) and soluble cue (i.e. FBS). The data therefore suggest that for HeLa cells, the FBS gradient determines the direction of migration while the cells maintain an intrinsic migration speed on the adhesive tracks.
We have also assessed the migration of the macrophage cell line J774. In the absence of adhesive and soluble gradients, these cells migrate at much higher speeds (38.55 ± 17.88 μm/hr) than HeLa cells (16.47 ± 4.28 μm/hr) but with less persistency i.e. J774 cells change direction (directionality of 0.059 ± 0.091), measured as the ratio of displacement to track length more often than HeLa cells (0.57 ± 0.12) on microcontact printed fibronectin tracks. J774 cells are also less contact dependent than HeLa cells. The displacement of J774 cells was higher on the PEG regions (102.20 ± 54.73 μm) than on fibronectin tracks (42.95 ± 39.90 μm). In addition, J774 cells appeared to adapt to different surface chemistry by changing their mechanism of migration from mesenchymal to amoeboid. In conclusion, adhesive cues are not as important to J774 cells as they are for HeLa cells. Both cell types preferentially migrated towards the direction of the soluble cue.
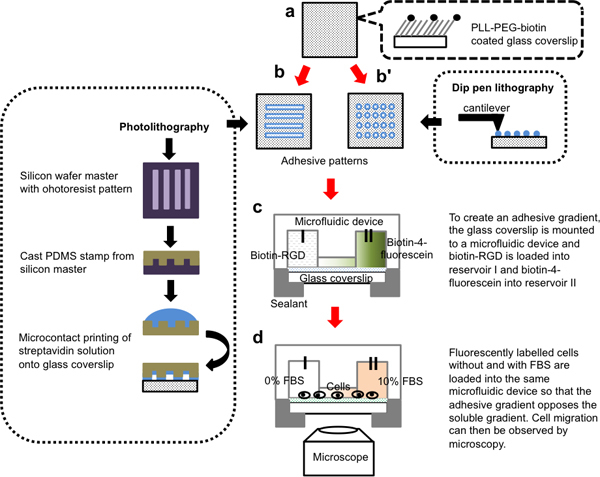 Figure 1. Schematic of the fabrication process of a microfluidic setup that allows the imaging of migrating cells responding to opposite gradients of adhesive versus soluble cues.a. Glass coverslips are passivated with biotinylated PLL-PEG. b. Adhesive patterns containing fluorescent streptavidin are printed onto the biotinylated surface either as lines using microcontact printing (b) or dots using dip pen lithography (b'). c. The modified surface is attached to the bottom of the microfluidic device. Gradients of immobilized biotin-RGD are created via reservoir I and II. d. Cells and a soluble chemoattractant gradient of 0-10% FBS are loaded into the microfluidic device. After cells adhere to the surface, live cell migration over RGD gradients on streptavidin tracks and in the presence of FBS gradients can be imaged in the channel connecting the two reservoirs.
Figure 1. Schematic of the fabrication process of a microfluidic setup that allows the imaging of migrating cells responding to opposite gradients of adhesive versus soluble cues.a. Glass coverslips are passivated with biotinylated PLL-PEG. b. Adhesive patterns containing fluorescent streptavidin are printed onto the biotinylated surface either as lines using microcontact printing (b) or dots using dip pen lithography (b'). c. The modified surface is attached to the bottom of the microfluidic device. Gradients of immobilized biotin-RGD are created via reservoir I and II. d. Cells and a soluble chemoattractant gradient of 0-10% FBS are loaded into the microfluidic device. After cells adhere to the surface, live cell migration over RGD gradients on streptavidin tracks and in the presence of FBS gradients can be imaged in the channel connecting the two reservoirs.
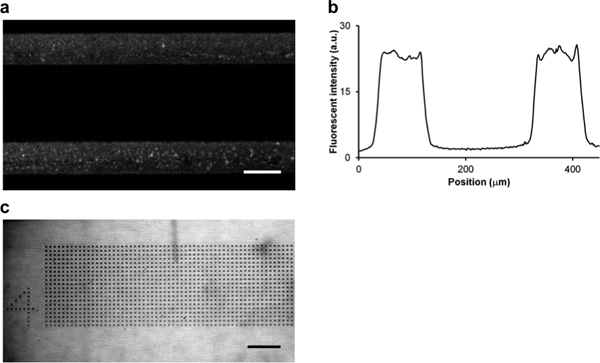 Figure 2. Cell adhesive patterns (lines and dots) can be generated with two different printing techniques.a. Fluorescent image of microcontact printed streptavidin-AlexaFluor350 tracks (Abs: 346 nm, Em: 442 nm; grey) on PLL-PEG-coated glass coverslips. The microcontract printing mask was designed as lines that were 100 μm wide with 290 μm center-to-center space. b. The average fluorescent intensity of microcontact printed streptavidin tracks, as shown in a, from five separate surfaces as cross-sections from top to bottom. The average width of the track is 90 ± 6 μm and the average center-to-center spacing is 293 ± 2 μm. c. A brightfield image of streptavidin-containing dots generated with dip pen lithography. The diameters of the dots ranged from 6 - 9 μm with 15 μm center-to-center space. Scale bars in a andc are 100 μm.
Figure 2. Cell adhesive patterns (lines and dots) can be generated with two different printing techniques.a. Fluorescent image of microcontact printed streptavidin-AlexaFluor350 tracks (Abs: 346 nm, Em: 442 nm; grey) on PLL-PEG-coated glass coverslips. The microcontract printing mask was designed as lines that were 100 μm wide with 290 μm center-to-center space. b. The average fluorescent intensity of microcontact printed streptavidin tracks, as shown in a, from five separate surfaces as cross-sections from top to bottom. The average width of the track is 90 ± 6 μm and the average center-to-center spacing is 293 ± 2 μm. c. A brightfield image of streptavidin-containing dots generated with dip pen lithography. The diameters of the dots ranged from 6 - 9 μm with 15 μm center-to-center space. Scale bars in a andc are 100 μm.
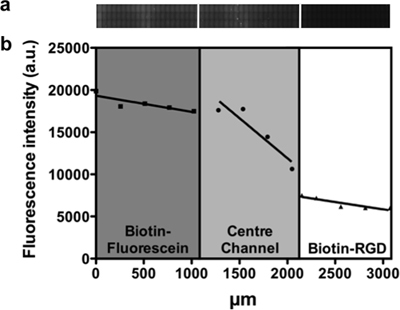 Figure 3. Biotin-4-fluorescein gradient (0 μg/μl - 0.03 μg/μl) on a glass surfaces uniformly coated with streptavidin. Biotin-4-fluorescein and biotin-RGD were loaded in either reservoir of the microfluidic device (sticky-Slide Chemotaxis 3D, Ibidi) that are connected by a 1 mm long channel. After a 1 hr incubation, the device was rinsed with PBS and refilled with PBS. a. 60 mosaic images were taken with a total image length 3,072 μm and b the fluorescence intensity was measured across the length of the entire mosaic. Linear trend lines were fitted to the fluorescence intensity to indicate the RGD gradient in the channel.
Figure 3. Biotin-4-fluorescein gradient (0 μg/μl - 0.03 μg/μl) on a glass surfaces uniformly coated with streptavidin. Biotin-4-fluorescein and biotin-RGD were loaded in either reservoir of the microfluidic device (sticky-Slide Chemotaxis 3D, Ibidi) that are connected by a 1 mm long channel. After a 1 hr incubation, the device was rinsed with PBS and refilled with PBS. a. 60 mosaic images were taken with a total image length 3,072 μm and b the fluorescence intensity was measured across the length of the entire mosaic. Linear trend lines were fitted to the fluorescence intensity to indicate the RGD gradient in the channel.
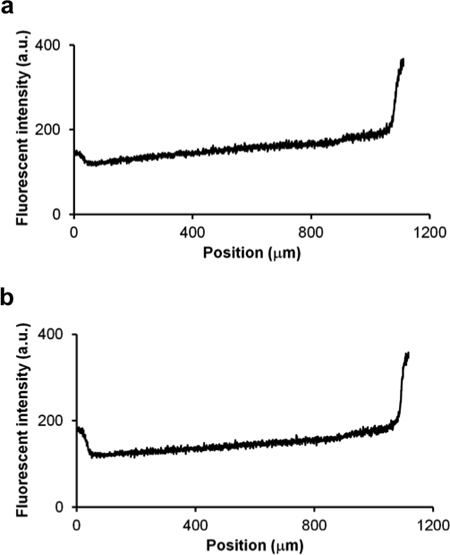 Figure 4. The fluorescence intensity of a fluorescein gradient (0 μM - 1 μM) in the microfluidic channel a immediately after setup (T = 0 hr) and b after a 16 hr incubation. PBS without and with 1 μM fluorescein was loaded from the left and right reservoir of the microfluidic device (sticky-Slide Chemotaxis 3D, Ibidi), respectively. The microfluidic channel is 1 mm in length and is found at position 50 to 1,050 μm. The gradient was stable for at least 16 hr.
Figure 4. The fluorescence intensity of a fluorescein gradient (0 μM - 1 μM) in the microfluidic channel a immediately after setup (T = 0 hr) and b after a 16 hr incubation. PBS without and with 1 μM fluorescein was loaded from the left and right reservoir of the microfluidic device (sticky-Slide Chemotaxis 3D, Ibidi), respectively. The microfluidic channel is 1 mm in length and is found at position 50 to 1,050 μm. The gradient was stable for at least 16 hr.
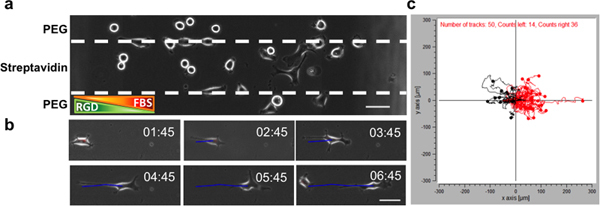 Figure 5. HeLa cell migration in a microfluidics chamber (sticky-Slide Chemotaxis 3D, Ibidi) with opposing adhesive gradient (immobilized RGD on streptavidin tracks) and soluble gradient (0-10% FBS).a. A representative image of cells in a microfluidics chamber with opposing gradients. Scale bar is 50 μm. b Time-lapse images of a cell taken with an epifluorescence microscope (Nikon Ti-E). Images were taken at 15 min intervals for 20 hr and cell trajectory (blue line) obtained via cell centroid positioning (ImageJ manual tracking plugin). Scale bar is 50 μm. c. Cell trajectory from images as shown in b. Cell trajectories that migrated towards the higher concentration of soluble FBS are shown in red (Ntraces = 36); cell trajectories that migrated towards higher concentrations of adherent RGD are shown in black (Ntraces = 14). Click here to view larger figure.
Figure 5. HeLa cell migration in a microfluidics chamber (sticky-Slide Chemotaxis 3D, Ibidi) with opposing adhesive gradient (immobilized RGD on streptavidin tracks) and soluble gradient (0-10% FBS).a. A representative image of cells in a microfluidics chamber with opposing gradients. Scale bar is 50 μm. b Time-lapse images of a cell taken with an epifluorescence microscope (Nikon Ti-E). Images were taken at 15 min intervals for 20 hr and cell trajectory (blue line) obtained via cell centroid positioning (ImageJ manual tracking plugin). Scale bar is 50 μm. c. Cell trajectory from images as shown in b. Cell trajectories that migrated towards the higher concentration of soluble FBS are shown in red (Ntraces = 36); cell trajectories that migrated towards higher concentrations of adherent RGD are shown in black (Ntraces = 14). Click here to view larger figure.
Discussion
In this protocol, we used a commercially available microfluidic device (sticky-Slide Chemotaxis 3D from Ibidi) to study the effects of chemically modified surfaces and chemoattractant gradients on cell migration. This microfluidic setup does not require flow because the gradient is established by diffusion along the length of the channel. This is important because flow could differentially affect slowly migrating cells such as fibroblasts that form stable focal adhesions and fast migrating cells like most leukocytes that form small focal complexes and synapses26. Shear stress from laminar flow may affect the stability of cell adhesion to adhesive cues and cell chemotaxis towards soluble cues27,28. We found differences in migration behavior between slow moving HeLa cells and fast moving J774 cells thereby showing that the microfluidic setup is applicable for both adherent and less adherent cell types. Such comparisons also revealed interesting difference between cell types in adhesion and migration responses to surface patterns that contained both adhesive cues (RGD peptides or fibronectin) and anti-fouling regions (PEG areas).
Surface patterns were created with microcontact printing29 and dip-pen lithography30,31. Microcontact printing is a simple and relatively fast, reproducible patterning process. However, the design and fabrication of the PDMS stamp, which is achieved by photolithography, is more involved and time consuming. It also introduces its own limitations in that small features (< 5 μm) are difficult to achieve and depend on the aspect ratio of the pattern. In brief, microcontact printing is ideal for large patterns up to hundreds of micron size and in experiments, where the same patterns need to created many times. On the other hand, dip pin lithography can be used to generate micron to nanoscaled patterns. It is more suited to printing dots than lines, although lines can be fabricated. Dip pin lithography affords greater freedom in pattern designs, as there is no need for pre-fabricated masks, masters or stamps. However, the speed of the patterning process is slow, making printing of large areas cumbersome and access to a specialized instrument is crucial. The two patterning methods are relevant for consideration because the size and shape of adhesive cue patterns influence the organization and distribution of biomechanical machineries that regulate cell migration32,33. Additionally, the limitation of the commercial microfluidic device employed here is that the channel length (1 μm) cannot be altered and so one cannot control the maximum steepness of the adhesive and soluble gradients. If steeper gradients are necessary, one would need to fabricate a microfluidic device with a shorter channel between the two reservoirs. A key advantage of the protocol described here is the modular design so that different surfaces, surface chemistries, patterning approaches and microfluidic devices could be combined for a wide variety of live cell imaging experiments.
Disclosures
We have no commercial interests to disclose.
Acknowledgments
The authors acknowledge funding from the Australian Research Council and National Health and Medical Research Council of Australia and also like to thank theAustralian National Fabrication Facility for the SU-8 master for the microcontact printing. SHN is supported by the Ministry of Higher Education Malaysia and Universiti Sains Malaysia.
References
- Pulsipher A, Yousaf MN. Surface Chemistry and Cell Biological Tools for the Analysis of Cell Adhesion and Migration. Chem. BioChem. 2010;11:745–753. doi: 10.1002/cbic.200900787. [DOI] [PubMed] [Google Scholar]
- Hall A. The cytoskeleton and cancer. Cancer Metastasis Reviews. 2009;28:5–14. doi: 10.1007/s10555-008-9166-3. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nature Reviews Cancer. 2011;11:573–587. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harbor Perspectives in Biology. 2011;3 doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngalim SH, Magenau A, Saux GLe, Gooding JJ, Gaus K. How do cells make decisions: engineering micro- and nanoenvironments for cell migration. Journal of Oncology. 2010;2010:363106. doi: 10.1155/2010/363106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy DC, Wilke-Mounts SJ, Hocking DC. Chimeric fibronectin matrix mimetic as a functional growth- and migration-promoting adhesive substrate. Biomaterials. 2011;32:2077–2087. doi: 10.1016/j.biomaterials.2010.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T, Xu X, Hereld D. Chemotaxis, chemokine receptors and human disease. Cytokine. 2008;44:1–8. doi: 10.1016/j.cyto.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Jin T. Imaging G-protein Coupled Receptor (GPCR)-mediated Signaling Events that Control Chemotaxis of Dictyostelium Discoideum. J. Vis. Exp. 2011. p. e3128. [DOI] [PMC free article] [PubMed]
- Chung BG, et al. A Gradient-generating Microfluidic Device for Cell Biology. J. Vis. Exp. 2007. p. e271. [DOI] [PMC free article] [PubMed]
- Rhoads DS, Guan JL. Analysis of directional cell migration on defined FN gradients: role of intracellular signaling molecules. Exp. Cell Res. 2007;313:3859–3867. doi: 10.1016/j.yexcr.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, et al. Simple haptotactic gradient generation within a triangular microfluidic channel. Lab on a Chip. 2010;10:2130–2138. doi: 10.1039/b924222h. [DOI] [PubMed] [Google Scholar]
- Li Jeon N, et al. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat. Biotech. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- Joanne Wang C, et al. A microfluidics-based turning assay reveals complex growth cone responses to integrated gradients of substrate-bound ECM molecules and diffusible guidance cues. Lab on a Chip. 2008;8:227–237. doi: 10.1039/b713945d. [DOI] [PubMed] [Google Scholar]
- Frequently Asked Questions (FAQ) :: ibidi [Internet] ibidi GmbH; 2010. Available from: http://www.ibidi.com/service/faq.html. [Google Scholar]
- Pujic Z, Mortimer D, Feldner J, Goodhill GJ. Assays for eukaryotic cell chemotaxis. Combinatorial Chemistry & High Throughput Screening. 2009;12:580–588. doi: 10.2174/138620709788681952. [DOI] [PubMed] [Google Scholar]
- Thery M, Piel M. Adhesive micropatterns for cells: a microcontact printing protocol. Cold Spring Harbor protocols. 2009;2009:pdb prot5255. doi: 10.1101/pdb.prot5255. [DOI] [PubMed] [Google Scholar]
- Shen K, Qi J, Kam LC. Microcontact Printing of Proteins for Cell Biology. J. Vis. Exp. 2008. p. e1065. [DOI] [PMC free article] [PubMed]
- Shin Y, et al. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat. Protocols. 2012;7:1247–1259. doi: 10.1038/nprot.2012.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D, Xia Y, Whitesides GM. Soft lithography for micro- and nanoscale patterning. Nat. Protocols. 2010;5:491–502. doi: 10.1038/nprot.2009.234. [DOI] [PubMed] [Google Scholar]
- Huang Y, Agrawal B, Sun D, Kuo JS, Williams JC. Microfluidics-based devices: New tools for studying cancer and cancer stem cell migration. Biomicrofluidics. 2011;5:13412. doi: 10.1063/1.3555195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillborg H, et al. Crosslinked polydimethylsiloxane exposed to oxygen plasma studied by neutron reflectometry and other surface specific techniques. Polymer. 2000;41(00):6851–6863. [Google Scholar]
- Liu L, Ratner BD, Sage EH, Jiang S. Endothelial Cell Migration on Surface-Density Gradients of Fibronectin, VEGF, or Both Proteins. Langmuir. 2007;23:11168–11173. doi: 10.1021/la701435x. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. The Journal of Cell Biology. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazzam F, DeLano FA, Zweifach BW, Schmid-Schönbein GW. The leukocyte response to fluid. Proceedings of the National Academy of Sciences. 1997;94:5338–5343. doi: 10.1073/pnas.94.10.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker GM, et al. Effects of flow and diffusion on chemotaxis studies in a microfabricated gradient generator. Lab on a Chip. 2005;5:611–618. doi: 10.1039/b417245k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A, Reinhoudt DN, Huskens J. Microcontact Printing: Limitations and Achievements. Adv. Mater. 2009;21:2257–2268. [Google Scholar]
- Salaita K, Wang Y, Mirkin CA. Applications of dip-pen nanolithography. Nat. Nano. 2007;2:145–155. doi: 10.1038/nnano.2007.39. [DOI] [PubMed] [Google Scholar]
- Wouters D, Schubert US. Nanolithography and nanochemistry: probe-related patterning techniques and chemical modification for nanometer-sized devices. Angewandte Chemie (International ed. in English) 2004;43:2480–2495. doi: 10.1002/anie.200300609. [DOI] [PubMed] [Google Scholar]
- Xia N, et al. Directional control of cell motility through focal adhesion positioning and spatial control of Rac activation. FASEB J. 2008;22:1649–1659. doi: 10.1096/fj.07-090571. [DOI] [PubMed] [Google Scholar]
- Oakes PW, Beckham Y, Stricker J, Gardel ML. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. The Journal of Cell Biology. 2012;196:363–374. doi: 10.1083/jcb.201107042. [DOI] [PMC free article] [PubMed] [Google Scholar]