

Abstract
Purpose
Sterile corneal infiltrates can cause pain, blurred vision, and ocular discomfort in silicone hydrogel contact-lens users. The current study investigates the potential for the synthetic lymphocyte functional antigen-1 (LFA-1) antagonist lifitegrast (SAR 1118) to block corneal inflammation using a murine model.
Methods
The role of LFA-1 (CD11a/CD18) was examined either in CD18−/− mice, by intraperitoneal injection of anti-CD11a, or by topical application of lifitegrast. Corneal inflammation was induced by epithelial abrasion and exposure to either tobramycin-killed Pseudomonas aeruginosa or Staphylococcus aureus in the presence of a 2-mm-diameter punch from a silicone hydrogel contact lens. After 24 h, corneal thickness and haze were examined by in vivo confocal microscopy, and neutrophil recruitment to the corneal stroma was detected by immunohistochemistry.
Results
Neutrophil recruitment to the corneal stroma and development of stromal haze were significantly impaired in CD18−/− mice or after injection of anti-CD11a. Topical lifitegrast also inhibited P. aeruginosa- and S. aureus–induced inflammation, with the optimal application being a 1% solution applied either 2 or 3 times prior.
Conclusion
As LFA-1-dependent neutrophil recruitment to the corneal stroma can be blocked by topical lifitegrast, this reagent could be used in combination with antibiotics to prevent leukocyte infiltration to the corneal stroma in association with contact-lens wear.
Introduction
The estimated number of contact lens wearers exceeds 34 million in the United States and 140 million worldwide.1 Silicone hydrogel lenses are among the most commonly used, as they are relatively inexpensive and more broadly tolerated than hard lenses. However, silicone hydrogel use also increases the risk for sterile corneal infiltrates, including contact-lens peripheral ulcers (CLPU) and contact-lens-associated red eye (CLARE).1–4 Patients with CLPU or CLARE experience a syndrome of severe pain, redness, blurred vision, and ocular discomfort, and may discontinue wearing contact lenses.
Although the cause of sterile corneal infiltrates has yet to be formally demonstrated, a major risk factor for developing corneal infiltrates is isolation of bacteria from lenses or lens-care products, but not from the cornea (reviewed in Refs.2,3). A recent study showed significantly increased bacterial bioburden from silicone hydrogel lenses from patients experiencing corneal infiltrates compared with asymptomatic wearers, which isolated gram-positive and gram-negative bacteria, including Staphylococcus and Pseudomonas species.5 As many of the patients with corneal infiltrates also showed defects in the corneal epithelium by fluorescein staining,5 there is a need for an agent that protects the corneal surface and prevents leukocyte infiltration.
We have used murine models to identify potential causes of corneal inflammation and demonstrated that when the epithelium is abraded, activation of toll-like receptors (TLR)4 and TLR2 by killed Pseudomonas aeruginosa and Staphylococcus aureus, respectively, leads to production of proinflammatory and chemotactic cytokines in the cornea, neutrophil infiltration, and loss of corneal clarity demonstrated by confocal imaging.6–9
This model of corneal infiltrates is consistent with reports of bacterial isolation from lenses and lens cases of contact-lens wearers with corneal infiltrates.3,10 Together with biopsy data showing neutrophil infiltration,11 it seems reasonable to predict that bacterial cell wall components contribute to development of sterile infiltrates by activating TLRs on the corneal epithelium and by resident macrophages.
In the current study, we examine the role of lymphocyte functional antigen-1 (LFA-1 and CD11a/CD18), which is an integrin receptor expressed on leukocytes that binds to intercellular adhesion molecule-1 (ICAM-1) expressed on vascular endothelial cells, as the LFA-1/ICAM-1 interaction is an essential step in leukocyte extravasation from the capillary endothelial cells into surrounding tissues.12 In the cornea, ICAM-1 is important in neutrophil recruitment to the corneal stroma in P. aeruginosa keratitis,13 and in wound-healing models, leukocyte migration in the corneal stroma involves LFA-1/ICAM-1 interactions on neutrophils and keratocytes, respectively.14,15 As leukocytes are also important in sterile inflammatory responses associated with contact lens wear,11,16 specific inhibition of LFA-1/ICAM-1 interactions is a potential target to limit corneal inflammation.
We also examine the role of LFA-1 in corneal inflammation using lifitegrast (SAR 1118), which is a small-molecule LFA-1 antagonist that binds the CD11a subunit of LFA-1, thereby blocking the interaction between LFA-1 and ICAM-1.17 Topical application of lifitegrast to the corneal surface of healthy adults is safe and well tolerated,18 and has shown efficacy in canine keratoconjunctivitis sicca and in a rat model of diabetic retinopathy.19,20 SAR 1118 was also found to improve signs and symptoms of Dry Eye Disease in a recent study of 230 patients.21 In the current study, we demonstrate that lifitegrast, has potent anti-inflammatory activity on corneal inflammation induced by antibiotic-killed P. aeruginosa and S. aureus in the presence of a silicone hydrogel lens. Lifitegrast therefore has a potential clinical application in preventing and treating contact-lens-associated corneal infiltrates and epithelial defects caused by gram-positive or gram-negative bacteria.
Methods
Source of bacterial strains
P. aeruginosa strain ATCC 19660 was obtained from ATCC, and S. aureus strain 8325-4 was a gift from Dr. Richard O'Callaghan (University of Mississippi). Bacteria were grown overnight (18 h) in a brain–heart infusion broth, and aliquots from these stationary cultures were diluted 1:100, and grown until OD650=0.2, which we found to be 1×108 CFU/mL. For antibiotic treatment, bacteria were centrifuged, washed once with phosphate-buffered saline (PBS), and resuspended at 2×109 bacteria/mL in 0.3% Tobramycin in PBS (Sigma). Bacterial killing was confirmed by the absence of growth on TSB agar plates.
Mouse strains
C57BL/6 and CD18−/− mice (6–8 weeks old) were obtained from the Jackson Laboratory. All mice were maintained in pathogen-free conditions in microisolator cages, and were treated in accordance with the guidelines provided in the ARVO statement for the Use of Animals in Ophthalmic and Vision Research. The animal protocol was also approved by the Case Western Reserve University IACUC.
Mouse model of contact-lens-associated corneal inflammation
Mice were anesthetized by intraperitoneal injection of 0.4 mL 2,2,2-tribromoethanol. The corneal epithelium was abraded using 3 parallel scratches with a 26-gauge needle. Five microliters bacterial suspension containing 1×107 organisms was placed on the corneal surface, and a 2-mm-diameter punch from a Lotrafilcon™ CIBA VISION silicone hydrogel contact lens was placed on top as described.9 Contact lenses remained on the anesthetized mice for 2 h, after which time they were removed as the animals came out of anesthesia.
To block LFA-1, 150 μg (10 mg/kg, 15 g/per mouse weight) anti-CD11a (muM17; EBioscience) or control rat IgG2a was injected into the peritoneal cavity of C57BL/6 mice 1 day before inducing inflammation. For mice treated topically with lifitegrast (SAR 1118; SARcode Bioscience), the agent was prepared as a 0.1%, 1%, or 5% solution in PBS, and 5 μL was placed on the intact corneal surface either before or after inflammation.
In vivo confocal microscopy analysis of corneal thickness and haze
In vivo analysis of cellular infiltration was accomplished using a Nidek Confoscan™ as described.9 Briefly, anesthetized mice were immobilized, and the cornea was examined using a 40×objective with a transparent gel (Genteal; Novartis Ophthalmics). A series of 1-μm images of the entire cornea was captured using NAVIS™ software, and stromal thickness (area between basal epithelium and corneal endothelium) was measured directly using NAVIS™ software. To measure corneal reflectivity, the light intensity readout of each 1–2-μm image of the corneal stroma was exported into Prism (Graph Pad Software), and the total area under the curve was then calculated as previously described.9
Immunohistochemistry
Eyes were snap-frozen in liquid nitrogen, and 5-μm sections were incubated 2 h with anti-neutrophil antibody NIMP-R14 diluted 1:100 in 1% fetal calf serum/tris buffered saline (TBS) (1% FCS/TBS). After washing, corneal sections were incubated with FITC-conjugated rabbit anti-rat antibody (Vector Laboratories) diluted 1:200 in 1% FCS/TBS. Slides were mounted in Vectashield containing DAPI (Vector Laboratories), and the number of neutrophils in each section was examined by fluorescence microscopy and quantified by direct counting. This approach has been used extensively in our laboratory for studies of corneal inflammation.6–9
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay to detect apoptotic cells in the cornea
The terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay is based on the ability of terminal transferase (TdT) to catalyze attachment of fluorescein dUTP to free 3′OH ends of genomic DNA. Frozen 5-μm corneal sections were incubated with the TUNEL reaction mixture containing TdT and fluorescein-dUTP, and the incorporated fluorescein was detected by fluorescence microscopy. Corneal sections were treated with reagents according to the manufacturer's directions (Roche), counterstained with DAPI to show the nuclei, and the sections were examined by fluorescence microscopy.
Mass spectrometry to detect lifitegrast
Corneas were dissected, frozen at −80°C, and shipped to ICON Development Sciences, LLC, for liquid chromatography–mass spectrometry (LC-MS) analysis using known standards to identify and quantify lifitegrast in the tissue.
Statistics
Statistical analyses were performed using ANOVA with Tukey post hoc analysis (Prism; Graph Pad Software). Statistical significance was defined as a P value <0.05.
Results
Anti-LFA-1 antibody inhibits neutrophil recruitment to the corneal stroma
Neutrophils comprise most of the cellular infiltrate at this time point in response to killed P. aeruginosa, which we showed activates the TLR4 pathway,9,22 or killed S. aureus, which activates the TLR2 pathway.8,23 We therefore examined the effect of LFA-1 blockade on neutrophil recruitment to the corneal stroma. Corneas were abraded, and tobramycin-killed P. aeruginosa or S. aureus were placed on the ocular surface and covered by a 2-mm-diameter punch from a silicone hydrogel contact lens.
Figure 1A shows that mice given anti-CD11a before treatment with tobramycin-killed P. aeruginosa had significantly fewer neutrophils and lower corneal haze and thickness compared with mice given control IgG. In addition, anti-CD11a reduced corneal haze and thickness to the level of trauma controls (abraded corneas given topical PBS), although neutrophil numbers remained higher than trauma controls.
FIG. 1.

Effect of blocking lymphocyte functional antigen-1 (LFA-1) on Pseudomonas aeruginosa- and Staphylococcus aureus-induced corneal inflammation. C57BL/6 mice were treated i.p. with anti-LFA-1 or control rat IgG2a, and corneal inflammation was induced by tobramycin-killed P. aeruginosa (A) or S. aureus (B) in the presence of a contact lens punch. (C) Corneas of CD18−/− and C57BL/6 mice were abraded, and treated with killed P. aeruginosa. After 24 h, corneal thickness and haze were quantified by Confoscan analysis, and neutrophil infiltration to the corneal stroma was determined by direct counting of immunostained corneal sections. Data are mean±SD of 5 mice per group, and are representative of 2 repeat experiments for each organism.
Similarly, there were significantly increased neutrophil numbers, stromal thickness, and haze in mice treated with tobramycin-killed S. aureus compared with PBS-treated corneas (Fig. 1B). In contrast, these indicators of corneal inflammation were significantly lower in anti-CD11a-treated mice. The percent of blood neutrophils was unchanged after anti-CD11a antibody treatment (data not shown), indicating that this antibody did not deplete systemic neutrophil numbers. As a complementary approach, corneas of CD18−/− mice were abraded, and killed P. aeruginosa were added to the ocular surface as before. After 24 h, corneas were examined as before. As shown in Fig. 1C, stromal neutrophils, haze, and thickness were significantly lower than in C57BL/6 mice.
Together, these findings demonstrate that LFA-1 interactions play an essential role in lipopolysaccharide (LPS)- and tobramycin-killed P. aeruginosa- and S. aureus-induced corneal inflammation.
Concentration of lifitegrast in the cornea after topical application
To ascertain the amount of lifitegrast absorbed by the intact cornea after topical application, 5 μL 5%, 1%, or 0.1% lifitegrast was applied to the intact cornea 3 times over 19 h (15, 3, and 1 h before euthanasia). Corneas were examined by LC-MS-MS analysis. As shown in Fig. 2, lifitegrast was detected in corneas treated with each concentration; however, rather than finding a dose–response, a significantly higher concentration of lifitegrast was detected after topical application of 1% compared with 0.1% and 5% S lifitegrast.
FIG. 2.
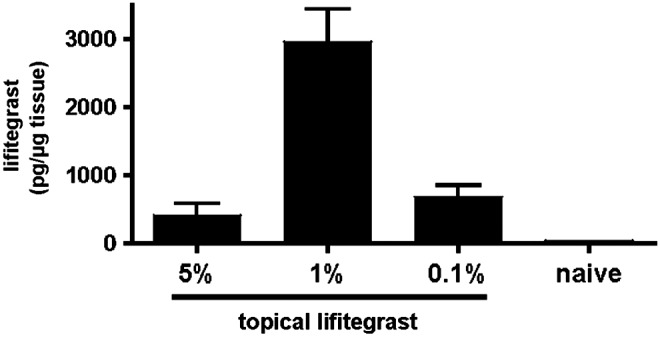
Pharmacokinetics of SAR 1118 in the cornea. A single drop (5 μL) of lifitegrast (5%, 1%, or 0.1%) was applied to the intact cornea 15, 3, and 1 h before euthanasia and corneal dissection. The presence of lifitegrast was determined by liquid chromatography–mass spectrometry. Data are mean±SD of 5 mice per group.
Lifitegrast inhibits P. aeruginosa- and S. aureus-induced corneal inflammation
To determine the effect of lifitegrast on corneal inflammation, 5 μL of 5%, 1%, or 0.1% antagonist was added topically to the intact cornea at 15, 3, and 1 h before inducing inflammation with tobramycin-killed P. aeruginosa as described above. As shown in Fig. 3A, Expt. 1, neutrophil numbers were significantly lower in mice treated with 5%, 1%, or 0.1% lifitegrast compared with PBS controls. Similarly, corneal haze was significantly lower in mice given 1% or 0.1% lifitegrast compared with PBS controls. In Expt. 2, mice given 1% lifitegrast had significantly lower neutrophil numbers and corneal haze compared with controls, whereas there was no effect of the other concentrations, and no effect on corneal thickness. Table 1 shows the results of 8 experiments where lifitegrast was added topically either before or after inflammation. Prior treatment either twice (−15, −3 h), or 3 times as described above showed significant inhibition of neutrophil infiltration at 5%, 1%, or 0.1% lifitegrast; however, the 1% solution was consistently more effective than 5% or 0.1% lifitegrast, with higher percent inhibition and P values of 0.001–0.005. In contrast, a single topical application of lifitegrast before and after inducing inflammation did not significantly inhibit neutrophil recruitment (Table 1, Expts. 6–8). These findings are also consistent with the concentration study (Fig. 3) showing maximal lifitegrast in the corneas after topical application of 1% solution.
FIG. 3.

Effect of lifitegrast on (A) P. aeruginosa- and (B) S. aureus-induced corneal inflammation. C57BL/6 mice were treated with 5%, 1%, or 0.1% lifitegrast as described in the legend to Fig. 2, and tobramycin-killed P. aeruginosa or S. aureus was added to induce inflammation. After 24 h, neutrophil numbers and corneal thickness were assessed. Data are mean±SD of 5 mice per group, and 2 representative experiments are shown.
Table 1.
Effect of Lifitegrast (SAR 1118) on Corneal Inflammation
| Expt | Inoculum | Group | Number of mice | Time of applicationsa | Neutrophils/section | % inhibition | P |
|---|---|---|---|---|---|---|---|
| 1 | PA | Vehicle | 3 | −15 h, −3 h, −1 h | 348.0 | ||
| 5% SAR 1118 | 3 | 195.7 | 46 | 0.01 | |||
| 2 | PA | Vehicle | 4 | −15 h, −3 h | 376.5 | ||
| 1% SAR 1118 | 3 | 171.3 | 53 | 0.001 | |||
| 0.1% SAR 1118 | 3 | 158.7 | 48 | 0.001 | |||
| 3 | PA | Vehicle | 5 | −15 h, −3 h, −1 h | 359.8 | ||
| 5% SAR 1118 | 5 | 271.2 | 23 | 0.01 | |||
| 1% SAR 1118 | 5 | 211.8 | 42 | 0.001 | |||
| 0.1% SAR 1118 | 5 | 241.4 | 35 | 0.01 | |||
| 4 | PA | Vehicle | 3 | −15 h, −3 h | 385.3 | ||
| 0.1% SAR 1118 | 3 | 275.0 | 36 | 0.015 | |||
| 1% SAR 1118 | 4 | 162.0 | 62 | 0.004 | |||
| 5% SAR 1118 | 35 | 0.016 | |||||
| 5 | SA | Vehicle | 4 | −15 h, −3 h | 242.0 | ||
| 1% SAR 1118 | 4 | 112.0 | 53 | 0.005 | |||
| 6 | PA | Vehicle | 3 | −1 h, +1 h | 373.3 | 0 | |
| 1% SAR 1118 | 3 | 336.7 | 0.398 | ||||
| 7 | PA | Vehicle | 5 | −1 h, +1 h | 245.8 | 28 | |
| 0.1% SAR 1118 | 5 | 175.8 | 0.206 | ||||
| 8 | PA | Vehicle | 4 | −1 h, +1 h | 445.3 | 0 | |
| 5% SAR 1118 | 3 | 494.3 | −0.227 |
before or after corneal abrasion and stimulation with P. aeruginosa or S. aureus.
PA, Pseudomonas aeruginosa; SA, Staphylococcus aureus; NS, not significant (P>0.05).
To determine if lifitegrast inhibits corneal inflammation induced by tobramycin-killed S. aureus, mice were given 1% lifitegrast topically at 15, 3, and 1 h before abrading the cornea and inducing inflammation with tobramycin-killed S. aureus. As shown in Fig. 3B, neutrophil numbers, corneal haze, and corneal thickness were significantly lower in lifitegrast compared with control mice, indicating that lifitegrast also inhibits corneal inflammation induced by killed S. aureus.
Lifitegrast does not induce apoptosis of corneal epithelial cells in vivo
To determine if topical application of 1% lifitegrast induces apoptosis of corneal epithelial cells, mice were treated with 1% lifitegrast before inducing corneal inflammation by killed S. aureus, and corneal sections were examined, immunostained for neutrophils, and stained by TUNEL assay, which detects apoptotic cells. Sections were counterstained with DAPI to detect the cell nuclei. Figure 4A shows representative images of inhibition of neutrophil infiltration in mice treated with 1% lifitegrast. As shown in Fig. 4B, TUNEL-positive cells were detected in the corneal stroma of lifitegrast and control corneas. The reduced number of TUNEL-positive cells in lifitegrast-treated corneas, together with observations from our past studies,9 indicates that these cells are neutrophils. However, the absence of TUNEL-positive corneal epithelial cells in lifitegrast-treated corneas demonstrates that lifitegrast does not induce apoptosis in corneal epithelial cells. TUNEL-positive cells were detected in the corneal stroma, corresponding to the presence of neutrophils.
FIG. 4.
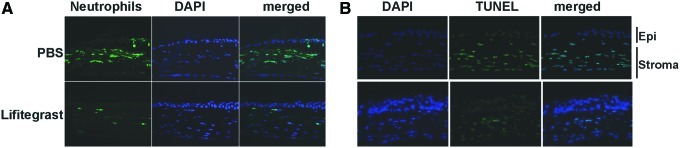
Effect of 1% SAR 1118 on S. aureus-induced neutrophil recruitment and on epithelial cell apoptosis. Corneas were treated with 1% SAR 1118 before inducing inflammation by incubating 2 h with tobramycin-killed S. aureus in the presence of a contact lens. After 24 h, eyes were processed for histology, and 5-μm sections were immunostained with NIMP-R14 and counterstained with DAPI (A), or were stained with TUNEL reagents, counterstained with DAPI. Images are representative of 5 mice per group (B). Epi, corneal epithelium. Original magnification is×400. Color images available online at www.liebertpub.com/jop
Discussion
The I domain of the CD11a subunit is the binding region of LFA-1 integrin, which interacts with the first Ig-like domain of ICAM-1.24 The binding site is discontinuous in the primary structure; however, crystallographic and mutagenesis studies revealed the tertiary structure, which showed that the key residues are presented along one side of the ICAM-1 first domain.24 These observations were the basis for developing small-molecule antagonists, including lifitegrast.17,25 This molecule is soluble in aqueous solution at concentrations >100 mg/mL, and topical administration of this antagonist to diabetic rats showed that lifitegrast reached the retina, and inhibits retinal leukostasis and vascular leakiness.20 The potential for using this antagonist in topical application was shown in a recent study for healthy adults, where it appeared to be both safe and well tolerated.18 Lifitegrast was also found to improve the signs and symptoms of dry eye with no adverse effects.21 These findings are consistent with our observations that lifitegrast is present in the cornea after topical application, and that there is no apoptotic effect on corneal epithelial cells.
We also demonstrated using lifitegrast, anti-CD11a, and CD11b (CD18)−/− mice that LFA-1 interactions are essential for leukocyte recruitment to the corneal stroma, and the development of subclinical changes in the corneal stroma, including increased corneal thickness and increased reflectivity or haze. The results are consistent with clinical descriptions of sterile corneal infiltrates. Earlier studies showed that LFA-1 and ICAM-1 are expressed in the inflamed human cornea26; LFA-1 has an important role in leukocyte recruitment during wound healing14; and ICAM-deficient mice have impaired leukocyte infiltration in murine models of P. aeruginosa and limbal and suture injuries, although not found in Herpes Simplex keratitis.13,27–29 We also found that 5% lifitegrast was consistently less effective than a 1% solution in reducing the inflammatory response, which appears to be due to less absorption into the mouse cornea (Fig. 2). However, it is important to note that in a large clinical trial, a dose-dependent response was observed in several clinical endpoints assessing efficacy in treating dry eye disease.21 The dissimilarity between those findings and ours may be a function of differences in corneal permeability between humans and mice.
Our previous studies demonstrated that killed S. aureus and P. aeruginosa activate the TLR2 and TLR4/MD-2 signaling cascade in the cornea, respectively, resulting in production of chemotactic and proinflammatory cytokines, which then mediate recruitment of neutrophils from limbal vessels to the corneal stroma, resulting in corneal inflammation.8,9 Proinflammatory cytokines also induce expression of adhesion molecules, including ICAM-1, on vascular endothelial cells. As neutrophils constitutively express LFA-1, this process is important for recruitment of these cells to the corneal stroma. Once present in the corneal stroma and exposed to bacterial products, neutrophils degranulate and release proteolytic enzymes such as matrix metalloproteinases (MMP8 and MMP9), which can cause tissue damage.30,31 Given that neutrophils are detected in patients with contact-lens-associated corneal inflammation,11 it is likely that neutrophils and other leukocytes contribute to the adverse events associated with contact-lens wear. This understanding of the sequence of events leading to corneal inflammation allows for a more targeted approach to blocking inflammation than using corticosteroids.32
One approach is to target TLR activation, and we reported that LPS- and killed P. aeruginosa-induced corneal inflammation can be blocked by the MD-2 antagonist Eritoran tetrasodium (E5564); however, this reagent did not inhibit corneal inflammation induced by TLR2 antagonists.9 We also demonstrated that neutralizing chemokine activity or PECAM-1 inhibits corneal inflammation,7,33 and that blocking TLR signaling by MAP kinase inhibitors or short-chain ceramide also inhibits corneal inflammation.23,34 In the current study, LFA-1 antagonism clearly blocks neutrophil recruitment to the cornea. Although we did not examine cytokine production after LFA-1 blockade (data not shown), we anticipate that any reduction in chemokines or proinflammatory cytokines would be due to the lower number of neutrophils at the site, as these cells produce numerous cytokines.35
In summary, results presented in the current study clearly demonstrate that LFA-1 antagonism inhibits corneal inflammation induced by antibiotic-treated S. aureus and P. aeruginosa. Furthermore, topical application of lifitegrast before inflammation significantly reduces this immune response without inducing epithelial cell apoptosis, thereby demonstrating the proof of concept for the use of lifitegrast in combination with antibiotics in prophylactic and possibly therapeutic applications for contact-lens-associated corneal infiltrates.
Acknowledgments
The authors wish to thank Scott Howell and Catherine Doller in the Visual Sciences Core Facility. This research was supported by the NIH grants RO1EY14362 (E.P.) and P30EY11373 (E.P.), and a sponsored research agreement with SARcode Corporation. Additional support for these studies was provided by The Research to Prevent Blindness Foundation and the Ohio Lions Eye Research Foundation.
Author Disclosure Statement
E.P. is the recipient of an Alcon Research Institute award. E.P. received a sponsored research award from SARcode Bioscience. T.R.G. and C.A.O. are affiliated with the same company.
References
- 1.Stapleton F. Keay L. Jalbert I. Cole N. The epidemiology of contact lens related infiltrates. Optom. Vis. Sci. 2007;84:257–272. doi: 10.1097/OPX.0b013e3180485d5f. [DOI] [PubMed] [Google Scholar]
- 2.Szczotka-Flynn L. Debanne S.M. Cheruvu V.K., et al. Predictive factors for corneal infiltrates with continuous wear of silicone hydrogel contact lenses. Arch. Ophthalmol. 2007;125:488–492. doi: 10.1001/archopht.125.4.488. [DOI] [PubMed] [Google Scholar]
- 3.Szczotka-Flynn L. Diaz M. Risk of corneal inflammatory events with silicone hydrogel and low dk hydrogel extended contact lens wear: a meta-analysis. Optom. Vis. Sci. 2007;84:247–256. doi: 10.1097/OPX.0b013e3180421c47. [DOI] [PubMed] [Google Scholar]
- 4.Robboy M.W. Comstock T.L. Kalsow C.M. Contact lens-associated corneal infiltrates. Eye Contact Lens. 2003;29:146–154. doi: 10.1097/01.ICL.0000072830.41886.1E. [DOI] [PubMed] [Google Scholar]
- 5.Szczotka-Flynn L. Lass J.H. Sethi A., et al. Risk factors for corneal infiltrative events during continuous wear of silicone hydrogel contact lenses. Invest. Ophthalmol. Vis. Sci. 2010;51:5421–5430. doi: 10.1167/iovs.10-5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson A.C. Heinzel F.P. Diaconu E., et al. Activation of toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest. Ophthalmol. Vis. Sci. 2005;46:589–595. doi: 10.1167/iovs.04-1077. [DOI] [PubMed] [Google Scholar]
- 7.Khatri S. Lass J.H. Heinzel F.P., et al. Regulation of endotoxin-induced keratitis by PECAM-1, MIP-2, and toll-like receptor 4. Invest. Ophthalmol. Vis. Sci. 2002;43:2278–2284. [PubMed] [Google Scholar]
- 8.Sun Y. Hise A.G. Kalsow C.M. Pearlman E. Staphylococcus aureus-induced corneal inflammation is dependent on Toll-like receptor 2 and myeloid differentiation factor 88. Infect. Immun. 2006;74:5325–5332. doi: 10.1128/IAI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Y. Pearlman E. Inhibition of corneal inflammation by the TLR4 antagonist Eritoran tetrasodium (E5564) Invest. Ophthalmol. Vis. Sci. 2009;50:1247–1254. doi: 10.1167/iovs.08-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szczotka-Flynn L.B. Pearlman E. Ghannoum M. Microbial contamination of contact lenses, lens care solutions, and their accessories: a literature review. Eye Contact Lens. 2010;36:116–129. doi: 10.1097/ICL.0b013e3181d20cae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holden B.A. Reddy M.K. Sankaridurg P.R., et al. Contact lens-induced peripheral ulcers with extended wear of disposable hydrogel lenses: histopathologic observations on the nature and type of corneal infiltrate. Cornea. 1999;18:538–543. [PubMed] [Google Scholar]
- 12.Ley K. Laudanna C. Cybulsky M.I. Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 13.Hobden J.A. Masinick S.A. Barrett R.P. Hazlett L.D. Proinflammatory cytokine deficiency and pathogenesis of Pseudomonas aeruginosa keratitis in aged mice. Infect. Immun. 1997;65:2754–2758. doi: 10.1128/iai.65.7.2754-2758.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z. Burns A.R. Smith C.W. Lymphocyte function-associated antigen-1-dependent inhibition of corneal wound healing. Am. J. Pathol. 2006;169:1590–1600. doi: 10.2353/ajpath.2006.060415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagen D. Laubinger S. Li Z., et al. ICAM-1 mediates surface contact between neutrophils and keratocytes following corneal epithelial abrasion in the mouse. Exp. Eye Res. 2010;91:676–684. doi: 10.1016/j.exer.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holden B.A. La Hood D. Grant T., et al. Gram-negative bacteria can induce contact lens related acute red eye (CLARE) responses. CLAO J. 1996;22:47–52. [PubMed] [Google Scholar]
- 17.Gadek T.R. Burdick D.J. McDowell R.S., et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. 2002;295:1086–1089. doi: 10.1126/science.295.5557.1086. [DOI] [PubMed] [Google Scholar]
- 18.Semba C.P. Swearingen D. Smith V.L., et al. Safety and pharmacokinetics of a novel lymphocyte function-associated antigen-1 antagonist ophthalmic solution (SAR 1118) in healthy adults. J. Ocul. Pharmacol. Ther. 2011;27:99–104. doi: 10.1089/jop.2009.0105. [DOI] [PubMed] [Google Scholar]
- 19.Murphy C.J. Bentley E. Miller P.E., et al. The pharmacologic assessment of a novel lymphocyte function-associated antigen-1 antagonist (SAR 1118) for the treatment of keratoconjunctivitis sicca in dogs. Invest. Ophthalmol. Vis. Sci. 2011;52:3174–3180. doi: 10.1167/iovs.09-5078. [DOI] [PubMed] [Google Scholar]
- 20.Rao V.R. Prescott E. Shelke N.B., et al. Delivery of SAR 1118 to the retina via ophthalmic drops and its effectiveness in a rat streptozotocin (STZ) model of diabetic retinopathy (DR) Invest. Ophthalmol. Vis. Sci. 2010;51:5198–5204. doi: 10.1167/iovs.09-5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Semba C.P. Torkildsen G.L. Lonsdale J.D., et al. A phase 2 randomized, double-masked, placebo-controlled study of a novel integrin antagonist (SAR 1118) for the treatment of dry eye. Am. J. Ophthalmol. 2012;153:1050–1060.e1. doi: 10.1016/j.ajo.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y. Karmakar M. Roy S., et al. TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J. Immunol. 2010;185:4272–4283. doi: 10.4049/jimmunol.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adhikary G. Sun Y. Pearlman E. C-Jun NH2 terminal kinase (JNK) is an essential mediator of Toll-like receptor 2-induced corneal inflammation. J. Leukoc. Biol. 2008;83:991–997. doi: 10.1189/jlb.1107783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimaoka M. Xiao T. Liu J.H., et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keating S.M. Clark K.R. Stefanich L.D., et al. Competition between intercellular adhesion molecule-1 and a small-molecule antagonist for a common binding site on the alphal subunit of lymphocyte function-associated antigen-1. Protein Sci. 2006;15:290–303. doi: 10.1110/ps.051583406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldberg M.F. Ferguson T.A. Pepose J.S. Detection of cellular adhesion molecules in inflamed human corneas. Ophthalmology. 1994;101:161–168. doi: 10.1016/s0161-6420(94)31370-4. [DOI] [PubMed] [Google Scholar]
- 27.Jung H.W. Jung C.R. Choi B.K., et al. Herpesvirus infection of ICAM-1-deficient mice. Curr. Eye Res. 2004;29:201–208. doi: 10.1080/02713680490504650. [DOI] [PubMed] [Google Scholar]
- 28.Zhu S.N. Dana M.R. Expression of cell adhesion molecules on limbal and neovascular endothelium in corneal inflammatory neovascularization. Invest. Ophthalmol. Vis. Sci. 1999;40:1427–1434. [PubMed] [Google Scholar]
- 29.Moromizato Y. Stechschulte S. Miyamoto K., et al. CD18 and ICAM-1-dependent corneal neovascularization and inflammation after limbal injury. Am. J. Pathol. 2000;157:1277–1281. doi: 10.1016/S0002-9440(10)64643-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlson E.C. Lin M. Liu C.Y. Kao W.W. Perez V.L. Pearlman E. Keratocan and lumican regulate neutrophil infiltration and corneal clarity in lipopolysaccharide-induced keratitis by direct interaction with CXCL1. J. Biol. Chem. 2007;282:35502–35509. doi: 10.1074/jbc.M705823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin M. Jackson P. Tester A.M., et al. Matrix metalloproteinase-8 facilitates neutrophil migration through the corneal stromal matrix by collagen degradation and production of the chemotactic peptide Pro-Gly-Pro. Am. J. Pathol. 2008;173:144–153. doi: 10.2353/ajpath.2008.080081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearlman E. Johnson A. Adhikary G., et al. Toll-like receptors at the ocular surface. Ocul. Surf. 2008;6:108–116. doi: 10.1016/s1542-0124(12)70279-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin M. Carlson E. Diaconu E. Pearlman E. CXCL1/KC and CXCL5/LIX are selectively produced by corneal fibroblasts and mediate neutrophil infiltration to the corneal stroma in LPS keratitis. J. Leukoc. Biol. 2007;81:786–792. doi: 10.1189/jlb.0806502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Y. Fox T. Adhikary G. Kester M. Pearlman E. Inhibition of corneal inflammation by liposomal delivery of short-chain, C-6 ceramide. J. Leukoc. Biol. 2008;83:1512–1521. doi: 10.1189/jlb.0108076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mantovani A. Cassatella M.A. Costantini C. Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]