

Abstract
The ST2L receptor for interleukin 33 (IL-33) mediates pulmonary inflammation and immune system–related disorders, such as asthma and rheumatoid arthritis. At present, very little is known about the molecular regulation of ST2L expression. Here we found that FBXL19, an ‘orphan’ member of the Skp1–Cullin–F-box family of E3 ubiquitin ligases, selectively bound to ST2L to mediate its polyubiquitination and elimination in the proteasome. Degradation of ST2L involved phosphorylation of ST2L at Ser442 catalyzed by the kinase GSK3β. Overexpression of FBXL19 abrogated the proapoptotic and inflammatory effects of IL-33 and lessened the severity of lung injury in mouse models of pneumonia. Our results suggest that modulation of the IL-33–ST2L axis by ubiquitin ligases might serve as a unique strategy for lessening pulmonary inflammation.
Interleukin 33 (IL-33), a relatively recently identified member of the IL-1 family of cytokines, is an endogenous proinflammatory danger signal released from injured or dying host cells1,2. Initially, IL-33 was identified as a nuclear factor expressed in endothelial cells3; however, subsequent studies have found that this cytokine is a highly potent distress signal released from necrotic cells after trauma or infection4,5. IL-33 is sufficient to elicit severe allergic inflammation and induce a sepsis-like state that leads to substantial pulmonary impairment6–8. Those effects seem to be driven by activation of the pathways of the transcription factors NF-κB and AP-1 linked to higher expression of genes encoding the lipopolysaccharide (LPS) receptor TLR, the coreceptor CD14 and the adaptor MyD88 (refs. 9,10). Inhibition of IL-33 by administration of neutralizing antibody to IL-33 (anti-IL-33) or of IL-33 decoy receptors attenuates lung inflammation in mouse models7,11. IL-33-deficient mice also show both less mortality and cytokine release in the LPS-induced model of sepsis12, which suggests that endogenous IL-33 has a critical role as a proximal effector in sepsis. Those results contrast with another study demonstrating that exogenous IL-33 attenuates sepsis by enhancing the influx of neutrophils to the site of infection13. Such disparate results among studies could be explained by the model systems used and route of IL-33 administration. Nevertheless, these observations as a whole suggest that modulation of the axis of IL-33 and its receptor might serve as a useful strategy for limiting the severity of pulmonary inflammation.
ST2, a member of the family of IL-1 receptors, has been identified as the receptor for IL-33 and consists of two main isoforms: a soluble, secreted form (sST2) and a transmembrane, long form (ST2L)14,15. These isoforms, sST2 and ST2L, have diametrically opposed effects on inflammatory responses. The secreted form, sST2, binds to IL-33 and functions as an inhibitor of IL-33 signals and thus has anti-inflammatory properties11,16. In contrast, ST2L is the cognate receptor for IL-33 and is actively expressed by effector cells of the immune response and has a critical role in triggering inflammation17–19. ST2L is a classic type I membrane receptor that contains three extracellular immunoglobulin G (IgG)-like domains, a transmembrane domain and an intracellular Toll–IL-1 receptor domain. ST2L localizes on the surface of a variety of cell types, including lung epithelia and endothelia20. IL-33 induces secretion of IL-8 by airway epithelial and pulmonary endothelial cells20. As the proinflammatory effects of extracellular IL-33 are achieved through ligation to ST2L, studies assessing the regulation of ST2L protein expression may idenitfy important molecular cues that serve as a basis for the abrogation of IL-33 signaling.
The ubiquitin-proteasome system degrades most intracellular proteins, including membrane-surface receptors21–24. Three enzyme complexes (E1, E2 and E3) are involved in linking ubiquitin chains onto target proteins25. Among the families of E3 ubiquitin ligases, the Skp1–Cullin-1–F-box protein (SCF) ligase complex is one of the largest26; in this complex, the F-box protein is the substrate-recognition component. Over 60 F-box proteins have been identified, but only a few, such as β-Trcp and FBXW7, are well characterized. F-box proteins have two main domains: an F-box motif and a substrate-binding motif. F-box proteins use their F-box motif to bind to Skp1 and assemble the SCF ligase complex, whereas the substrate-binding motif is used for recognition and interaction with phosphorylated substrates27. Through an in silico search, the ‘orphan’ F-box protein FBXL19, has been identified28. FBXL19 has considerable sequence similarity to other SCF proteins28; however, the verification of FBXL19 as an SCF E3 subunit, its regulation and molecular targets remain to be determined. Here we found that FBXL19 targeted the IL-33–ST2L axis, selectively mediating the ubiquitination and degradation of ST2L to limit IL-33-induced pulmonary inflammation. Phosphorylation of ST2L mediated by the glycogen synthase kinase GSK3β provided a phosphodegron molecular signal (phosphorylation-dependent degradation motif) for the binding of ST2L to FBXL19. Our results might serve as a basis for the development of new approaches to lessen the severity of inflammation through the use of components of the ubiquitin-proteasomal machinery to modify the availability of an indispensable receptor linked to sepsis-induced lung injury.
RESULTS
FBXL19 mediates ubiquitination and degradation of ST2L
In the process of investigating the role of IL-33–ST2L in airway inflammation, we first assessed the stability of ST2L. Treatment of MLE12 mouse lung epithelial cells with the protein-biosynthesis inhibitor cycloheximide (20 μg/ml) diminished ST2L mass in a time-dependent manner and demonstrated that endogenous ST2L had a half-life of ~5 h (Fig. 1a,b). To identify which pathway is involved in the degradation of ST2L, we exposed MLE12 cells to inhibitors of proteasomes (MG-132) or lysosomes (leupeptin) before cycloheximide treatment. MG-132 (20 μM) attenuated the degradation of ST2L, but leupeptin (100 μM) did not (Fig. 1a,b). Overexpressed mouse ST2L with a green fluorescent protein tag localized in the plasma membrane and cytoplasm, but it did not localize in lysosomes (data not shown). These results suggested that degradation of ST2L was mediated by the proteasome. As ubiquitination is a sorting signal for targeting to the proteasome, we next investigated if ST2L is modified by ubiquitination. We immunoprecipitated ubiquitin from cell lysates, followed by immunoblot analysis of ST2L; this demonstrated that ST2L was polyubiquitinated (Fig. 1c) and that the ubiquitin chain had, at least in part, the classic Lys48 linkage (Supplementary Fig. 1a). Furthermore, over-expression of hemagglutinin-tagged ubiquitin induced degradation of ST2L and total ubiquitination of proteins, as assessed in whole-cell lysates (Fig. 1d). These results indicated that ST2L was degraded by the ubiquitin proteasomal machinery in a lung epithelial cell line.
Figure 1.

FBXL19 mediates the degradation of ST2L via the ubiquitinproteasome system. (a,b) Immunoblot analysis of ST2L and β-actin (loading control throughout) in lysates of MLE12 cells treated with cycloheximide (CHX) alone (top) or together with leupeptin (middle) or MG-132 (bottom; a), and half-life (t1/2) of ST2L, determined by densitometry of the results in a (b). (c) Immunoprecipitation (IP) of proteins from lysates of MLE12 cells with IgG or anti-ubiquitin (Ub), followed by immunoblot analysis (IB) of the precipitates with anti-ST2. Ub(n), polyubiqutination. (d) Immunoblot analysis of ST2L, hemagglutinin-tagged proteins (HA) and β-actin in lysates of MLE12 cells transfected with various concentrations (below lanes) of plasmid encoding hemagglutinin-tagged ubiquitin (HA-Ub). (e) Immunoblot analysis of ST2L, V5-tagged proteins and β-actin in lysates of MLE12 cells transfected with plasmid (below lanes) encoding β-galactosidase (lacZ; control) or various V5-tagged F-box proteins (FBX). (f) Immunoblot analysis of ST2L, sST2, V5-tagged proteins and β-actin in lysates of MLE12 cells transfected with various concentrations (below lanes) of plasmid encoding FBXL19-V5. (g,h) Immunoblot analysis of ST2L, V5-tagged proteins and β-actin in lysates of HBEpCs (g) and HPAECs (h) transfected as in f. (i) Immunoblot analysis of ST2L and β-actin in lysates of MLE12 cells transfected with control (shCtrl) or FBXL19-specific (shFBXL19) shRNA, then left untreated (–) or treated with cycloheximide (+). (j) Immunoblot analysis of ST2L, V5-tagged proteins and β-actin in lysates of MLE12 cells transfected control plasmid (Vec) or plasmid encoding FBXL19-V5 in the presence of dimethyl sulfoxide (DMSO) or MG-132. (k) Wild-type (WT) FBXL19 (top) and variants of FBXL19 with truncation of the NH2 terminus (below) containing (N254) or lacking (N305) the F-box motif. LRRs, leucine-rich repeats. (l) Immunoblot analysis of ST2L, V5-tagged proteins and β-actin in lysates of MLE12 cells left untransfected (–) or transfected with plasmids encoding wild-type or mutant FBXL19 as in k. Data are from one experiment representative of three.
F-box proteins in the SCF E3 ligase complex recognize and bind specific substrates27. Using an unbiased screen, we began to identify which F-box protein targets ST2L. We overexpressed more than 30 F-box proteins, then assessed the abundance of ST2L. Among five such F-box proteins, only ectopic expression of V5-tagged FBXL19 (FBXL19-V5) resulted in lower abundance of immunoreactive ST2L (Fig. 1e). In addition, ectopic expression of FBXL19-V5 diminished ST2L protein in a dose dependent manner, but sST2 did not (Fig. 1f). In primary cultures of human bronchial epithelial cells (HBEpCs) and human pulmonary artery endothelial cells (HPAECs), over-expression of FBXL19 also resulted in less ST2L protein (Fig. 1g,h). Knockdown of FBXL19 with short hairpin RNA (shRNA) targeting FBXL19 attenuated the degradation of ST2L (Fig. 1i), concomitant with lower expression of FBXL19 mRNA and protein (Supplementary Fig. 2). The FBXL19-V5–mediated degradation of ST2L was attenuated by proteasomal blockade with MG-132 (Fig. 1j). An FBXL19 variant lacking the F-box motif was unable to bind Skp1 (Supplementary Fig. 3). Transfection of plasmid encoding wild-type FBXL19-V5 or an FBXL19 variant with truncation at the NH2 terminus that still contained the F-box motif resulted in lower ST2L expression; however, overexpression of an FBXL19 variant that lacked the F-box motif had no effect on ST2L protein (Fig. 1k,l). Together these results indicated that FBXL19 is a component of an SCF complex that mediates proteasomal degradation of ST2L.
IL-33 diminishes ST2L expression
Next we assessed ligand-induced degradation of ST2L, because turnover of cell-surface receptors in response to engagement of ligands is important in controlling receptor expression and intracellular signaling29. Treatment of MLE12 cells with IL-33 diminished the signal intensity of ST2L on the cell surface, as assessed by immunoblot analysis (Fig. 2a), flow cytometry (showing ST2L on the surface of control cells, but a left shift of the curve (less STL2) after IL-33 treatment; Fig. 2b) and fluorescence microscopy (without permeabilization; Fig. 2c). To investigate if the lower abundance of cell-surface ST2L was due to internalization and/or degradation of the receptor, we examined the trafficking of ST2L and ST2L protein mass in whole-cell lysates after treatment with IL-33. IL-33 induced the internalization of ST2L (data not shown) and decreased ST2L mass in MLE12 cells in a dose- and time-dependent manner (Fig. 2d,e). In addition, treatment with IL-33 resulted in lower expression of ST2L protein in primary cultures of human small-airway epithelial cells or rat alveolar type II epithelial cells (Supplementary Fig. 1b), which indicated a more widespread ability of IL-33 to rapidly trigger degradation of ST2L in lung epithelium via binding of the ligand to its receptor. IL-33 had no effect on the expression of the related receptor IL-1R1 (Fig. 2a,f), and IL-1β had no effect on ST2L expression (Fig. 2g), which suggested that IL-33 specifically induced the degradation of ST2L.
Figure 2.

IL-33 mediates the degradation of ST2L. (a) Immunoblot analysis of ST2L and IL-1R1 among cell surface proteins isolated from MLE12 cells treated for 2 h with IL-33 (0–50 ng/ml). (b) Flow cytometry analysis (left) of ST2L on the surface of MLE12 cells treated for 2 h with IL-33 (0–50 ng/ml); gray shaded curve indicates control cells (staining with IgG). Right, quantification of the results at left (frequency of surface protein calculated from geometric mean of fluorescence intensity). *P < 0.01, compared with treatment with 0 ng/ml of IL-33 (unparied t-test). (c) Microscopy of ST2L on the surface of MLE12 cells treated for 2 h with vehicle (Veh) or IL-33 (10 ng/ml), assessed by immunofluorescence staining (without permeabilization) with fluorescein isothiocyanate–labeled anti-ST2 (green) alone (left) or together with the DNA-intercalating dye DAPI (red; right). Scale bars, 5 μm. (d,e) Immunoblot analysis of ST2L and β-actin in lysates of MLE12 cells treated for 2 h with IL-33 at a concentration of 0–50 ng/ml (d) or treated for 0–2 h with IL-33 at a concentration of 10 ng/ml (e). (f) Immunoblot analysis of IL-1R1 and β-actin in lysates of MLE12 cells treated as in d. (g) Immunoblot analysis of ST2L and β-actin in lysates of MLE12 cells treated for 2 h with IL-1β (0–50 ng/ml). Data are from one experiment representative of three (a,b,d–g; error bars (b), s.d.) or are representative of three independent experiments (c).
FBXL19 mediates IL-33-induced degradation of ST2L
To identify which proteolytic pathway is involved in the degradation of ST2L, we exposed cells to MG-132 or leupeptin before treatment with IL-33. Pretreatment with MG-132 attenuated the IL-33-induced degradation of ST2L, but pretreatment with leupeptin did not (Fig. 3a,b), which suggested that the IL-33-induced degradation of ST2L was mediated by the proteasome. Further, IL-33 induced polyubiquitination of ST2L in a time-dependent manner, as assessed by immunoprecipitation with anti-ubiquitin and immunoblot analysis of ST2L (Fig. 3c). Thus, IL-33-induced degradation of ST2L was mediated by the ubiquitin-proteasome pathway. Knockdown of FBXL19 by transfection of FBXL19-specific shRNA attenuated IL-33-induced ubiquitination and degradation ST2L, and the effects of this were reversed by overexpression of FBXL19-V5 (Fig. 3d–f). These results suggested that the IL-33-induced degradation of ST2L was mediated by FBXL19 and was perhaps acting as part of a feedback control mechanism to regulate steady-state amounts of the receptor for IL-33.
Figure 3.

FBXL19 mediates the IL-33-induced degradation of ST2L via the ubiquitin-proteasome system. (a,b) Immunoblot analysis of ST2L and β-actin in lysates of MLE12 cells given no pretreatment (top) or pretreated with leupeptin (100 μM; middle) or MG-132 (20 μM; bottom) and then treated with various concentrations (below lanes) of IL-33 (a), and densitometry analysis of band intensity in a (b). (c) Immunoassay of lysates of MLE12 cells treated for 0–2 h with IL-33 (10 ng/ml), assessed by immunoprecipitation with anti-ubiquitin and immunoblot analysis of precipitates (top) and input lysates (bottom) with anti-ST2 or β-actin. (d) Immunoprecipitation (as in c) and immunoblot analysis of ST2L, V5-tagged proteins and β-actin in lysates of MLE12 cells transfected for 72 h with plasmid encoding control or FBXL19-specific shRNA alone or with plasmid encoding FBXL19-V5, then left untreated or treated for 2 h with IL-33 (10 ng/ml). (e,f) Immunoblot analysis of ST2L, V5-tagged proteins and β-actin in lysates of MLE12 cells transfected as in d (Ctrl, control plasmid), then treated for 0–2 h with IL-33 (10 ng/ml; e), and densitometry analysis of band intensity in e (f). Data are from one experiment representative of three.
GSK3β regulates phosphorylation and degradation of ST2L
Protein phosphorylation can serve as a molecular signal for ubiquitination by the E3 enzyme complex27. To investigate whether the degradation of ST2L was dependent on its phosphorylation, we first determined if ST2L was phosphorylated in response to treatment with IL-33. Immunoprecipitation indicated that IL-33 induced serine- phosphorylation of both endogenous ST2L (Fig. 4a) and over-expressed Flag-tagged mouse ST2L (Fig. 4b) in a time-dependent manner. GSK3β regulates the phosphorylation and degradation of proteins29,30. ST2L contains a consensus sequence motif (Ser442-X-X-X-Ser446, where ‘X’ indicates any amino acid) for phosphorylation by GSK3β30,31. Therefore, we next determined whether GSK3β has a role in the phosphorylation and degradation of ST2L. Treatment of MLE12 cells with IL-33 induced the tyrosine-phosphorylation of GSK3β (an active form; Fig. 4c). Transfection of plasmid encoding wild-type GSK3β or a constitutively active form of GSK3β induced the phosphorylation and degradation of ST2L (Fig. 4d). Knockdown of GSK3β by through the use of small interfering RNA (siRNA) targeting GSK3β effectively attenuated IL-33-induced serine-phosphorylation of ST2L (Fig. 4e), which suggested that the IL-33-induced phosphorylation of ST2L was mediated by GSK3β. Further, knockdown or inhibition of GSK3β was sufficient to abrogate the effects of GSK3β on the IL-33-induced degradation of ST2L (Fig. 4f,g). To investigate whether activation of GSK3β regulated the binding of FBXL19 to ST2L, we transfected MLE12 cells with plasmid encoding Flag-tagged mouse ST2L, then treated the cells with the GSK3β inhibitor TWS119 before transfecting them with plasmid encoding FBXL19-V5. Inhibition of GSK3β blocked the binding of FBXL19-V5 to Flag-tagged ST2L (Fig. 4h). Furthermore, knockdown of GSK3β blocked the IL-33-induced ubiquitination of ST2L (Fig. 4i). To identify sites in ST2L for phosphorylation by GSK3β, we transfected cells with plasmid encoding either of two candidate ST2L variants, ST2L(S442A) and ST2L(S547A), that contain substitutions at putative sites for phosphorylation by GSK3β. Although IL-33 induced the degradation of wild-type ST2L and ST2L(S547A), it did not alter the steady-state amount of the immunoreactive ST2L(S442A) mutant (Fig. 5a). Recombinant GSK3β phosphorylated Flag-tagged mouse ST2L but not ST2L(S442A) (Fig. 5b). Treatment with IL-33 did not induce serine-phosphorylation (data not shown) or ubiquitination (Fig. 5c) of the ST2L(S442A) mutant. Further, when we cotransfected cells with plasmid encoding FBXL19-V5 plus plasmid encoding Flag-tagged ST2L or ST2L(S442A), the ST2L(S442A) variant showed less interaction with FBXL19-V5, as assessed by coimmunoprecipitation (Fig. 5d). Hence, the association of FBXL19 with ST2L required a critical phosphorylation-acceptor site to mediate the ubiquitination and degradation of ST2L. These results suggested that GSK3β phosphorylated ST2L at Ser442 and regulated the stability of the receptor in lung epithelia.
Figure 4.

GSK3β phosphorylates and promotes the degradation of ST2L. (a,b) Immunoassay of lysates of MLE12 cells with endogenous ST2L expression (a) or overexpression of Flag-tagged mouse ST2L (ST2L-Flag; b), treated for various times (below lanes) with IL-33, assessed by immunoprecipitation with antibody to phosphorylated serine (p-Ser) and immunoblot analysis of precipitates (top) and input lysates (bottom) with anti-ST2 (a) or anti-Flag (b) and anti-β-actin. (c) Immunoblot analysis of GSK3β phosphorylated at Tyr216 (GSK3β-p-Y216) and total GSK3β in lysates of MLE12 cells treated for various times (below lanes) with IL-33. (d) Immunoassay of serine-phosphorylation of ST2L in MLE12 cells left untransfected (Veh) or transfected to overexpress wild-type GSK3β (WT) or a constitutively active form of GSK3β (Active), assessed by immunoprecipitation of lysates as in a and immunoblot analysis of precipitates (top) and input lysates (bottom) with anti-ST2, anti-GSK3β and β-actin. (e) Immunoassay of MLE12 cells transfected with control (siCtrl) or GSK3β-specific (siGSK3β) siRNA and left untreated or treated for 30 min with IL-33, assessed by immunoprecipitation as in a and immunoblot analysis as in d. (f) Immunoblot analysis of ST2L, GSK3β and β-actin in lysates of MLE12 cells transfected as in e and treated for 2 h with IL-33. (g) Immunoblot analysis of ST2L and β-actin in lysates of MLE12 cells treated with TWS119 before treatment for 2 h with IL-33. (h) Immunoassay of MLE12 cells transfected to overexpress Flag-tagged mouse ST2L, then treated with TWS119 and transfected for an additional 24 h with plasmid encoding FBXL19-V5, assessed by immunoprecipitation with anti-V5 and immunoblot analysis of precipitates and input lysates with anti-Flag. (i) Immunoassay of lysates of MLE12 cells transfected with control or GSK3β-specific siRNA, then treated for 2 h with IL-33, assessed by immunoprecipitation with anti-ubiquitin and immunoblot analysis of precipitates and input lysates with anti-ST2 and anti-β-actin. Data are from one experiment representative of three.
Figure 5.

Identification of a GSK3β-phosphorylation site in ST2L. (a) Immunoblot analysis of Flag-tagged proteins and β-actin in lysates of MLE12 cells transfected with plasmid encoding Flag-tagged wild-type ST2L (ST2L(WT)), ST2L(S442A) or ST2L(S547A), then treated for 0, 2 or 4 h with IL-33. (b) Immunoblot analysis of MLE12 cells transfected to express Flag-tagged wild-type ST2L or ST2L(S442A), followed by immunoprecipitation with anti-Flag and incubation of the immunoprecipitated complexes (top) and input lysates (below) for 1 h with (+) or without (–) recombinant active GSK3β and ATP, probed for phosphorylated serine (top) or GSK3β and Flag-tagged proteins (below). (c) Immunoassay of MLE12 cells transfected as in b, then treated for 2 h with IL-33, assessed by immunoprecipitation with anti-ubiquitin and immunoblot analysis of the precipitates and input lysates with anti-Flag. (d) Immunoassay of lysates of MLE12 cells transfected as in b, along with plasmid encoding FBXL19-V5, assessed by immunoprecipitation with IgG or anti-Flag and immunoblot analysis of the precipitates and input lysates with anti-V5. Data are from one experiment representative of three.
ST2L has a ubiquitination and docking site for FBXL19
To identify putative ubiquitin-acceptor sites in ST2L, we substituted several candidate lysine residues of mouse ST2L with arginine (substitutions in parentheses below). Of several mutants tested, only ST2L(K326R) showed stability in response to treatment with cycloheximide; wild-type ST2L and ST2L(K246R) did not (Fig. 6a,b). Overexpression of FBXL19-V5 resulted in lower expression of wild-type ST2L and ST2L(K246R) but not ST2L(K326R) (Fig. 6c). In vitro ubiquitination assays that included the full complement of reaction components (E1, E2, FBXL19, Skp1, Cullin-1, ubiquitin and ATP) and recombinant substrates showed that FBXL19 ubiquitinated wild-type ST2L and ST2L(K246R) but not ST2L(K326R) (Fig. 6d). Thus, Lys326 was a critical residue for site-specific polyubiquitination of mouse ST2L.
Figure 6.
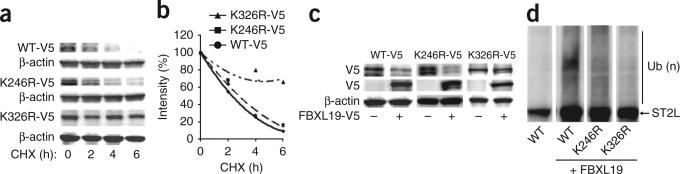
Identification of an ST2L ubiquitin-acceptor site and FBXL19-docking site. (a,b) Immunoblot analysis of V5-tagged proteins and β-actin in lysates of MLE12 cells transfected with plasmid encoding V5-tagged wild-type ST2L (WT-V5), ST2L(K246R) (K246R-V5) or ST2L(K326R) (K326R-V5), then treated for 0–6 h (below lanes) with cycloheximide (a), and analysis of half-lives by densitometry of the results in a (b). (c) Immunoblot analysis of V5-tagged proteins and β-actin in lysates of MLE12 cells transfected as in a (above blots) with or without cotransfection with plasmid encoding FBXL19-V5 (below lanes). (d) Immunoblot analysis of V5-tagged proteins among purified SCF complexes incubated with V5-tagged wild-type ST2L or ST2L(K326R) and the full complement of ubiquitination reaction components. Data are from one experiment representative of three.
Next we investigated where FBXL19 binds in the ST2L sequence (the docking site for FBXL19). We generated several deletion mutants of mouse ST2L with carboxy-terminal Flag tags (Supplementary Fig. 4a) and incubated those with lysates of HEK293 human embryonic kidney cells expressing histidine-tagged FBXL19. After precipitation with histidine-coated beads, only the Flag-tagged mutant ST2L(C328) with deletion at the carboxyl terminus showed very limited ability to interact with FBXL19 (Supplementary Fig. 4b), which suggested the FBXL19 bound to ST2L in the carboxyl terminus of ST2L.
FBXL19 blocks IL-33-mediated apoptosis
IL-33 is a proinflammatory cytokine that induces T helper type 2–associated immune responses1,18,19, enhances LPS-induced release of cytokines9,10,12,20 and promotes cell death32,33; however, its effect on distal lung epithelium has not been investigated. Cortactin, an oncogenic-like protein that regulates cell motility and proliferation, is degraded during cell death34. Overexpression of cortactin attenuates detachment-induced apoptosis in head and neck squamous-cell carcinoma cells35, whereas cortactin depletion can promote apoptosis36; hence, cortactin might regulate cellular lifespan. IL-33 induced serine-phosphorylation and degradation of cortactin in MLE12 cells (Fig. 7a,b), which suggested that this process might induce apoptosis. We measured apoptosis by staining with annexin V–propidium iodide, in which staining with both reagents indicates late apoptotic cells, and staining with annexin V plus lack of staining with propidium iodide indicates early apoptotic cells. This showed that treatment for 6 h with IL-33 (10 ng/ml) induced apoptosis of MLE12 cells (Fig. 7c). Overexpression of cortactin attenuated the IL-33-induced apoptosis of cells (Fig. 7d). Thus, these results suggested that IL-33-induced cell death might be mediated by degradation of cortactin. Notably, overexpression of FBXL19-V5 attenuated the IL-33-induced degradation of cortactin in MLE12 cells (Fig. 7e,f), HBEpCs and HPAECs (Supplementary Fig. 5a) and apoptosis of MLE12 cells (Fig. 7g) and HBEpCs (Supplementary Fig. 5b). IL-33 induces the release of cytokines such as IL-8 and IL-6 from human lung epithelial and endothelial cells20. We found that ectopically expressed FBXL19-V5 abrogated the IL-33-induced release of IL-8 from HBEpCs and of IL-6 from HPAECs (Supplementary Fig. 5c). IL-33 also increases vascular endothelial permeability, a key proinflammatory effect17. Notably, overexpression of FBXL19-V5 blocked the IL-33-induced barrier disruption and stress-fiber formation of HPAECs (Supplementary Fig. 5d,e). These data together suggested that although endogenous FBXL19 normally mediated the effects of IL-33 on the ubiquitination of ST2L (Fig. 3d) and steady-state turnover of ST2L (Fig. 3e), supra-physiological concentrations of the F-box protein might impair IL-33 signaling by depleting the availability of its cognate receptor.
Figure 7.

FBXL19 attenuates IL-33-induced degradation of cortactin and apoptosis. (a) Immunoassay of lysates of MLE12 cells treated for 0–60 min (below lanes) with IL-33, assessed by immunoprecipitation with antibody to phosphorylated serine, followed by immunoblot analysis of precipitates and input lysates with anti-cortactin. (b) Immunoblot analysis of cortactin and β-actin in lysates of MLE12 cells treated with IL-33. (c) Apoptosis of MLE12 cells treated for 6 h with vehicle, IL-33 or LPS, assessed by flow cytometry. Numbers in quadrants indicate percent cells in each throughout; right half of each plot indicates apoptotic cells throughout. PI, propidium iodide. (d) Apoptosis of MLE12 cells transfected with vector only (Vec) or transfected with plasmid encoding cortactin (right), then treated for 6 h with IL-33, assessed as in c. (e,f) Immunoblot analysis of cortactin, V5-tagged proteins and β-actin in MLE12 cells transfected with vector only or plasmid encoding FBXL19-V5, then left untreated or treated for 6 h with IL-33 (e), and densitometry of band intensity of the cortactin blots in e (f). *P < 0.05 and **P < 0.01 (unpaired t-test). (g) Apoptosis of MLE12 cells transfected with vector only (left two plots) or plasmid encoding FBXL19-V5 (right two plots), then left untreated (Ctrl and FBXL19-V5) or treated for 6 h with IL-33 (IL-33 and FBXL19-V5 + IL-33), assessed as in c. Data are from one experiment representative of three (error bars (f), s.d.).
FBXL19 prevents endotoxin-induced acute lung injury
Intratracheal administration of LPS for 24 h, at a dose of 1 mg per kg body weight, or of Pseudomonas aeruginosa (strain PA103) for 24 h, at a dose 4 × 104 colony-forming units per mouse, to C57/BL6 mice resulted in a higher concentration of IL-33 in bronchoalveolar lavage (BAL) fluid than that in mice treated with PBS (Fig. 8a). To investigate the effects of FBXL19 on endotoxin-induced lung injury, we treated mice by intratracheal administration of a lentiviral vector encoding a fusion of FBXL19 and red fluorescent protein or a control lentiviral vector encoding the red fluorescent protein only and analyzed in vivo expression of these constructs in lung tissue by fluorescence scanning (Fig. 8b). Overexpression of FBXL19 diminished ST2L expression in lung tissue to a degree similar to the effects of challenge with IL-33 (for 24 h at a dose of 4 μg per mouse) on ST2L expression in lung tissue (Supplementary Fig. 6a). Intratracheal challenge with IL-33 triggered apoptosis of lung epithelial cells, as measured by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL; ~42% of intratracheal LPS-induced cell death; Supplementary Fig. 6b,c). However, overexpression of FBXL19 in mice blocked the cell death (Fig. 8c,d) and inflammatory cellular infiltration (Supplementary Fig. 6d) induced by intratracheal challenge with IL-33. Furthermore, lentiviral overexpression of FBXL19 effectively attenuated the pulmonary inflammation, as assessed histologically (Fig. 8e,f), and the alveolar protein leakage (Fig. 8g) induced by LPS or P. aeruginosa (strain PA103), and also resulted in less induction of IL-6 (Fig. 8h) and tumor necrosis factor (Fig. 8i) in BAL fluid by LPS or P. aeruginosa (strain PA103). In vivo knockdown of FBXL19 with lentivirus expressing FBXL19-specific shRNA promoted the intratracheal LPS–mediated induction of IL-6 in BAL fluid (Supplementary Fig. 7a) and augmented appearance of pulmonary infiltrates, as assessed histologically (Supplementary Fig. 7b,c). This technique substantially diminished steady-state FBXL19 mRNA in vivo (Supplementary Fig. 7d). To investigate whether the effects of FBXL19 were specific to IL-33–ST2L, we examined effects of overexpression of FBXL19 on IL-1R1 expression and IL-1β signaling. Overexpression of FBXL19-V5 had no effect on IL-1R1 expression or IL-1β-induced signaling, such as activation of the kinase Erk1-Erk2 (Supplementary Fig. 8a,b). The administration of FBXL19 had no effect on inflammatory cellular infiltration induced by intratracheal IL-1β (Supplementary Fig. 8c). These results suggested that FBXL19 blocked the integrity of the IL-33–ST2L axis by selectively promoting the degradation of a key receptor to abrogate sepsis-induced lung injury.
Figure 8.

FBXL19 diminishes endotoxin-induced acute lung injury. (a) Enzyme-linked immunosorbent assay of IL-33 in BAL fluid (BALF) from C57/BL6 mice (n = 4–6 per group) challenged intratracheally for 24 h with PBS, LPS (1 mg per kg body weight) or P. aeruginosa strain PA103 (4 × 104 colony-forming units per mouse). (b) Fluorescence scanning microcopy (top) and light microcopy (bottom) of lentiviral expression in lung tissue from mice (n = 4 per group) left untreated (Ctrl) or given intratracheal administration of control lentivirus (Lenti-ctrl) or lentivirus encoding FBXL19 (Lenti-FBXL19). (c) TUNEL assay of apoptosis in lung tissues from C57/BL6 mice (n = 4 per group) infected intratracheally with control lentivirus or lentivirus encoding FBXL19 and given intratracheal administration of PBS or IL-33 (4 μg per mouse); arrows indicate TUNEL+ cells. Scale bars, 10 μm. (d) Quantification of the apoptotic cells in c (five areas per slide; four slides per group). (e) Microcopy of hematoxylin and eosin–stained lung tissues from mice (n = 4–6 per group) infected intratracheally with control lentivirus or lentivirus encoding FBXL19 and given intratracheal administration of PBS, LPS or P. aeruginosa strain PA103 (doses as in a). Scale bars, 12.5 μm. (f) Quantification of influx of the inflammatory cells into alveolar spaces in e. (g–i) Protein assay of total protein (g) and enzyme-linked immunosorbent assay of IL-6 (h) and tumor necrosis factor (TNF; i) in BAL fluid from mice treated as in e. *P < 0.05 and **P < 0.01 (unpaired t-test). Data are representative of four (a–d) or six (e–i) experiments (error bars (a,b,f–i), s.d.).
DISCUSSION
The ligation of IL-33 to its receptor ST2L is crucial, as it has an important role in the pathogenesis of immune system–related disorders, such as asthma and rheumatoid arthritis, as well as septic lung injury6,37,38. Understanding the regulation of ST2L expression is essential for identifying the molecular targets that mediate proinflammatory signaling by IL-33. Here we have shown that ST2L is a phosphorylated receptor regulated by IL-33 at the post-translational level through its steady-state processing by F-box protein FBXL19, a prototypical SCF subunit that was both sufficient and required to mediate ubiquitination and degradation of ST2L in lung epithelia. The ubiquitination activity of FBXL19 was facilitated by GSK3β, which generated a phosphodegron-like molecular signature in ST2L, leading to its proteasomal degradation. Hence, we used FBXL19 to modulate the IL-33–receptor axis. Indeed, ectopic expression of FBXL19 attenuated a previously unrecognized effect of IL-33 on apoptosis and, notably, limited the severity of endotoxin- and P. aeruginosa–induced inflammatory lung injury. Whether our results might translate to newer approaches for the treatment of pneumonia remains speculative, but they indicate that the delivery of small-molecule agonists to enhance or mimic the actions of FBXL19 in the IL-33–ST2L axis might be a means of more precisely lessening inflammation.
The internalization, mono- or polyubiquitination and degradation of cytokine receptors are well-described processes for feedback regulation of ligand-induced signaling events29; however, the internalization and post-translational modification of ST2L has not been studied so far, to our knowledge. Our results have shown that under steady-state conditions, ST2L was gradually internalized and degraded (half-life, ~5 h) and exceeded the stability of some surface receptors, such as the G protein–coupled estrogen receptor11. After ligation of IL-33 to endogenous ST2L, however, surface expression rapidly decreased, consistent with the processing of the related cytokine receptors IL-5R23, IL-17R39, IL-2Rβ21 and the common γ-chain22 after ligand engagement. Some type I receptors that undergo endocytosis and degradation also have ubiquitin-binding motifs in their cytosolic tails that serve as recognition signals for the SCF β-Trcp ligase40. Likewise, FBXL19 interacted with the carboxy-terminal region of ST2L to facilitate polyubiquitination at Lys326. Whether ST2L has a distinct ubiquitin-dependent endocytosis–like motif that engages an F-box protein or interacts with endocytic adaptors through ubiquitin-binding domains to promote proteasomal sorting requires further evaluation.
There are very limited data on the role of F-box proteins in the ubiquitination and degradation of cytokine receptors, although several RING-finger E3 ligases, such as TRAF6 (ref. 39), c-Cbl22 and RNF41 (ref. 24), regulate the turnover of cytokine receptors. Phosphorylation is an initial step for the recruitment of many F-box proteins to interact with target substrates and transfer ubiquitin to those target substrates, and the molecular activity of FBXL19 resembles the canonical recognition of phosphodegrons by SCF proteins27. For example, activation of protein kinase C enhances the phosphorylation and degradation of the receptor IL-8RB41, and kinases of the Jak family control the ubiquitination and degradation of the receptor IL-5R23 and the receptor for leptin24. The phosphorylation of substrates by GSK3β is directed in part by the minimal-recognition motif Ser-X-X-X-Ser. Here, activation of GSK3β induced the phosphorylation of ST2L at Ser442 to facilitate docking of FBXL19 in the receptor, which thereby mediated the ubiquitination and degradation of ST2L. These results support the emerging proposal of a role for GSK3β in controlling the lifespan of surface receptors with divergent roles, such as DR5 (ref. 42) and the receptor for prolactin43.
IL-33 is an important distress signal or ‘alarmin’ released from damaged endothelial or epithelial cells that potently induces the activation of inflammasomes44 and promotes the release of cytokines (such as IL-5, IL-6, IL-8 and IL-13) from a variety of cell types44, including epithelial and endothelial cells20. Mounting data also suggest that IL-33 regulates cellular lifespan, as shown by the ability of inhibition of IL-33 to provide protection against cisplatin-induced apoptosis of kidney tubule epithelial cells32. IL-33 also enhances the apoptosis of human eosinophils mediated by the lectin Siglec-8 (ref. 33). Our study has supported those published observations by showing that IL-33 induced apoptosis of lung epithelium and that this effect was controlled by alteration of receptor concentrations.
Our study has highlighted the finding that manipulation of the proteolytic processing of a surface receptor by ubiquitin-mediated degradation was sufficient to profoundly attenuate cytokine-mediated pulmonary inflammation by endotoxin-containing pathogens. It has been shown that downregulation of ST2L by siRNA blocks the effects of IL-33-induced release of cytokines from pulmonary epithelial and endothelial cells20, which emphasizes the biological role of the ligation of IL-33 to its receptor in the lung. Although abrogation of IL-33 signaling through the use of knockout mice, neutralizing antibodies or sST2 decoy receptors has been described before, identifying the substrate-recognition specificity of FBXL19 might represent a more elegant strategy for attenuating the IL-33–ST2L pathway. Typically, F-box proteins (such as FBW7 and β-Trcp) have many divergent substrates; thus, we cannot exclude the possibility of other molecular targets, and we do not mean to imply that ST2L is the only substrate for FBXL19. Nevertheless, our fundamental observation was that FBXL19 attenuated IL-33-mediated activity by removing ST2L, but not sST2 or IL-1R1, on the cell surface, a critical initial step that led to profound biological consequences. Future studies should focus on high-throughput screening of peptide mimics of FBXL19 to attenuate inflammation in bacterial pneumonia that exploits molecular interactions in the IL-33–ST2L→GSK3β→FBXL19 pathway.
ONLINE METHODS
Cells and reagents
MLE12 cells (American Type Culture Collection) were cultured at 37 °C in an atmosphere of 5% CO2 with HITES medium (RPMI-1640 medium modified to contain hydrocortisone, insulin, transferrin, estradiol and selenium) containing 10% FBS and antibiotics. HEK293 cells were cultured in DMEM containing 10% FBS and antibiotics. Primary culture of human bronchial cells (HBEpCs; Lonza) and human pulmonary artery endothelial cells (HPAECs; Lonza) was done with medium supplemented with growth factors provided by Lonza. Fluorescein isothiocyanate–conjugated monoclonal anti-ST2 (DJ8) was from MD Bioscience. Polyclonal anti-ST2 (ab25877) was from Abcam. Human and mouse recombinant IL-33 were from R&D Systems. Anti-V5 (2F11F7), mammalian expressional plasmid pcDNA3.1/His-V5-topo, and Escherichia coli Top10 competent cells were from Life Technologies. Anti-cortactin (H222), anti-GSK3β (27C10) and anti-ubiquitin (P4D1) were from Cell Signaling. Cycloheximide, leupeptin, LPS, anti-Myc (9E10) and anti-Flag (M2) were from Sigma. MG-132 was from EMD Chemicals. Immunobilized protein A/G beads and control IgG (sc2025, sc2027 and sc2028) and anti-FBXL19 (P-13) were from Santa Cruz Biotechnology. Antibody to GSK3β phosphorylated at Tyr216 (13A) was from BD Bioscience. All commercially available materials of the highest grade were used.
Plasmid and siRNA transfection
The cDNA encoding wild-type mouse ST2L or human FBXL19 and mutants of those were inserted into the vector pCDNA3.1/V5-His-Topo (Invitrogen). The cDNA encoding mouse ST2L with a carboxy-terminal Flag tag was inserted into the vector pacAd5 (University of Iowa Gene Transfer Vector Core). The primers for these were as follows: ST2L forward, CACCATGATTGACAGACAGAGAATGG; ST2L reverse, AAAGTGTTTCAGGTCTAAG; ST2L3xFlag reverse, CTACTTGTCATCG TCATCCTTGTAATCGATATCATGATCTTTATAATCACCGTCATGGT CTTTGTAGTCTCGAAAGTGTTTCAGGTCTAA; ST2L(K246R) forward, CCTGTTCAGCTTGCTTTGGCAGAGGCTCTCACT; ST2L(K246R) reverse, GGACAAGTCGAACGAAACCGTCTCDCGAGAGTGA; ST2L(K326R) forward, CACCATAAGGCTGAGAAGGAGACAACCAATTGATCAC; ST2L(K326R) reverse, GTGATCAATTGGTTGTCTCCTTCTCAGCCTTA TGGTG; FBXL19 forward, CACCATGGGGAAGGCTGAGGGTGTC; and FBXL19 reverse, GCTGTCCTTGAGAAGCAGCTT. Overexpression of plasmids or cellular transfection of siRNA in MLE12 cells was facilitated with the Amaxa nucleofector system. Lipofectamine2000 was used for transfection of plasmids into HEK293 cells according to the instruction of the manufacture (Life Technologies).
Isolation of cell surface proteins, preparation of protein extracts and immunoblot analysis
Proteins on the cell surface were isolated with a cell surface protein–isolation kit with biotin labeling, according to the manufacturer's instructions (Pierce Biotechnology). Cells or cell-surface protein were lysed in lysis buffer (Cell Signaling). Equal amounts of total protein from each sample were separated by SDS-PAGE and transferred to nitrocellulose, then incubated with primary antibody, followed by secondary antibody.
Coimmunoprecipitation
Equal amounts of protein from each sample were incubated with primary antibody before precipitation with protein A/G beads or were incubated overnight with histidine-coated beads. Precipitates were rinsed and eluted by boiling in SDS sample buffer.
Immunostaining
MLE12 cells were cultured in glass-bottomed dishes and were fixed for 20 min with 4% paraformaldehyde. Cells were made permeable for 1 min in 0.1% Triton-100 for analysis of the localization of intracellular ST2L and lysosomes. Cells were exposed to primary antibody, followed by incubation with fluorescence-labeled secondary antibody. A Zeiss LSM 510 confocal microscope was used for immunofluorescence cell imaging.
In vitro translation of cDNA of mouse ST2L wild-type and mutants
A TnT in vitro translation system was used according to the manufacturer's instructions (Promega) for In vitro transcription and translation. This mammalian-based system expresses soluble, functional proteins that are post-translationally modified. Translated V5-tagged wild-type and mutant mouse ST2L were analyzed by immunoblots probing the V5 tag.
Flow cytometry
MLE12 cells were collected with mild trypsinization. Cell death was assessed by two-color analysis of binding of annexin V–fluorescein isothiocyanate and uptake of propidium iodide. ST2L expression on cell surface was assessed with fluorescein isothiocyanate–labeled anti-ST2 (DJ8; MD Bioscience) with a FACSCalibur (Becton Dickinson).
In vitro ubiquitin conjugation assay
ST2L was ubiquitinated in a reaction mixture containing synthesized V5-tagged wild-type and mutant mouse ST2L, 50 mM Tris (pH 7.6), 5 mM MgCl2, 0.6 mM DTT, 2 mM ATP, 1.5 ng/μl E1 (Boston Biochem), 10 ng/μl Ubc5, 10 ng/μl Ubc7, 1 μg/μl ubiquitin (EMD Chemicals), 1 μM ubiquitin aldehyde, histidine-purified recombinant Cullin-1, Skp1 and Rbx1, plus FBXL19 immunoprecipitated from HEK293 cells. Mixtures were assessed by immunoblot analysis of the V5 tag.
Animals
All mice were housed in the University of Pittsburgh Animal Care Facility in accordance with institutional guidelines and guidelines of the US National Institutes of Health. Veterinary care of these mice and related animal experiments was approved by the University of Pittsburgh Animal Resources Center. C57/BL6 mice were given intratracheal administration of LPS (1 mg per kg body weight) or P. aeruginosa (strain PA103; 1 × 104 colony-forming units per mouse) . After 24 h, BAL fluid was collected for cytokine analysis by enzyme-linked immunosorbent assay. The cDNA encoding human FBXL19 was inserted into the pLVX-IRES-tdTomato vector (Clontech); lentiviral vectors encoding FBXL19 or FBXL19-specific shRNA and their controls were generated with a lentivirus packaging system (Clontech). C57/BL6 mice were given intratracheal administration of those lentivirus vectors (1 × 109 plaque-forming units per mouse) for 5 d before intratracheal inoculation of LPS or PA103 (doses described above) for 24 h. BAL fluid was collected for cytokine assay and lung tissues were immunoscanned and then fixed for staining with hematoxylin and eosin. For analysis of the effect of IL-33 on apoptosis in lung tissues, C57/BL6 were given intratracheal administration of IL-33 (4 μg per mouse) for 24 h and lung tissues were fixed, followed by TUNEL assay. For analysis of the effect of IL-1β on lung inflammation, C57/BL6 were given intratracheal administration of mouse IL-1β (4 μg/mouse) for 24 h and lung tissues were fixed for staining with hematoxylin and eosin.
Statistical analysis
A two-way analysis of variance or an unpaired t-test was used for statistical analysis, with P values of less than 0.05 considered indicative of significance.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Wallace for technical assistance. This study is based on work supported in part by the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. Supported by the US Department of Veterans Affairs (Merit Review Award), the US National Institutes of Health (R01 HL01916 to Y.Z. and R01 HL096376, R01 HL097376 and R01 HL098174 to R.K.M.) and the American Heart Association (12SDG9050005 to J.Z.). The contents presented do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
AUTHOR CONTRIBUTIONS
J.Z. and Y.Z. designed the study, did experiments, analyzed the data and wrote the manuscript; J.W., R.K.Mi. and D.F.M. did experiments; B.B.C. and T.C. cloned FBXL19; B.B.C. assisted with animal experiments; C.Z. provided reagents; and R.K.Ma. assisted Y.Z. with direction and study design and provided reagents and editorial assistance with this manuscript.
Supplementary information is available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Kurowska-Stolarska M, Hueber A, Stolarski B, McInnes IB. Interleukin-33: a novel mediator with a role in distinct disease pathologies. J. Intern. Med. 2011;269:29–35. doi: 10.1111/j.1365-2796.2010.02316.x. [DOI] [PubMed] [Google Scholar]
- 2.Smith DE. IL-33: a tissue derived cytokine pathway involved in allergic inflammation and asthma. Clin. Exp. Allergy. 2011;40:200–208. doi: 10.1111/j.1365-2222.2009.03384.x. [DOI] [PubMed] [Google Scholar]
- 3.Küchler AM, et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am. J. Pathol. 2008;173:1229–1242. doi: 10.2353/ajpath.2008.080014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lüthi AU, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J. Biol. Chem. 2009;284:19420–19426. doi: 10.1074/jbc.M901744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammad H, et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, et al. Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem. Biophys. Res. Commun. 2009;386:181–185. doi: 10.1016/j.bbrc.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Ohno T, et al. Paracrine IL-33 stimulation enhances lipopolysaccharide-mediated macrophage activation. PLoS ONE. 2011;6:e18404. doi: 10.1371/journal.pone.0018404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espinassous Q, et al. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J. Immunol. 2009;183:1446–1455. doi: 10.4049/jimmunol.0803067. [DOI] [PubMed] [Google Scholar]
- 10.Joshi AD, et al. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunol. 2011;11:52. doi: 10.1186/1471-2172-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin H, et al. Adenovirus-mediated overexpression of soluble ST2 provides a protective effect on lipopolysaccharide-induced acute lung injury in mice. Clin. Exp. Immunol. 2011;164:248–255. doi: 10.1111/j.1365-2249.2011.04326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oboki K, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. USA. 2011;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alves-Filho JC, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2011;16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 14.Li H, et al. The cloning and nucleotide sequence of human ST2L cDNA. Genomics. 2000;67:284–290. doi: 10.1006/geno.2000.6269. [DOI] [PubMed] [Google Scholar]
- 15.Trajkovic V, Sweet MJ, Xu D. T1/ST2–an IL-1 receptor-like modulator of immune responses. Cytokine Growth Factor Rev. 2004;15:87–95. doi: 10.1016/j.cytogfr.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Sweet MJ, et al. A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J. Immunol. 2001;166:6633–6639. doi: 10.4049/jimmunol.166.11.6633. [DOI] [PubMed] [Google Scholar]
- 17.Choi YS, et al. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. 2009;114:3117–3126. doi: 10.1182/blood-2009-02-203372. [DOI] [PubMed] [Google Scholar]
- 18.Kurowska-Stolarska M, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J. Immunol. 2008;181:4780–4790. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 19.Kurowska-Stolarska M, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009;183:6469–6477. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 20.Yagami A, et al. IL-33 mediates inflammatory responses in human lung tissue cells. J. Immunol. 2011;185:5743–5750. doi: 10.4049/jimmunol.0903818. [DOI] [PubMed] [Google Scholar]
- 21.Rocca A, Lamaze C, Subtil A, Dautry-Varsat A. Involvement of the ubiquitin/proteasome system in sorting of the interleukin 2 receptor beta chain to late endocytic compartments. Mol. Biol. Cell. 2001;12:1293–1301. doi: 10.1091/mbc.12.5.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gesbert F, Malarde V, Dautry-Varsat A. Ubiquitination of the common cytokine receptor γc and regulation of expression by an ubiquitination/deubiquitination machinery. Biochem. Biophys. Res. Commun. 2005;334:474–480. doi: 10.1016/j.bbrc.2005.06.121. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Moczygemba M, Huston DP, Lei JT. JAK kinases control IL-5 receptor ubiquitination, degradation, and internalization. J. Leukoc. Biol. 2007;81:1137–1148. doi: 10.1189/jlb.0706465. [DOI] [PubMed] [Google Scholar]
- 24.Wauman J, De Ceuninck L, Vanderroost N, Lievens S, Tavernier J. RNF41 (Nrdp1) controls type 1 cytokine receptor degradation and ectodomain shedding. J. Cell Sci. 2011;124:921–932. doi: 10.1242/jcs.078055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jadhav T, Wooten MW. Defining an embedded code for protein ubiquitination. J. Proteomics Bioinform. 2009;2:316. doi: 10.4172/jpb.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J. Biosci. 2006;31:137–155. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 27.Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 28.Katoh M, Katoh M. Identification and characterization of FBXL19 gene in silico. Int. J. Mol. Med. 2004;14:1109–1114. [PubMed] [Google Scholar]
- 29.Carpenter G, Cohen S. Epidermal growth factor. J. Biol. Chem. 1990;265:7709–7712. [PubMed] [Google Scholar]
- 30.Leng S, et al. Glycogen synthase kinase 3 β mediates high glucose-induced ubiquitination and proteasome degradation of insulin receptor substrate 1. J. Endocrinol. 2010;206:171–181. doi: 10.1677/JOE-09-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou C, et al. LPS impairs phospholipid synthesis by triggering β-transducin repeat-containing protein (β-TrCP)-mediated polyubiquitination and degradation of the surfactant enzyme acyl-CoA:lysophosphatidylcholine acyltransferase I (LPCAT1). J. Biol. Chem. 2011;286:2719–2727. doi: 10.1074/jbc.M110.192377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akcay A, et al. IL-33 exacerbates acute kidney injury. J. Am. Soc. Nephrol. 2011;22:2057–2067. doi: 10.1681/ASN.2010091011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Na HJ, Hudson SA, Bochner BS. IL-33 enhances Siglec-8 mediated apoptosis of human eosinophils. Cytokine. 2012;57:169–174. doi: 10.1016/j.cyto.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YR, Kori R, John B, Tan TH. Caspase-mediated cleavage of actin-binding and SH3-domain-containing proteins cortactin, HS1, and HIP-55 during apoptosis. Biochem. Biophys. Res. Commun. 2001;288:981–989. doi: 10.1006/bbrc.2001.5862. [DOI] [PubMed] [Google Scholar]
- 35.Timpson P, et al. Aberrant expression of cortactin in head and neck squamous cell carcinoma cells is associated with enhanced cell proliferation and resistance to the epidermal growth factor receptor inhibitor gefitinib. Cancer Res. 2007;67:9304–9314. doi: 10.1158/0008-5472.CAN-07-0798. [DOI] [PubMed] [Google Scholar]
- 36.Clark ES, et al. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene. 2009;28:431–444. doi: 10.1038/onc.2008.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu D, et al. IL-33 exacerbates autoantibody-induced arthritis. J. Immunol. 2010;184:2620–2626. doi: 10.4049/jimmunol.0902685. [DOI] [PubMed] [Google Scholar]
- 38.Xu D, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. USA. 2008;105:10913–10918. doi: 10.1073/pnas.0801898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rong Z, et al. Interleukin-17F signaling requires ubiquitination of interleukin-17 receptor via TRAF6. Cell. Signal. 2007;19:1514–1520. doi: 10.1016/j.cellsig.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen CQ, Yin H, Lee BH, Chiorini JA, Peck AB. IL17: potential therapeutic target in Sjogren's syndrome using adenovirus-mediated gene transfer. Lab. Invest. 2011;91:54–62. doi: 10.1038/labinvest.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mueller SG, Schraw WP, Richmond A. Activation of protein kinase C enhances the phosphorylation of the type B interleukin-8 receptor and stimulates its degradation in non-hematopoietic cells. J. Biol. Chem. 1995;270:10439–10448. doi: 10.1074/jbc.270.18.10439. [DOI] [PubMed] [Google Scholar]
- 42.Rottmann S, Wang Y, Nasoff M, Deveraux QL, Quon KCA. TRAIL receptor-dependent synthetic lethal relationship between MYC activation and GSK3beta/FBW7 loss of function. Proc. Natl. Acad. Sci. USA. 2005;102:15195–15200. doi: 10.1073/pnas.0505114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plotnikov A, et al. Oncogene-mediated inhibition of glycogen synthase kinase 3β impairs degradation of prolactin receptor. Cancer Research. 2008;68:1354–1361. doi: 10.1158/0008-5472.CAN-07-6094. [DOI] [PubMed] [Google Scholar]
- 44.Oboki K, Nakae S, Matsumoto K, Saito H. IL-33 and airway inflammation. Allergy Asthma Immunol. Res. 2011;3:81–88. doi: 10.4168/aair.2011.3.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.