

Abstract
Schizophrenia is a common psychiatric disorder and caused by a combination of environmental, social and genetic factors. Histone deacetylases (HDACs) can translate epigenetic effects to the genome by modifying chromatin structure and gene expression. Inappropriate activity of HDACs is associated with cancer, cardiovascular and neurological diseases, and HDAC inhibitors are shown to improve the derivation of induced pluripotent stem (iPS) cells and to modulate cell lineage differentiation during brain development. We demonstrate that one of the HDAC genes, HDAC9, is hemizygously deleted in a small proportion of schizophrenia patients, and is widely expressed in mouse brain including areas where the neuropathology of schizophrenia is found. High levels of expression are observed in the hippocampus, layers II/III and V of the cerebral cortex, prefrontal and medial prefrontal cortex, piriform and cingulum cortex, basolateral amygdaloid nuclei and choroid plexus. HDAC9 protein is found in the cell body as well as in nerve fibers. Importantly, HDAC9 is not expressed in adult neural stem cells, glia, astrocytes, or oligodendrocytes, but expressed exclusively in post-mitotic and mature neurons. Our data suggest that HDAC9 may play a crucial role in neuronal function of adult brain.
Keywords: Adult neural stem cells, copy number variation, HDAC9, histone deacetylase, neuron-specific expression, schizophrenia
Introduction
Schizophrenia is a neurodevelopmental disorder and associated by environmental, social and genetic risk factors. Environmental factors may modify gene expression without affecting DNA sequences. One of the epigenetic mechanisms that imprint environmental cues onto the genome is carried out by two classes of enzymes: histone acetyltransferases and histone deacetylases (HDACs) [1]. Histone acetyltransferases acetylate whereas HDACs deacetylate the lysine residues of the histone proteins [2,3].
Histone proteins are elementary components of nucleosomes which form the fundamental unit of chromatin. They are subject to post-translational modifications including phosphorylation, methylation, ubiquitinyation and acetylation. Generally, increased acetylation leads to more relaxed chromatin structure and higher gene expression, whereas deacetylation of the lysine residues by HDACs results in more compact chromatin structure and decreased gene expression. Therefore, HDACs are largely considered as transcriptional repressors [2,4-6]. Since the purification of the first HDAC in 1996 [7], a total of 18 HDAC genes have been identified. HDAC proteins are grouped into 4 classes based on their similarity to yeast proteins [8]. Class I consists of HDAC1, 2, 3 and 8, class II includes HDAC4, 5, 6, 7, 9, and 10, and class III members are SirT1 – 7. HDAC11 is in class IV on its own because of its distinctive structure.
Mouse Hdac1 gene is crucial for early embryonic development, and germ-line deletion of Hdac1 causes early embryonic lethality. Although embryonic stem (ES) cell proliferation and embryoid body formation are unaffected by loss of either Hdac1 or Hdac2, Hdac1-deficient embryoid bodies are significantly smaller, showed spontaneous rhythmic contraction, and increased expression of both cardiomyocyte and neuronal markers. This suggests that loss of Hdac1 function promotes cardiomyocyte and neuronal differentiation of ES cells [9]. Interestingly, treatment of embryonic neural progenitor cells with a HDAC inhibitor trichostatin A (TSA) increases neuronal differentiation and decreases astrocyte differentiation when cultured in a minimal, serum-free medium, lacking any induction or growth factor. Also, neurons derived from TSA treatment appear to develop normal electrophysiological membrane properties characteristics [10]. Meanwhile, dysregulation of HDACs is shown to associate with a variety of pathological processes, including neurological disorders, inflammation, cardiovascular dysfunction and cancer. For example, Upregulation of HDAC5 and HDAC9 expression is associated with high-risk medulloblastoma [11]. HDAC9 also has a crucial role in musculoskeletal and cardiac function [12,13] and targeted deletion of HDAC9 leads to cardiac hypertrophy [14].
In the central nervous system, aberrant epigenetic regulation has been reported in psychiatric disorders including schizophrenia and depression [15-18]. HDAC5 may act as a central integrator of chronic drug addiction and stress. Chronic exposure to cocaine or stress decreases HDAC5 function in the nucleus accumbens, and increases histone acetylation and transcription of HDAC5 target genes, whereas loss of HDAC5 causes hypersensitive responses to chronic cocaine or stress [19]. Additionally, in a recent genetic association study, a SNP (rs1063639) of HDAC4 is linked schizophrenia in a Korean population [20]. However, the T/C substitution in the SNP (rs1063639) is synonymous, and does not alter the amino acid at aa855 position, and it is yet to be determined whether this synonymous substitution influences mRNA transcription or stability. Gavin and colleagues demonstrate that cultured lymphocytes from schizophrenic patients have lower levels of histone H3 acetylation [5,17,21], and HDAC inhibitors valproic acid (VPA) and TSA can induce GAD1 mRNA expression [17]. GAD1 encodes glutamic acid decarboxylase, GAD67, an enzyme synthesizing gamma-aminobutyric acid (GABA). GABA is a major inhibitory amino acid neurotransmitter and is commonly defective in schizophrenic patients. This suggests that higher HDAC activity may be associated with the cause of schizophrenia and HDAC inhibitor(s) may be of therapeutic potential for schizophrenia. However, postmortem studies did not reveal overt differences in chromatin modification in the prefrontal cortex of schizophrenia patients [22]. On the other hand, deletion rather than duplication of HDAC9 is reported in schizophrenic patients recently [23]. Therefore, the extent of involvement and the mechanisms by which HDACs are involved in neurological disorders largely remain unclear, and the knowledge of HDAC expression in the brain is fundamental to understand roles of HDAC function in neurological diseases.
Existing data on the expression of HDAC9 in the brain is extremely controversial. For example, high levels of HDAC9 expression were detected in the brain and skeletal muscle by Northern hybridization when the gene was initially cloned [13]. However, using a systematic approach of 40-mer oligonucleotide probes, Hdac9 mRNA expression was only just above the background level in rat brain by in situ hybridization [24]. On the other hand, out of 12 HDAC genes analyzed using relative quantification, HDAC9a and HDAC9b were the most highly expressed HDACs in normal brain [25]. This controversy highlights an urgent need to clarify HDAC9 expression in the brain.
We present here evidence that HDAC9 gene is deleted in a small portion of schizophrenia patients. We carried out an immunohistochemical study with antibodies against HDAC9 and found wide expression of Hdac9 protein in mouse brain with high levels in the hippocampus and cerebral cortex, where neuropathologies are found in schizophrenia. The expression is exclusive to the post-mitotic neurons, suggesting HDAC9 may be crucial for the function of mature neurons.
Materials and methods
Copy number variations (CNV)
The recruitment and assessment of cases and controls and the methods of CNV analysis are as fully described [26]. In brief, all subjects gave informed consent. The studies were approved by the appropriate research ethics committee. Diagnoses of schizophrenia according to DSM4 criteria were reached by consensus between two trained psychiatrists with information from direct interview and case notes. The 3,391 schizophrenic patients and 3,181 ancestrally matched controls [26] were analysed for CNVs on the UCSC human genome database (hg18) for all HDAC genes (http://genome.ucsc.edu/cgi-bin/hgGateway).
Immunohistochemistry
Two-month-old wild type mice were killed humanely by cervical dislocation. Mouse brains were dissected and embedded in OCT, and then freshly frozen on dry ice. Fresh mouse brain was sectioned at 12 μm with the aid of a Cryostat (Leica CM1850), and sections were mounted on slides and stored at -20oC for further analysis. Antigen retrieval was done in a citrate buffer [27,28]. Sections were then incubated overnight with a rabbit polyclonal primary antibody against a synthetic peptide EVPVGLEPISPLDLRT derived from the N-terminus of HDAC9 (ab18970, ABcam, 1:200) on its own, or in combination with a second cell-type specific primary antibody for double labelling. Cell-type antibodies used were Ki67 (TEC-3) from Dako (1:50), CNPase antibody from ABcam (1:1000), mouse anti-GFAP (1:1000) and anti-NeuN (1:400) from Chemicon. All secondary antibodies were purchased from Molecular Probe (Donkey anti-rabbit and anti-mouse), with a dilution of the 1:1000 for the red, and 1:400 for the green antibody. Control experiments were performed in parallel in the absence of the primary antibody. Images were captured by an Axiovert 40CFL microscope with AxioVision Rel. 4.5 software (Zeiss).
Results
HDAC9 is deleted in a small subset of schizophrenic population
We analyzed the CNVs in 3,391 schizophrenic patients and in 3,181 ancestrally matched controls [26] for all HDAC genes, and identified three schizophrenic patients to carry single copy deletion at chr7:17674100-18634682; chr7: 18374606-18498502 and chr7:18595436-18707719, respectively. These deletions commonly involve the HDAC9 gene (Figure 1). In addition, one mental retardation patient was shown to carry a single copy of chr22: 48938557-49139096, which encodes HDAC10 and ~10 other genes (not shown). In the control group, only one individual was identified to harbor HDAC CNV, with 3 copies of chr17:39196329-40203723 which encodes HDAC5 gene together with >20 other genes. However, no CNV was found for the HDAC1-4, HDAC6-8, HDAC11 and SIRT1-7 genes in controls or schizophrenic patients. These data suggest that CNV of HDAC genes is more common in schizophrenic cases (4/3,391 cases) than in controls (1/3,181 controls), and deletion of HDAC9 may be associated with a small subset of schizophrenic population.
Figure 1.
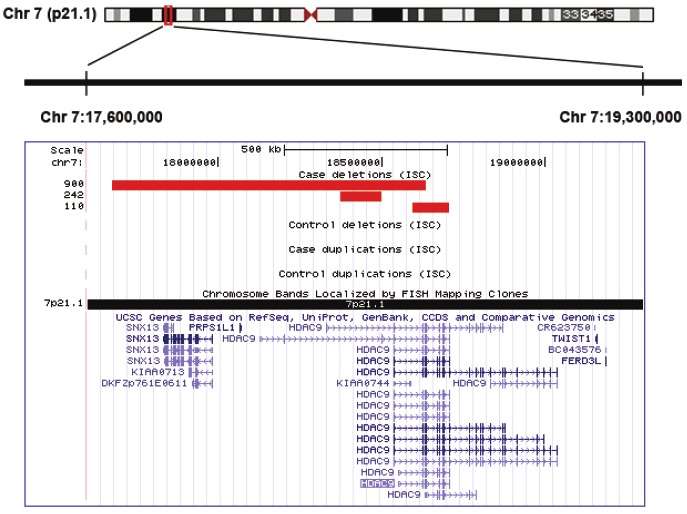
HDAC9 gene deletion in schizophrenia. (A). Schematic presentation of 1.7 Mb chromosomal region at 7p21.1 (Chr7:17,600,000-19,300,000), which encodes HDAC9 and flanking genes. (B). A genome-wide survey of rare CNV showed deletions in three schizophrenic patients out of the 3,391 cases, with none in 3,181 controls. The deleted regions highlighted in red.
Expression of Hdac9 in mouse brain
To examine neuroanatomical expression of Hdac9, we carried out an immunohistochemical study with mouse adult brain sections. Hdac9 was widely, but not uniformly, expressed in the mouse brain (Figure 2). For example, little expression was detected in the anterior olfactory nuclei posterior (Figure 2C), medial habenular nuclei (Figure 2K) or subcommisural organ SCO (Figure 2L). However, high Hdac9 expression was found in all fields of hippocampus (Figure 2F, K, L), layers II/III and V of cerebral cortex (Figure 2E), prefrontal cortex (Figure 2A) and medial prefrontal cortex (Figure 2B), piriform (Figure 2C-D) and cingulum (Figure 2H) cortex, basolateral amygdaloid nuclei (Figure 2D), and choroid plexus (Figure 2K-L).
Figure 2.
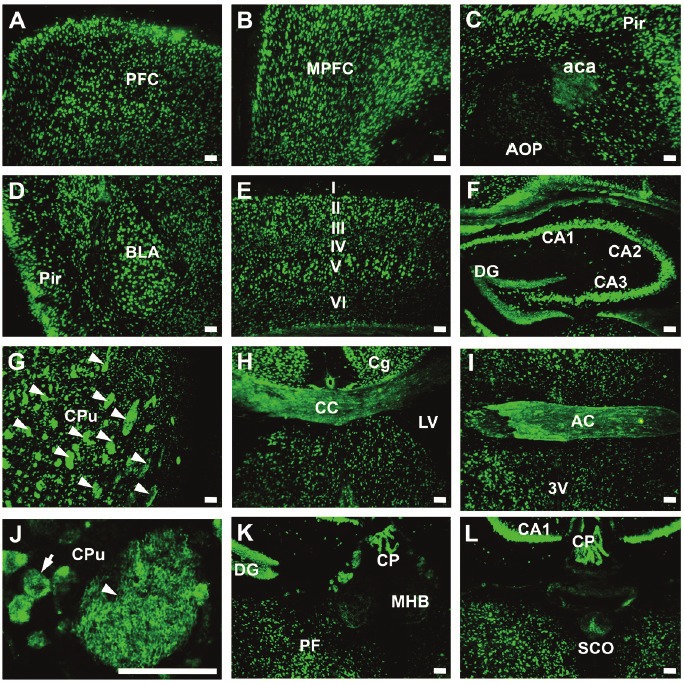
Neuroanatomic expression of Hdac9 in the mouse brain. Immunofluorescent staining was carried out on brain sections with anti-HDAC9, and green fluorescence showed the expression of Hdac9. Abbreviations: 3V- third ventricle; AC – anterior commissure; aca – anterior commissure, anterior; AOP – anterior olfactory nucleus, posterior; BLA – basolateral amygdaloid nucleus, anterior; CA1-CA3 – fields of Ammon’s horn of the hippocampus; CC – corpus callosum; Cg – cingulum; CP – choroid plexus; CPu – caudate putamen; DG – dentate gyrus; I-VI – six layers of the cerebral cortex; LV – lateral ventricle; MHB – medial habenular nucleus; MPFC – medial prefrontal cortex; PF – parafascicular thalamic nucleus; PFC – prefrontal cortex; Pir – piriform cortex; SCO – subcommisural organ. Bars=50um.
Hdac9 is not expressed in oligodendrocytes
Using anti-HDAC9 antibody, we also observed an intense immunofluorescent staining in the corpus callosum (Figure 2H), anterior commissure (Figure 2I), and caudate putamen (Figure 2G). In contrast to the punctuate distribution of Hdac9 in neuronal cell bodies (arrow, Figure 2J), high magnification showed bundles of fibre-like staining pattern in the caudate putamen (arrowhead, Figure 2J). To examine whether Hdac9 is expressed in oligodendrocytes, we carried out double immunostaining with an antibody again 2',3'-cyclic nucleotide 3' phosphodiesterase (CNP), a marker for oligodendroglial cells. Hdac9 did not co-label with CNP in the dorsal lateral cerebral cortex (Figure 3A-C) or in the retrosplenial gruanular cortex (RSG) (Figure 3D-F), showing that Hdac9 was not expressed in oligodendrocytes.
Figure 3.
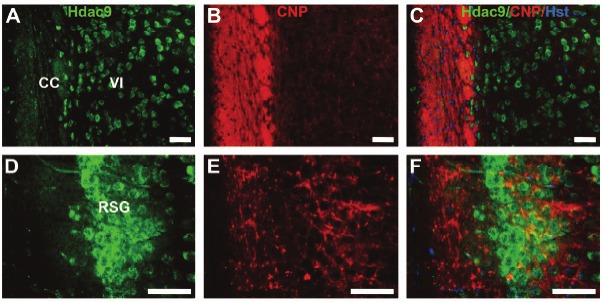
Hdac9 is not expressed in oligodendrocytes. Brain sections were doubly immunostained with anti-HDAC9 (green) and anti-CNP (red) for oligodendrocytes. Images were taken from dorsal lateral cerebral cortex with layer VI and corpus collasum (CC) in A-C and from retrosplenial granular cortex (RSG) in D-F. Bars=50um.
Hdac9 is not expressed in astrocytes or glia
To further examine cell-type expression of the Hdac9, we performed double immunostaining with anti-GFAP, a marker for glia and astrocyes. No co-labelling of Hdac9 and GFAP was found in the cerebral cortex, hippocampus or other brain regions (Figure 4), showing Hdac9 was not expressed in astrocytes or glia.
Figure 4.
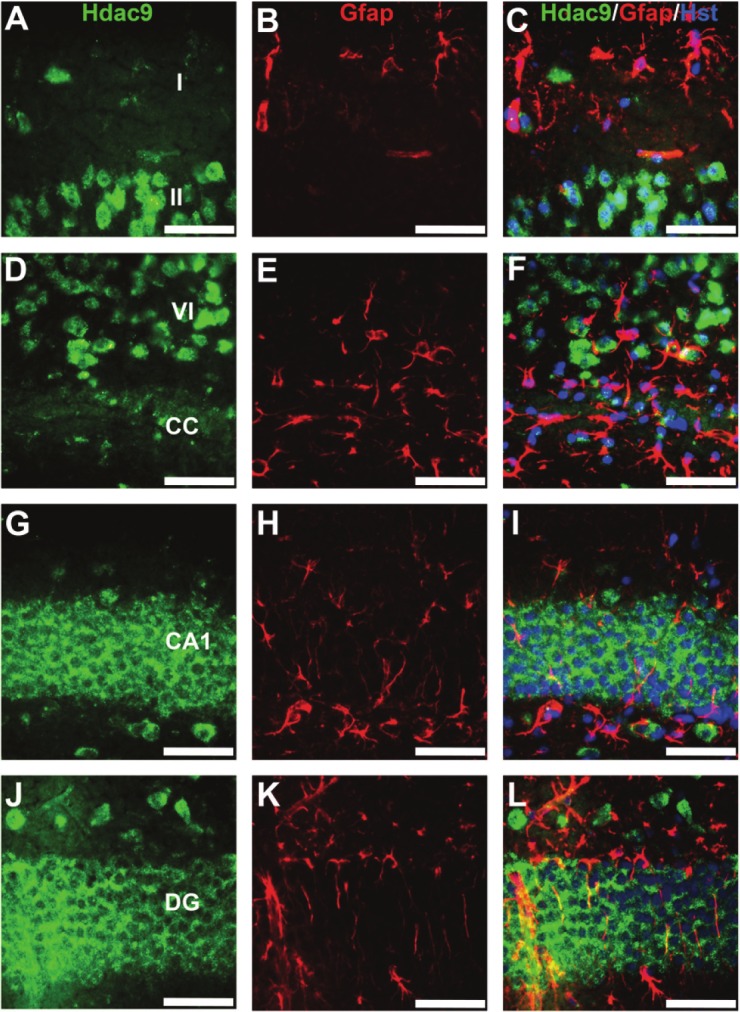
Hdac9 is not expressed in GFAP-positive cells. Brain sections were double immunolabeled with anti-HDAC9 (green) and anti-GFAP (red). Images were taken from layers I-II (A-C) or VI (D-F) regions of cerebral cortex, or CA1 (G-I) or dentate gyrus (J-L) regions of the hippocampus. CA1 – field of CA1 Ammon’s horn; CC – corpus callosum; DG – dentate gyrus. Bars=50um.
Hdac9 is expressed in post-mitotic neurons, but not in adult neural stem cells
Next, we co-immunostained brain sections for Hdac9 with NeuN, a marker for mature neurons (Figure 5). Hdac9 was highly co-labeled with NeuN in the hippocampal granular neurons (Figure 5A-F), in the pyramidal neurons of the cerebral cortex (Figure 5G-I) and other brain regions (not shown).
Figure 5.

HDAC9 is expressed in NeuN-positive neurons. Double immunostaining was carried out with anti-HDAC9 (green) and NeuN (red) for mature neurons, and counter-stained with Hoechst (blue) for nuclei. Images were taken from the hippocampal (A-F) or cortical (G-I) regions. Arrows in A-C indicated newborn nerons which were not labelled by NeuN but strongly expressing Hdac9 in the subgranular zone. DG – dentate gyrus. Bars=50um.
Adult neural stem cells are localized in the subgranular zone of the hippocampus, subventricular zone and rostral migratory stream. We then examined Hdac9 expression in adult neural stem cells by co-labelling with Ki67, a marker for proliferating cells. No co-expression of Hdac9 and Ki67 was found in the mouse brain (Figure 6). Thus, Hdac9 is not expressed in adult neural stem cells, but, expressed exclusively in post-mitotic and mature neurons.
Figure 6.
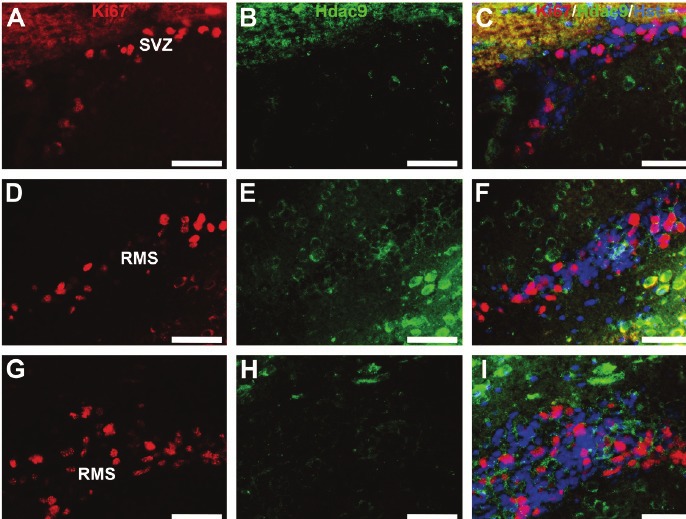
Hdac9 is not expressed in Ki67-positive adult neural stem cells. Co-immunolabeling was performed on mouse brain sections with anti-HDAC9 (green) and Ki67 (red) for proliferating cells. Images were sampled from the subventricular zone (SVZ) in A-C, and from different regions of the rostral migration stream (RMS) in D-I. Bars=50um.
Discussion
Schizophrenia is associated with environmental, social and genetic factors. Non-genetic factors may modify genome at gene expression level through epigenetic mechanisms. The nucleosome is a fundamental unit of chromatin structure and function, which consists of ~147 bp DNA wrapped by 8 histone proteins. The acetylation status of the histone proteins is crucial for maintaining appropriate gene expression levels. Deacetylation of the N-terminal tail of histones via HDACs leads to compact chromatin structure and reduced gene expression. We have shown here that the HDAC9 gene is deleted in a small subset of schizophrenic patients, and Hdac9 protein is enriched in brain areas where neuropathology of schizophrenia is found.
HDAC9 is relatively under-investigated among all HDACs. High levels of HDAC9 mRNA were initially detected in the brain extract by Northern hybridization [13], and later by quantitative analyses in normal human brain samples [25]. Surprisingly, Hdac9 mRNA expression was almost undetectable in rat brain by in situ hybridization using 40-mer oligonucleotide probes [24]. It is unknown whether this is due to the species difference, probes and/or experimental conditions. We found a wide expression of Hdca9 protein in adult mouse brain, which supports Zhous’ [13] and Lucio-Eterovic’s [25] observations. The observed expression was specific, because little expression was detected in the anterior olfactory nuclei posterior, medial habenular nuclei or subcommisural organ. High levels of Hdac9 expression were found in choroid plexus, hippocampus, cerebral cortex, prefrontal cortex, medial prefrontal cortex, piriform/cingulum cortex and basolateral amygdaloid nuclei of adult mouse brain.
It is worth to note that different HDAC may be involved in the generation of different brain cell types. For example, Sirt1, a class III HDAC, appeared to suppress neurogenesis but promote astroglial lineage differentiation. Direct activation of Sirt1 or mild oxidation suppressed proliferation of neural progenitor cells and directed them into astroglial lineage at the expense of the neuronal lineage. In utero shRNA-mediated knockdown of Sirt1 in neural progenitor cells prevented oxidation-mediated suppression of neurogenesis [29]. On the other hand, we reported here that Hdac9 protein was primarily associated with neurons, and not with oligodendrocytes, astrocytes or glia. Similarly, Hdac2-5 and Hdac10-11 were previously found to express highly in neurons of rat brain. However, Hdac2–5 and Hdac11 also were found in oligodendrocytes [24], suggesting that they might play a role in myelination during CNS development [30]. Together with our data, these findings suggest that different HDAC genes may be required for different lineage differentiation of brain cells, and HDAC9 and HDAC10 are the neuron-specific ones among all HDACs. This makes it more intriguing to investigate the roles of HDAC9 and HDAC10 in neurons. Importantly, an alternatively spliced and C-terminal truncated form of HDAC9 that lacks a catalytic domain, the histone deacetylase-related protein (HDRP), has been reported to be protective against apoptosis in cultured neurons [12,31], and HDAC9 is required for dendritic growth [32].
Abnormal dendritic density and synaptic dysfunction are associated with neurodevelopmental disorders including autism and schizophrenia. A SNP of HDAC4 is associated with schizophrenia [20], and HDAC4 may play an important part in the peripheral and central nervous system. The HDAC9 gene is recently found to have varied copy numbers in schizophrenia [23], which makes further study of this protein very important. We present here evidence that the HDAC9 gene is hemizygously deleted in a small proportion (~0.1%) of schizophrenia patients, and that the Hdac9 protein is highly expressed in brain areas including hippocampus and cerebral cortex, where neuropathology of schizophrenia is found. Interestingly, a nonselective HDAC inhibitor VPA has been shown to reverse schizophrenia-like behavior in several mouse models [33,34], and has been explored in human subjects [35], although its efficacy is yet to be confirmed. Furthermore, HDAC inhibitors have shown promise in animal models of depression [36] and in humans patients when used in conjunction with antipsychotics [37,38].
Schizophrenia is a neurodevelopmental disorder and roles of HDAC9 during brain development are currently unknown. We showed that the Hdac9 protein in mouse brain is expressed exclusively in post-mitotic/mature neurons, not in adult neural stem cells, suggesting that HDAC9 expression may not be good for stem cells. In support of this, epigenetic factors are found to modulate derivation iPS cells from somatic cells. Pre-treatment of somatic cells with 5-aza-dC against DNA methyltransferase or TSA against HDACs was shown to improve the efficiency of reprogramming process via nuclear cloning [39] or via iPS cell generation [40-41]. Lower levels of histone acetylation were also observed in iPS cells derived from three transcription factors (OCT4, SOX3, KLF4) without c-MYC, and supplementation of HDAC inhibitor TSA was able to significantly improve the iPS cells for their developmental potential [42]. Similarly, another HDAC inhibitor VPA improved reprogramming efficiency by more than 100-fold [40]. TSA was also shown to increase Nkx2.5 expression in iPS cell lines and induces myocardial differentiation in mouse iPS cells [43]. Therefore, manipulation of HDAC activity may become an important tool in regenerative medicine.
Intriguingly, the HDAC9 protein is translocated from nucleus to cytoplasm during postnatal development, and this process can be induced by an increase of spontaneous firing activity in cultured mouse cortical neurons [32]. We showed that Hdac9 is not expressed in neural stem cells but expressed in post-mitotic neurons in adults. Follow-up studies on neuroanatomical changes in genetically modified Hdac9 animals will enhance our understanding of the roles of Hdac9 in brain development and function. Our discovery that HDAC gene deletion is associated with a small subset of schizophrenic patients suggests that alternative compounds, such as specific HDAC activators, shall also be investigated for their effects in neurons and in animal models.
Acknowledgement
The work is funded by SULSA Transgenic Facility and Saudi Arabian Ministry of Higher Education via King Abdullah Program for Scholarships.
Declaration of conflicts of interest
Authors declared no conflict of interest.
References
- 1.Bertrand P. Inside HDAC with HDAC inhibitors. Eur J Med Chem. 2010;45:2095–2116. doi: 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- 2.Khan S, Khan U. Role of histone acetylation in cell physiology and diseases: An update. Clin Chim Acta. 2010;411:1401–1411. doi: 10.1016/j.cca.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 3.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 4.DeRuijter A, VanGennip A, Caron H, Kemp S, VanKuilenburg A. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gavin D, Sharma R. Histone modifications, DNA methylation, and schizophrenia. Neurosci Biobehav Rev. 2010;34:882–888. doi: 10.1016/j.neubiorev.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seto E, Yang X. Regulation of histone deacetylase activities and functions by phosphorylation and dephosphorylation. In: Ralph AB, Edward AD, editors. Handbook of Cell Signaling. Second Edition. San Diego: Academic Press; 2010. pp. 2379–2388. [Google Scholar]
- 7.Taunton J, Hassig C, Schreiber S. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 8.Verdin E, Dequiedt F, Kasler H. Class II histone deacetylases: Versatile regulators. Trends Genet. 2003;19:286–293. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 9.Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107:8242–8247. doi: 10.1073/pnas.1000478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balasubramaniyan V, Boddeke E, Bakels R, Küst B, Kooistra S, Veneman A, Copray S. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience. 2006;143:939–951. doi: 10.1016/j.neuroscience.2006.08.082. [DOI] [PubMed] [Google Scholar]
- 11.Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M, Taylor MD, von Deimling A, Pfister S, Witt O. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. 2010;16:3240–3252. doi: 10.1158/1078-0432.CCR-10-0395. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Chen H, Jaramillo E, Wang L, D'Mello SR. Histone deacetylase-related protein inhibits AES-mediated neuronal cell death by direct interaction. J Neurosci Res. 2008;86:2423–2431. doi: 10.1002/jnr.21680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou X, Marks P, Rifkind R, Richon V. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A. 2001;98:10572–10577. doi: 10.1073/pnas.191375098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang C, McKinsey T, Chang S, Antos C, Hill J, Olson E. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsankova N, Renthal W, Kumar A, Nestler E. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 16.Renthal W, Nestler E. Histone acetylation in drug addiction. Semin Cell Dev Biol. 2009;20:387–394. doi: 10.1016/j.semcdb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavin D, Kartan S, Chase K, Jayaraman S, Sharma R. Histone deacetylase inhibitors and candidate gene expression: An in vivo and in vitro approach to studying chromatin remodeling in a clinical population. J Psychiatr Res. 2009;43:870–876. doi: 10.1016/j.jpsychires.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Covington H, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takizawa D, Kakizaki S, Horiguchi N, Tojima H, Yamazaki Y, Ichikawa T, Sato K, Mori M. Histone deacetylase inhibitors induce cytochrome P450 2B by activating nuclear receptor constitutive androstane receptor. Drug Metab Dispos. 2010;38:1493–1498. doi: 10.1124/dmd.110.032854. [DOI] [PubMed] [Google Scholar]
- 20.Kim T, Park J, Kim H, Chung J, Kim J. Association of histone deacetylase genes with schizophrenia in korean population. Psychiatry Res. 2010;178:266–269. doi: 10.1016/j.psychres.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Gavin D, Rosen C, Chase K, Grayson D, Tun N, Sharma R. Dimethylated lysine 9 of histone 3 is elevated in schizophrenia and exhibits a divergent response to histone deacetylase inhibitors in lymphocyte cultures. J Psychiatry Neurosci. 2009;34:232–237. [PMC free article] [PubMed] [Google Scholar]
- 22.Akbarian S, Hsien-Sung H. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52:293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Tam G, Van de Lagemaat L, Redon R, Strathdee K, Croning M, Malloy M, et al. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochem Soc Trans. 2010;38:445–51. doi: 10.1042/BST0380445. [DOI] [PubMed] [Google Scholar]
- 24.Broide R, Redwine J, Aftahi N, Young W, Bloom F, Winrow C. Distribution of histone deacetylases 1-11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- 25.Lucio-Eterovic AK, Cortez MA, Valera ET, Motta FJ, Queiroz RG, Machado HR, Carlotti CG Jr, Neder L, Scrideli CA, Tone LG. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer. 2008;8:243. doi: 10.1186/1471-2407-8-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramos-Vara J. Principles and methods of immunohistochemistry. Methods Mol Biol. 2011;691:83–96. doi: 10.1007/978-1-60761-849-2_5. [DOI] [PubMed] [Google Scholar]
- 28.Shi S, Key M, Kalra K. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: An enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 1991;39:741–748. doi: 10.1177/39.6.1709656. [DOI] [PubMed] [Google Scholar]
- 29.Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schröter F, Ninnemann O, Siegert E, Bendix I, Brüstle O, Nitsch R, Zipp F, Aktas O. Sirt1 contributes critically to the redoxdependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- 30.Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D'Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugo N, Oshiro H, Takemura M, Kobayashi T, Kohno Y, Uesaka N, Song WJ, Yamamoto N. Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur J Neurosci. 2010;31:1521–1532. doi: 10.1111/j.1460-9568.2010.07218.x. [DOI] [PubMed] [Google Scholar]
- 33.Tremolizzo L, Doueiri MS, Dong E, Grayson DR, Davis J, Pinna G, Tueting P, Rodriguez-Menendez V, Costa E, Guidotti A. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 34.Flood D, Choinski M, Marino M, Gasior M. Mood stabilizers increase prepulse inhibition in DBA/2NCrl mice. Psychopharmacology (Berl) 2009;205:369–377. doi: 10.1007/s00213-009-1547-y. [DOI] [PubMed] [Google Scholar]
- 35.Glick I, Bosch J, Casey D. A double-blind randomized trial of mood stabilizer augmentation using lamotrigine and valproate for patients with schizophrenia who are stabilized and partially responsive. J Clin Psychopharmacol. 2009;29:267–271. doi: 10.1097/JCP.0b013e3181a443d0. [DOI] [PubMed] [Google Scholar]
- 36.Schroeder F, Lin C, Crusio W, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 37.Kelly D, Conley R, Feldman S, Yu Y, McMahon R, Richardson C. Adjunct divalproex or lithium to clozapine in treatment-resistant schizophrenia. Psychiatr Q. 2006;77:81–95. doi: 10.1007/s11126-006-7963-9. [DOI] [PubMed] [Google Scholar]
- 38.Wassef A, Dott S, Harris A, Brown A, O'Boyle M, Meyer W, et al. Randomized, placebocontrolled pilot study of divalproex sodium in the treatment of acute exacerbations of chronic schizophrenia. J Clin Psychopharmacol. 2000;20:357–361. doi: 10.1097/00004714-200006000-00011. [DOI] [PubMed] [Google Scholar]
- 39.Rybouchkin A, Kato Y, Tsunoda Y. Role of Histone Acetylation in Reprogramming of Somatic Nuclei Following Nuclear Transfer. Biol Reprod. 2006;74:1083–1089. doi: 10.1095/biolreprod.105.047456. [DOI] [PubMed] [Google Scholar]
- 40.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han J, Sachdev PS, Sidhu KS. A combined epigenetic and non-genetic approach for reprogramming human somatic cells. PLoS One. 2010;5:e12297. doi: 10.1371/journal.pone.0012297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Araki R, Hoki Y, Uda M, Nakamura M, Jincho Y, Tamura C, Sunayama M, Ando S, Sugiura M, Yoshida MA, Kasama Y, Abe M. Crucial Role of c-Myc in the Generation of Induced Pluripotent Stem Cells. Stem Cells. 2011;29:1362–70. doi: 10.1002/stem.685. [DOI] [PubMed] [Google Scholar]
- 43.Kaichi S, Hasegawa K, Takaya T, Yokoo N, Mima T, Kawamura T, Morimoto T, Ono K, Baba S, Doi H, Yamanaka S, Nakahata T, Heike T. Cell line-dependent differentiation of induced pluripotent stem cells into cardiomyocytes in mice. Cardiovasc Res. 2010;88:314–323. doi: 10.1093/cvr/cvq189. [DOI] [PubMed] [Google Scholar]