

Abstract
Amyotrophic lateral sclerosis (ALS) is characterized by predominant vulnerability and central degeneration of both corticospinal/corticobulbar motor neurons (CSMN; “upper motor neurons”) in cerebral cortex, and spinal/bulbar motor neurons (SMN; “lower motor neurons”) in spinal cord and brainstem. Increasing evidence indicates broader cerebral cortex pathology in cognitive, sensory, and association systems in select cases. It remains unclear whether widely accepted transgenic ALS models, in particular hSOD1G93A mice, undergo degeneration of CSMN and molecularly/developmentally closely related populations of nonmotor projection neurons [e.g., other subcerebral projection neurons (SCPN)], and whether potential CSMN/SCPN degeneration is specific and early. This relative lack of knowledge regarding upper motor neuron pathology in these ALS model mice has hindered both molecular-pathophysiologic understanding of ALS and their use toward potential CSMN therapeutic approaches. Here, using a combination of anatomic, cellular, transgenic labeling, and newly available neuronal subtype-specific molecular analyses, we identify that CSMN and related nonmotor SCPN specifically and progressively degenerate in hSOD1G93A mice. Degeneration starts quite early and presymptomatically, by postnatal day 30. Other neocortical layers, cortical interneurons, and other projection neuron populations, even within layer V, are not similarly affected. Nonneuronal pathology in neocortex (activated astroglia and microglia) is consistent with findings in human ALS cortex and in affected mouse and human spinal cord. These results indicate previously unknown neuron type-specific vulnerability of CSMN/sensory and association SCPN, and identify that characteristic dual CSMN and SMN degeneration is conserved in hSOD1G93A mice. These results provide a foundation for detailed investigation of CSMN/SCPN vulnerability and toward potential CSMN therapeutics in ALS.
Introduction
Independent, but presumed to be mechanistically related, degeneration of both “upper” [corticospinal/corticobulbar motor neurons (CSMN)] and “lower” [spinal/bulbar motor neurons (SMN)] components of motor neuron circuitry (Lefebvre et al., 1998; Gavrilina et al., 2008) distinguishes amyotrophic lateral sclerosis (ALS) from disorders characterized by marked vulnerability and degeneration of either SMN in spinal muscular atrophy (SMA) (Rösler et al., 2000; Beckman et al., 2001; Brown and Robberecht, 2001; Bruijn et al., 2004; Stewart et al., 2006; Turner and Talbot, 2008) or CSMN in hereditary spastic paraplegia (HSP) and sporadic primary lateral sclerosis (PLS) (Fink, 2002, 2006; Rainier et al., 2003). ALS is a “system degeneration disorder,” with progressive degeneration of both CSMN and SMN, with increasing evidence for involvement of broader cortical neuronal and widely distributed nonneuronal pathology (Lomen-Hoerth et al., 2003; Strong, 2008), including astrogliosis and activated microglia in the neocortex of select ALS patients (Graham et al., 2004; Brown, 2005; Stewart et al., 2006).
Both inherited (familial) and sporadic forms of ALS show similar cardinal features: clinical course and neuropathology (Bendotti and Carrì, 2004; Boillée and Cleveland, 2004; Bruijn et al., 2004; Pasinelli and Brown, 2006). Thus, transgenic mouse models generated based on genetics of even relatively rare inherited forms of ALS offer important tools with which to investigate both sporadic and familial ALS, if patient pathology is faithfully reflected (Gurney, 1997, 2000). Toxic “gain of function” mutation in the superoxide dismutase-1 (SOD1) gene, linked to ∼10–20% of familial ALS (∼1–2–5% of all cases) is known to result in an ALS-like SMN degeneration phenotype in hSOD1G93A transgenic mice (Gurney et al., 1994; Tu et al., 1996; Gurney, 1997; Kunst et al., 1997; Beckman et al., 2001; Wong et al., 2002; Bendotti and Carrì, 2004; Wengenack et al., 2004; Hegedus et al., 2007). In contrast to considerable investigation of SMN in hSOD1G93A and other models (Chiu et al., 1995), much less is known about cortical components [degeneration of CSMN and subcerebral projection neurons (SCPN) that constitute cognitive, association, and integrative sensory circuitry (Zang and Cheema, 2002; Lobsiger et al., 2005; Yamanaka et al., 2006), and whether widely distributed nonneuronal pathology observed in select ALS patients is recapitulated in hSOD1G93A mice].
Reports on hSOD1G93A mice have suggested that their neuropathology is restricted mainly to spinal and bulbar motor neuron degeneration, but not in cortex (Wong et al., 2002; Ralph et al., 2005; Niessen et al., 2006), potentially suggesting that hSOD1G93A mice do not represent the complete human ALS phenotype. The lack of data on CSMN is perhaps not surprising, since only ∼6000 CSMN exist per hemisphere in mice, intermixed with millions of other cortical pyramidal neurons in the same region and layer V of motor cortex. Identification of specific degeneration of CSMN and related SCPN is enabled with recently identified molecular markers and knowledge about CSMN/SCPN developmental relationship (Gurney, 2000; Arlotta et al., 2005; Molyneaux et al., 2005, 2007, 2009; Tomassy et al., 2010). A few studies report corticospinal tract involvement in hSOD1G93A mice (Zang and Cheema, 2002; Lobsiger et al., 2005; Yamanaka et al., 2006), although complexities noted above limited analyses. One study, using retrograde labeling from spinal cord at postnatal day 60 (P60), reported CSMN loss (Zang and Cheema, 2002), although it was difficult to assess the following: (1) whether observed reduction in retrogradely labeled neurons was due to abnormal retrograde axonal transport or true neuronal loss; (2) whether specific CSMN degeneration or broader cortical degeneration occurs; (3) whether observed abnormalities develop before P60; or (4) whether broader populations of SCPN/other neurons are affected.
To provide a foundation for increasingly detailed investigation of CSMN/SCPN biology in ALS and for critical analysis of cellular and molecular mechanisms of shared biology between CSMN/SCPN and SMN components of motor neuron circuitry, we investigated whether distinct and specific degeneration of CSMN (with additional but not yet clarified non-CSMN cortical degeneration), as observed in ALS patients (Rösler et al., 2000; Zanette et al., 2002; Stewart et al., 2006), is faithfully reproduced in hSOD1G93A mice. Our results allow increasingly detailed cellular and molecular investigation of the biology of CSMN and broader molecularly and developmentally related populations of SCPN in hSOD1G93A mice. Elucidating this CSMN/SCPN biology might potentially contribute toward development of preventive and/or reparative therapeutics for ALS and related CSMN/SCPN degenerative diseases.
Materials and Methods
Mice.
Male and female wild-type (WT), hemizygous transgenic mice overexpressing the wild-type human SOD1 gene (hSOD1WT), and hemizygous transgenic mice expressing the SOD1 gene containing the G93A mutation in (hSOD1G93A) were used (Fig. 1A). All mouse strains are on the C57BL/6 background, and the range of gene copy number, determined before surgery, was the same in hSOD1G93A and hSOD1WT mice (30 ± 2). Thy1-YFP-hSOD1G93A and Thy1-YFP-hSOD1WT mice were generated by breeding Thy1-YFP mice [gift from J. Sanes, Harvard University, Boston, MA (Bareyre et al., 2005)] and hemizygous hSOD1G93A or hSOD1WT transgenic mice, respectively. Thy1-YFP;WT mice were used as controls. All mouse studies were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and performed in accordance with institutional, federal, and National Institutes of Health guidelines.
Figure 1.
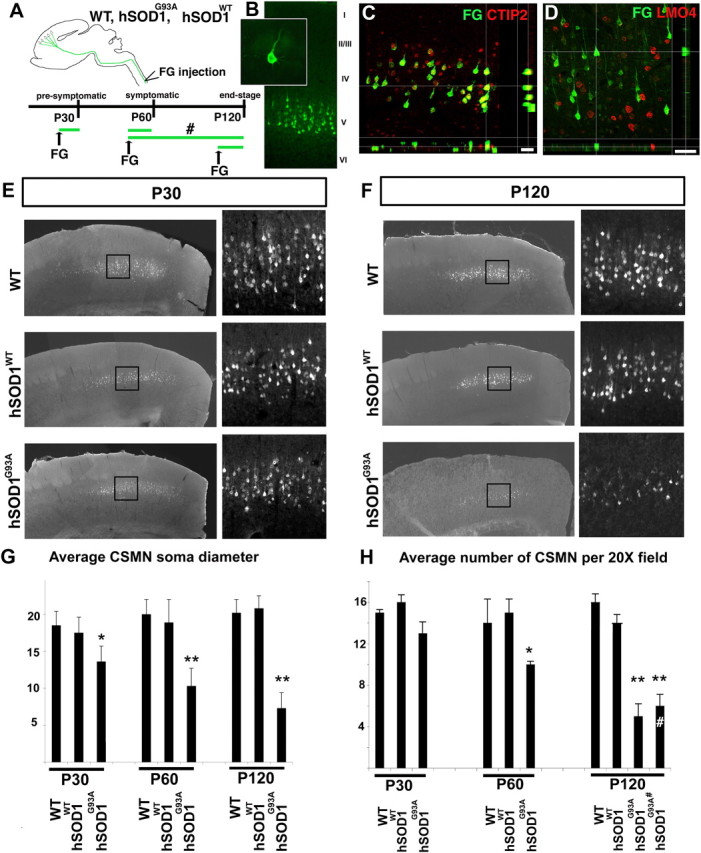
CSMN progressively degenerate in hSOD1G93A mice. A, Schematic diagram of experimental design. WT, hSOD1G93A, and hSOD1WT mice were retrogradely labeled by FluoroGold (FG) injections to be analyzed at P30 (presymptomatic), P60 (symptomatic), and P120 (end-stage). In a subset of experiments, CSMN were retrogradely labeled at P50 and analyzed at P120 (#). B, Retrogradely labeled CSMN reside in layer V of the motor cortex and display distinct CSMN morphology (inset). C, D, Confocal analysis reveals colocalization of the CSMN/SCPN-specific (within neocortex) transcription factor CTIP2 (C) and exclusion of the CSMN-excluded transcription factor LMO4 (D) in retrogradely labeled CSMN. E, F, Comparable and representative coronal sections of the motor cortex from WT, hSOD1WT, and hSOD1G93A mice at P30 (E) and at P120 (F). The boxed areas are enlarged in the adjacent panels to show retrogradely labeled CSMN. G, Bar graph representation of average CSMN soma diameter in WT, hSOD1WT, and hSOD1G93A mice at P30, P60, and P120. H, Bar graph representation of the average number of CSMN per 20× field in WT, hSOD1WT, and hSOD1G93A mice at P30, P60, and P120. CSMN that were retrogradely labeled at P50 and analyzed at P120 are labeled with “#”. Scale bars: C, 40 μm; D, 50 μm. Data are presented as mean ± SEM *p < 0.05; **p < 0.0001; G, H, Bonferroni test preceded by two-way ANOVA.
CSMN labeling and tissue collection.
CSMN were retrogradely labeled via stereotactic FluoroGold injections (2% FG, 250 nl/mouse) into the cervical region (C4–C6) of the corticospinal tract within the dorsal funiculus of the spinal cord at distinct, phenotypically distinguishable times defined previously (Gurney et al., 1994; Tu et al., 1996; Hall et al., 1998; Cleveland and Rothstein, 2001; Wengenack et al., 2004; Hegedus et al., 2007) as P20, “early”; P50, “symptomatic”; and P110, “end stage.” In a subset of experiments, mice injected at P50 were perfused at P120 to distinguish between genuine CSMN degeneration and potential appearance of reduced FG labeling due to defects in axonal transport (Fig. 1A, “#”). Ten days after FluoroGold injection, mice were deeply anesthetized and perfused with cold 0.1 m PBS supplemented with heparin, followed by cold 4% paraformaldehyde (PFA) in 0.1 m PBS. To further investigate whether degeneration of other neocortical projection neurons with equivalently long axons [most notably, interhemispheric callosal projection neurons (CPN)] occurs, in a subset of experiments dual CPN and CSMN retrograde labeling was performed in hSOD1G93A and WT mice at P30, and mice were perfused at P120. CPN were retrogradely labeled via stereotactic injection of green fluorescent microspheres into contralateral cortex (250 nl/mouse), and CSMN were retrogradely labeled via stereotactic injection of red fluorescent microspheres (250 nl/mouse) into the cervical region (C4–C6) of the corticospinal tract within the dorsal funiculus of the spinal cord. Brains were postfixed in 4% PFA overnight, and 40 μm thick coronal sections were cut on a Leica VT 1000S vibrating microtome.
Immunocytochemistry, Nissl staining, and in situ hybridization.
All immunocytochemical procedures were performed on every twelfth tissue section of 40-μm-thick coronal sections. The following primary antibodies were used: FG (1:500, Millipore Bioscience Research Reagents), CTIP2 (1:200, Calbiochem), LMO4 (1:200, Santa Cruz Biotechnology), cleaved caspase 3 (1:250, Cell Signaling), parvalbumin (PV; 1:500, Sigma), neuropeptide Y (NPY; 1:500, Immunostar), GFAP (1:200, Santa Cruz Biotechnology), CD68 (1:100, Millipore Bioscience Research Reagents), and FoxP2 (1:250, Abcam). Appropriate secondary fluorescent antibodies (1:500, Alexa 488 and 546, Invitrogen) were applied in blocking solution at room temperature, in the dark, for 2–4 h. CTIP2 and LMO4 immunocytochemical analysis were enhanced by increased primary antibody incubation (2 d) at 4°C, and 1:200 dilution was used for secondary fluorescent antibodies at 4°C overnight. Nuclear counterstain (ToPro; 1:5000 in 0.1 m PBS) was used to investigate nuclear pyknosis. Nissl staining was performed on every twelfth comparable section (40 μm thick, coronal) isolated from WT, hSOD1WT, and hSOD1G93A mice. Cry-mu, Igfbp4, Cux2, Fezf2, and PlexinD1 probes were prepared, and in situ hybridization analysis was performed as previously described (Molyneaux et al., 2005, 2009).
Imaging, quantification, and statistical analysis.
Sections were mounted on slides and analyzed using a Nikon E1000 fluorescence microscope equipped with an X-Cite 120 illuminator (EXF0), and images were acquired using a cooled charge-coupled device digital camera (Q Imaging Retiga EX). Confocal images were collected using a Bio-Rad Radiance 2100 Rainbow laser scanning confocal microscope based on a Nikon E800 microscope.
Quantitative analyses were performed on three matched rostrocaudal sections separated by ∼ 480 μm each [every ∼12 sections; 40 μm section thickness (supplemental Fig. 1, available at www.jneurosci.org as supplemental material)]. Three adjacent fields defined by the 20× objective encompass the mediolateral extent of retrogradely labeled CSMN; the same systematically located fields were used for analysis in all mice. CSMN were counted only if their soma and apical dendrite were both visualized in the same thick 40 μm section. Representative sections and the locations of the three systematically located fields used for quantification of CSMN number and soma diameter in WT (P30, n = 5; P60, n = 5; P120, n = 5), hSOD1WT (P30, n = 5; P60, n = 5; P120, n = 5), and hSOD1G93A (P30, n = 5; P60, n = 5; P120, n = 8) mice are shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material). For each mouse, a total of nine fields were counted (three fields in each of three sections), and numbers were averaged. All statistical analyses were performed with INSTAT software (version 3.0a; Graphpad); parametric and nonparametric multiple-comparison tests, such as Bonferroni and Tukey tests, were used where appropriate, with a minimum significance level set at p < 0.05.
Quantification of interneuron number was performed based on expression of PV and NPY. Three comparable 40 μm coronal sections that span the motor cortex (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) from WT (P120, n = 2) and hSOD1G93A (P120, n = 3) mice were analyzed. To perform unbiased and inclusive quantitative analysis, the cortex was divided into three equal sectors, mediolaterally/radially defined by equal 30° arcs. All neurons with both full cell bodies and immunopositivity for PV and NPY were counted in each sector. All counts were performed “blinded” to the genotype of the sample by two independent observers. Interobserver variability was extremely low, with <3% variation between independent measurements.
To investigate the corticospinal tracts of Thy1-YFP;hSOD1G93A mice (P30, n = 2; P60, n = 3; P120, n = 4) and Thy1-YFP;hSOD1WT mice (P30, n = 3; P60, n = 3; P120, n = 3), spinal cords were isolated intact, and the cervical and lumbar segments of the spinal cords were sectioned axially (50 μm section thickness), whereas the thoracic segment was sectioned sagittally (150 μm section thickness). All thoracic segments containing axons were mounted on slides, and images of all sections that contained corticospinal tract axons were digitally collapsed to obtain a two-dimensional image to indicate the whole extent of the corticospinal tract. Representative axial sections of cervical and lumbar spinal cord were also imaged.
Results
CSMN progressively degenerate in hSOD1G93A mice
We used a combination of anatomical and molecular approaches to identify, quantify, and assess potential vulnerability and progressive degeneration of CSMN. We retrogradely labeled CSMN by FG injection into the corticospinal tract within the dorsal funiculus of the C4–C6 region of the spinal cord (Fig. 1A). CSMN were retrogradely labeled at P20 for analysis at P30 (presymptomatic), at P50 for analysis at P60 (symptomatic), and at P100 for analysis at P120 (end-stage). In a subset of experiments designed to distinguish between genuine CSMN degeneration and potential appearance of reduced FG labeling due to defects in axonal transport, CSMN were retrogradely labeled at P50 and analyzed at P120 (Fig. 1A, “#”). P30, P60, and P120 times were chosen based on published and well established stages of disease progression with respect to both observed behavioral phenotypic changes and spinal motor neuron degeneration in hSOD1G93A mice (Gurney et al., 1994; Hegedus et al., 2007). CSMN identity was established unequivocally by retrograde labeling from axon projections in the spinal cord, and verified based on (1) neuronal location in the primary motor cortex, (2) distinctive large CSMN morphology, and (3) expression of multiple recently identified positive and negative molecular markers that are either specifically expressed or excluded by CSMN (and related SCPN in some cases). Retrogradely labeled CSMN were confirmed to reside in layer V of the motor cortex and displayed distinct somatodendritic morphology of CSMN: they have a large pyramidal cell body and a long apical dendrite (Fig. 1B). Retrogradely labeled neurons expressed CTIP2 (Arlotta et al., 2005), a CSMN-specific (and cortico-brainstem; together, subcerebral-specific) transcription factor in layer V (n = 150, 100%) (Fig. 1C), and did not express LMO4, a transcription factor excluded from CSMN in layer V (n = 120, 0%) (Fig. 1D), confirming their CSMN identity. Although both CTIP2 and LMO4 expression levels are highest during development, and reduce with age, they remain specific and detectable (Fig. 1C,D) (see Materials and Methods). Homogeneity in the background of the WT, hSOD1WT, and hSOD1G93A mice, and the consistent copy number of transgenic gene expression (30 ± 2) in hSOD1WT and hSOD1G93A mice resulted in highly reproducible timing of disease onset and progression, enabling detailed and reproducible qualitative and quantitative analysis of CSMN degeneration at specific times during disease progression.
Qualitative analysis of retrogradely labeled CSMN in layer V of motor cortex in P30, P60, and P120 WT, hSOD1WT, and hSOD1G93A mice reveals dramatic CSMN vulnerability and marked degeneration in P120 hSOD1G93A mice compared with WT and hSOD1WT controls, present but more subtle at P30 and P60 (Fig. 1E–H). Quantitative analysis of CSMN soma diameter and neuron number at P30, P60, and P120 indicates progressive degeneration of CSMN only in hSOD1G93A mice (Fig. 1G,H; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). CSMN soma diameter in hSOD1G93A mice significantly decreases, even during the “presymptomatic” stage (P30), and this decrease becomes even more striking with disease progression (Fig. 1G) (14 ± 2 μm at P30; 10 ± 2 μm at P60; 7 ± 2 μm at P120). In contrast, neither WT CSMN (19 ± 2 μm at P30; 20 ± 2 μm at P60; 20 ± 2 μm at P120) nor hSOD1WT CSMN (18 ± 2 μm at P30; 19 ± 3 μm at P60; 21 ± 2 μm at P120) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) show any reduction in soma diameter.
Quantitative analysis of the number of CSMN in motor cortex indicates a progressive and dramatic degeneration of CSMN in hSOD1G93A mice beginning by P30 (Fig. 1H) (13 ± 1 per 20× field at P30, n = 5 mice; 10 ± 1 at P60, n = 6 mice; 5 ± 2 at P120, n = 8 mice; 6 ± 1 at P120, injected at P50, n = 3 mice) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In contrast, CSMN of neither WT (15 ± 1 at P30, n = 5 mice; 14 ± 2 at P60, n = 5 mice; 16 ± 2 at P120, n = 6 mice) nor hSOD1WT mice (16 ± 1 at P30, n = 4 mice; 15 ± 2 at P60, n = 5 mice; 14 ± 1 at P120, n = 5 mice) demonstrate neuronal degeneration. At P120, hSOD1G93A mice show identical numbers of retrogradely labeled CSMN, regardless of the time of retrograde labeling (5 ± 2 at P120, injected at P100, n = 8 mice; 6 ± 1 at P120, injected at P50, n = 3 mice), strongly indicating that CSMN undergo genuine somatic degeneration, and that the reduced number of retrogradely labeled CSMN is not due to confounding variables related to retrograde transport. In contrast, rubrospinal neurons, located in the red nucleus, and projecting to the cervical spinal cord (and thus also retrogradely labeled from the cervical spinal cord FG injections) are normal in number and morphology in hSOD1G93A mice (as well as in hSOD1WT and WT mice) at all ages including P120, reinforcing that degeneration is specific to CSMN in motor cortex in hSOD1G93A mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
CSMN degeneration occurs via apoptosis
Investigation of CSMN death at all three stages of disease progression, using Nissl staining, analysis for nuclear pyknosis, and cleaved caspase-3 (CC-3) expression, reveals that CSMN death is apoptotic, and starts by P30 in hSOD1G93A mice (Fig. 2). Nuclear pyknosis is an early sign of apoptotic cell death (de Rivero Vaccari et al., 2006), and CC-3 expression is detected in cells undergoing active apoptosis, although the time it takes CC-3-positive neurons to ultimately die is reported to take substantially longer in vivo than in vitro (Yang et al., 1998; Arai et al., 2005). Progressive apoptosis in hSOD1G93A spinal motor neurons has been documented previously (Li et al., 2000; Przedborski, 2004; Wengenack et al., 2004), but CSMN were not investigated in detail. Here, all three analyses (Nissl, nuclear pyknosis, CC-3) reveal a wave of apoptotic CSMN death that starts by P30 in hSOD1G93A mice. In striking contrast, WT and hSOD1WT CSMN do not demonstrate neuronal death by any of these three modes of analysis at any stage investigated. At P30, apoptosis is visible by nuclear pyknosis (Fig. 2A) (40 ± 5% of hSOD1G93A CSMN, n = 290; 1 ± 1% of WT CSMN, n = 271; 3 ± 1% hSOD1WT CSMN, n = 264) (Fig. 2A,D) and reduced soma size (Fig. 1G), and CC-3 expression is detected in some CSMN in hSOD1G93A mice (2 ± 1%, n = 148), but not in WT or hSOD1WT. To further investigate whether nuclear pyknosis, and thus neuronal degeneration, might begin even earlier than P30, we quantified nuclear pyknosis of CTIP2-positive CSMN in layer V of motor cortex at P5, P10, and P15 compared with WT control CSMN at these same ages. These analyses reveal that 0% of WT CSMN display pyknotic nuclei at any of these early ages (P5, n = 105; P10, n = 101; P15, n = 110), whereas 2% of P5 hSOD1G93A CSMN display pyknosis, n = 114; increasing to 4 ± 1% at P10, n = 147; and to 7 ± 1% at P15, n = 112. At P60, the percentages of CSMN with pyknotic nuclei (64 ± 3%, n = 243) or expressing CC-3 (9 ± 2%, n = 140) are substantially increased (Fig. 2B,D,E). At P120, CSMN degeneration in hSOD1G93A mice is even more dramatic, with 7 ± 2% of CSMN expressing CC-3, 61 ± 3% with pyknotic nuclei, and near absence of large pyramidal neurons in layer V of motor cortex by Nissl staining (Fig. 2C). It is likely that CSMN detected at each age would include CSMN (1) already committed to die via apoptosis, (2) already dysfunctional and in the process of cell death, and (3) dead but not yet cleared from the tissue. These data suggest that many retrogradely labeled somata of CSMN detected in the cortex at P30, P60, and/or P120 are already less viable (likely already undergoing degeneration), but they are either not yet dead or not yet cleared from the tissue. Together, these results indicate by multiple independent criteria that progressive CSMN degeneration occurs in hSOD1G93A mice, beginning early during the classically presymptomatic stage, and mediated via apoptotic pathways.
Figure 2.
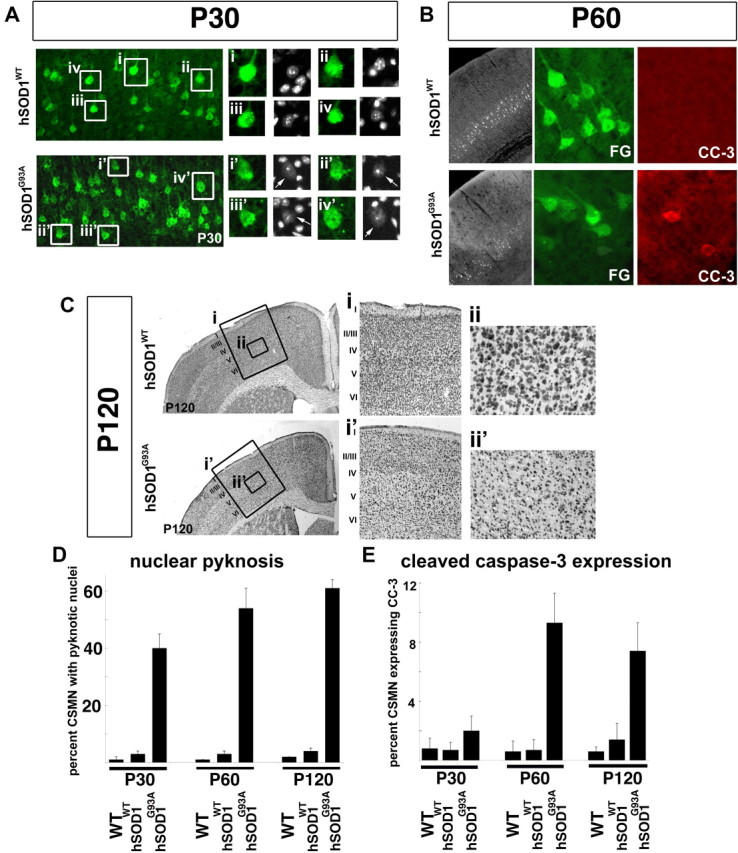
CSMN death occurs via apoptosis. A, At P30, hSOD1WT mice display healthy CSMN nuclei (i–iv), while apoptosis is visible by nuclear pyknosis in hSOD1G93A CSMN (i'–iv', arrows). The boxed areas (i–iv, i'–iv') in the low-magnification images are enlarged to the right. B, At P60, CC-3 expression was increased in many hSOD1G93A CSMN, but is not detected in hSOD1WT CSMN. C, At P120, Nissl stain analysis of motor cortex, and layer V in particular, reveals the normal complement of large pyramidal neurons in layer V of hSOD1WT motor cortex (i, ii) but a striking reduction in hSOD1G93A motor cortex (i', ii'). The boxed areas (i–ii') in the low-magnification images are enlarged to the right. D, Bar graph representation of the percentage of CSMN with pyknotic nuclei at P30, P60, and P120 in WT, hSOD1WT, and hSOD1G93A mice. E, Bar graph representation of the percentage of CSMN expressing CC-3, at P30, P60, and P120 in WT, hSOD1WT, and hSOD1G93A mice.
Broader degeneration of developmentally related nonmotor subcerebral projection neurons occurs in hSOD1G93A mice
To further investigate whether neocortical neuronal degeneration is specific to CSMN, or might involve other populations of cortical projection neurons and/or interneurons, we investigated the neuronal subtype identity and specificity of degeneration, using laminar and neuron type-specific molecular markers. We used a combination of immunocytochemical and in situ hybridization analyses in hSOD1G93A and hSOD1WT mice at P120. The results indicate that neuronal degeneration extends specifically to the developmentally related populations of motor and nonmotor, associative subcerebral projection neurons (SCPN), which include CSMN in the motor cortex. Neurons in other layers of the neocortex, and other neuron populations in layer V, are not affected.
Each molecular marker analyzed, both for CSMN/SCPN and for alternate neuron subtypes and lamina, confirms the specificity of CSMN/SCPN degeneration in the motor cortex of hSOD1G93A mice. Fezf2, a transcription factor required for CSMN/SCPN specification and development, and expressed at high levels in CSMN/SCPN throughout life (Molyneaux et al., 2005; Molyneaux et al., 2007), has normal expression in hSOD1WT mice at P120 (Fig. 3A). In contrast, Fezf2 expression in motor cortex of hSOD1G93A mice indicates dramatic loss of CSMN at P120 (Fig. 3Ai,ii). Interestingly, analysis of Fezf2 expression in nonmotor areas of neocortex indicate additional involvement, but less striking loss, of the broader population of SCPN (Fig. 3Aiii,iv), which contribute to nonmotor cognitive, association, and integrative sensory systems in the cortex. Cry-mu, which is expressed at high levels by CSMN during development, and at very low levels by a subset of corticofugal neurons located in layer VI (Molyneaux et al., 2005; Molyneaux et al., 2007), displays normal expression in hSOD1WT mice at P120 (Fig. 3Bi). In striking contrast, cry-mu expression confirms dramatic loss of CSMN in motor cortex in hSOD1G93A mice (Fig. 3Bii). With regard to non-CSMN/SCPN populations, PlexinD1 (Fig. 3C) and Cux2 (Fig. 3D), both expressed in neuron populations in superficial layers of the neocortex during development through adulthood (Molyneaux et al., 2007), do not indicate any difference in superficial layer neuron populations in hSOD1G93A versus hSOD1WT mice. To further differentiate true CSMN/SCPN neuronal loss from simple gene dysregulation, Igfbp4, expressed at high levels by CSMN during development, and at lower levels in layer II/III and layer VI neurons in the cortex (Molyneaux et al., 2007), displays normal expression in all populations in hSOD1WT mice at P120 (Fig. 3E). In contrast, in hSOD1G93A mice, Igfbp4 expression is sharply reduced in layer V (confirming CSMN/SCPN loss), while its expression is not affected in other neuron populations located in neocortical layers II/III and VI (Fig. 3E), further reinforcing the specificity of CSMN/SCPN degeneration in layer V of motor cortex. To even further consider whether neuronal degeneration is specific to CSMN/SCPN among the broader corticofugal populations (including, e.g., corticothalamic neurons), analysis of deepest layer VI of cortex for FoxP2 expression does not indicate any difference between hSOD1G93A and hSOD1WT mice (Fig. 3F). Similarly, LMO4, a transcription factor expressed by callosal projection neurons (inter-hemispheric, non-CSMN), interneurons, and other populations in layer V, but excluded from CSMN in layer V of the cortex (Arlotta et al., 2005; Molyneaux et al., 2005), is unchanged between hSOD1G93A and hSOD1WT mice (Fig. 3Gi,ii). In addition, dual retrograde labeling of CPN and CSMN at P30 in hSOD1G93A and WT mice with perfusion at P120 indicates 1) no loss of CPN in hSOD1G93A mice, 2) striking CSMN/SCPN-specific loss in hSOD1G93A mice (again, labeled early to exclude retrograde transport as a confounding variable), 3) specificity of CSMN degeneration in motor cortex of hSOD1G93A mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 3.
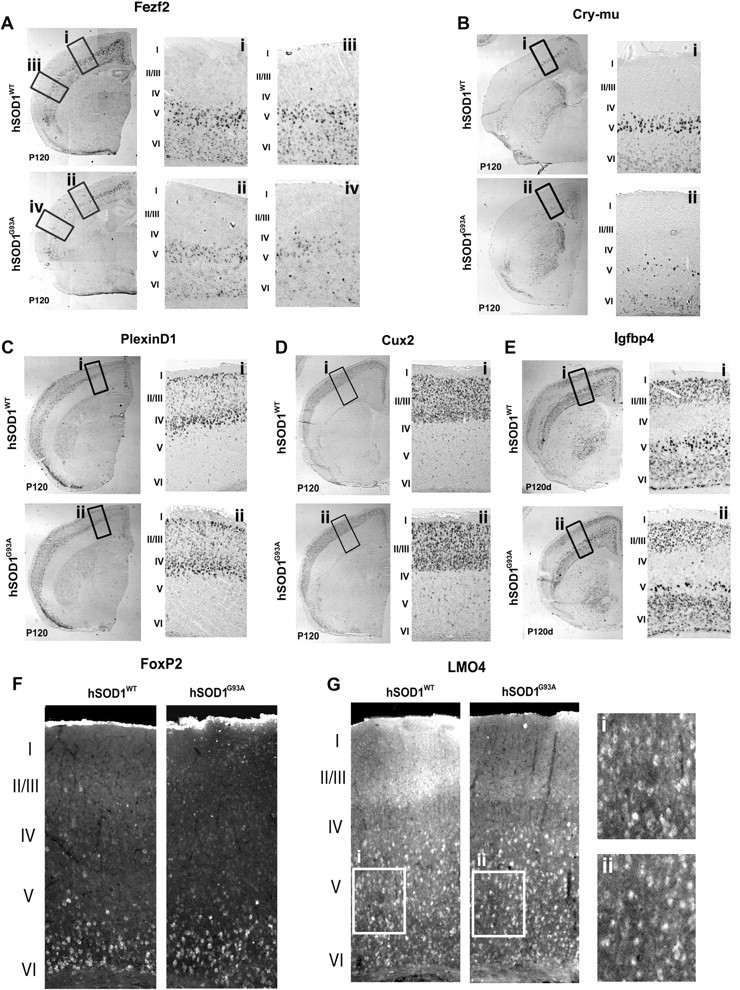
Degeneration is specific to CSMN and SCPN. A–E, In situ hybridization analysis of Fezf2 (A), Cry-mu (B), PlexinD1 (C), Cux2 (D), and Igfbp4 (E) on coronal sections of hSOD1WT and hSOD1G93A cortex at P120. The boxed areas (i–iv) in the low-magnification images are enlarged to the right; roman numerals indicate the layers of motor cortex. Fezf2 (A) is expressed at high levels in layer V of motor cortex (i) and nonmotor areas (iii) only in hSOD1WT mice. Fezf2 expression is dramatically reduced in the motor cortex (CSMN) (ii) and in the nonmotor areas of the cortex (SCPN) (iv) in hSOD1G93A mice. Cry-mu (B) is expressed at high levels in CSMN in hSOD1WT mice (i), and Cry-mu expression is dramatically reduced in hSOD1G93A mice (ii). PlexinD1 (C) and Cux2 (D), both expressed in neuron populations in superficial layers of the neocortex, show no difference between hSOD1WT and hSOD1G93A mice. Igfbp4 (E) is expressed at high levels by CSMN and at lower levels in layer II/III and VI neurons in the cortex; hSOD1WT mice display a normal complement of CSMN. In contrast, hSOD1G93A mice have many fewer Igfbp4-expressing neurons in layer V, while other neuron populations are not affected, further confirming the specificity of CSMN degeneration. F, FoxP2, expressed in the deepest layer VI of cortex, is unchanged between hSOD1WT and hSOD1G93A mice. G, LMO4 expression, normally expressed by layer V callosal projection neurons, interneurons, and other populations, but excluded from CSMN in layer V, is unchanged between hSOD1WT and hSOD1G93A mice at P120 (i, ii), reinforcing the specificity of CSMN degeneration in layer V of motor cortex in hSOD1G93A mice.
Interneurons are not similarly affected in hSOD1G93A mice
To investigate whether vulnerability and degeneration is restricted to CSMN/SCPN in the cortex of hSOD1G93A mice, or whether nonprojection neuron populations (interneurons) are affected, we examined a broad variety of interneurons based on the interneuron molecular markers parvalbumin and neuropeptide Y (NPY) in both hSOD1G93A (n = 3) and WT (n = 2) mice during the end-stage of disease progression. The results indicate that cortical interneuron populations are not apparently affected in hSOD1G93A mice (Fig. 4). The number of parvalbumin-expressing interneurons does not differ substantially or significantly between hSOD1G93A and WT mice, in any of the three mediolateral regions (Fig. 4A–C). Further, cellular morphologies of parvalbumin-positive cortical interneurons appear unchanged between WT and hSOD1G93A mice, without suggestion of neuronal degeneration (Fig. 4D,E). Similar results were obtained for NPY-expressing interneurons (data not shown). A prior report suggested the possibility of interneuron degeneration in the cortex based on reduced cortical inhibition in ALS patients (Ziemann et al., 1997), but our analysis in hSOD1G93A mice does not identify interneuron loss in these model mice. One recent study suggested increased cortical interneuron numbers in ALS mice, based on immunocytochemical analysis and applications of modeling (Minciacchi et al., 2009), but our results using broad mediolateral and rostrocaudal analysis do not identify such an increase. While it still remains possible that slight differences between the transgenic mice exist in localized areas, potentially secondary to degeneration of interneurons' CSMN/SCPN synaptic partners, we find no significant difference in interneurons more broadly between WT and hSOD1G93A mouse neocortex.
Figure 4.

Cortical interneuron populations do not similarly degenerate in hSOD1G93A mice. A–C, PV-positive interneurons were quantified in hSOD1G93A and WT mouse cortex at P120. The cortex was divided into three equal sectors and parvalbumin-positive neurons counted in each sector. A, PV immunocytochemistry in coronal sections of two representative examples of hSOD1G93A mouse cortex; one with the highest number of PV-positive interneurons (top) and one with the lowest number of PV-positive interneurons (bottom). B, PV immunocytochemistry in a coronal cortical section of a representative WT mouse. C, Bar graph displaying the total number of PV-positive neurons in each sector of the cortices from WT and hSOD1G93A mice. D, E, Cellular morphologies of interneurons were unchanged between hSOD1G93A (D) and WT mice (E), without suggestion of neuronal degeneration.
Astrogliosis and microgliosis are present late during CSMN/SCPN degeneration
Activated astrocytes have been implicated in disease pathogenesis in ALS and have been detected both in ALS patient brains and spinal cords, and in spinal cords of hSOD1G93A mice. To investigate whether astrogliosis (and, potentially, microglial activation) occurs in hSOD1G93A mouse neocortex, and whether astrogliosis and/or microgliosis is present relatively early or late, we assessed the presence of activated astrocytes and/or microglia in hSOD1G93A (P30, n = 3; P60, n = 3; P120, n = 4) and in hSOD1WT (P30, n = 2; P60, n = 2; P120, n = 3) mice using GFAP and CD68 immunocytochemical analysis, respectively. We find that astrogliosis and microgliosis occur specifically in hSOD1G93A mice (Fig. 5A–I, M) and not in hSOD1WT mice (Fig. 5J–L). It occurs mainly at late stages of disease progression in hSOD1G93A mice (Fig. 5G–I) and is not restricted to motor cortex (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). These results suggest that CSMN/SCMN vulnerability and degeneration is initiated before astrogliosis and microgliosis in the neocortex of hSOD1G93A mice.
Figure 5.
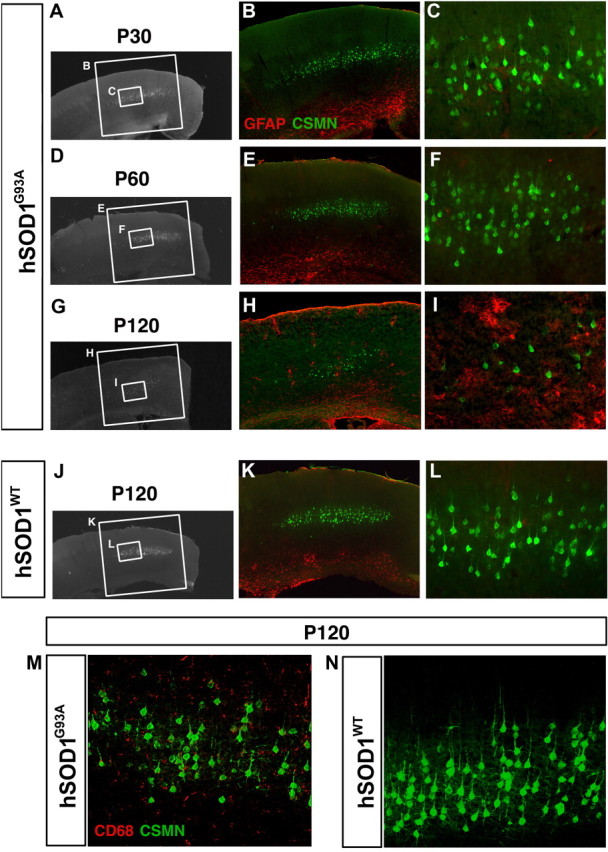
Astrogliosis and microgliosis is observed in hSOD1G93A mice. A–I, Astrogliosis in motor cortex of hSOD1G93A mice at P30 (A–C), P60 (D, E), and P120 (G–I). The boxed areas in the low-magnification images are enlarged to the right. Although progressive CSMN degeneration is observed in hSOD1G93A mice at P30 to P60 (B, C, E, F) and P120 (B, C, E, F, H, I), activated astrocytes were first detected by GFAP expression later, here shown at P120 (H, I). Astrogliosis was specific to hSOD1G93A mice, and hSOD1WT mice do not display activated astrocytes at P120 (J–L). Similarly, activated microglia were detected by CD68 expression only in hSOD1G93A cortex (M) and not in hSOD1WT cortex (N) at P120.
The corticospinal tract degenerates in hSOD1G93A mice
To investigate the time course of corticospinal tract (CST) degeneration in hSOD1G93A mice that is the predicted accompaniment of the CSMN degeneration we identified in these mice, we applied genetic CST labeling using hSOD1G93A, hSOD1WT, and WT mice crossed with cortical-specific Thy1-YFP mice (Bareyre et al., 2005). In these mice, multiple populations of cortical pyramidal neurons express yellow fluorescent protein (YFP), while only CST axons are labeled in the spinal cord. As reflected by the “lateral sclerosis” component of its original pathologically descriptive name (“sclerosis” of the “lateral”—main—corticospinal tract in primates), CST degeneration is central to ALS, as an easily visible component of the degeneration of CSMN, whose axons form the CST. Mice with cortical specific Thy1-YFP neuron labeling were used to generate Thy1-YFP;hSOD1WT and Thy1-YFP;hSOD1G93A mice by serial breeding (see Materials and Methods) (Fig. 6B). Results from Thy1-YFP;hSOD1G93A (P30, n = 2; P60, n = 3; P120, n = 3) (Fig. 6A,B) and Thy1-YFP;hSOD1WT mice (P30, n = 2; P60, n = 4; P120, n = 5) (Fig. 6B) indicate that corticospinal tract degeneration is specific to Thy1-YFP;hSOD1G93A mice. Degeneration is most dramatic between P60 and P120 (Fig. 6D–F). CST innervation of the lumbar spinal cord is essentially absent in Thy1-YFP;hSOD1G93A mice by the late stages of disease progression (Fig. 6Eiii), and there is substantial loss of the cervical corticospinal tract (Fig. 6Ei). These results indicate that CSMN somal degeneration that occurs quite early is followed by profound corticospinal tract degeneration by late stages in hSOD1G93A mice.
Figure 6.

The corticospinal tract degenerates in Thy1-YFP;hSOD1G93A mice. A, Breeding of hSOD1 transgenic mice with Thy1-YFP transgenic mice was used to visualize the CST in hSOD1G93A and hSOD1WT mice. Schematic drawing of cerebral cortex and spinal cord of Thy1-YFP transgenic mice, in which the CST is visualized by YFP fluorescence in the spinal cord. Cervical and lumbar segments of the spinal cord were investigated by single 50-μm-thick axial sections (i, iii). Sagittal sections were used for the thoracic cord; to identify the entire extent of the CST in the thoracic segment, all 150-μm-thick sagittal sections containing CST axons were digitally collapsed into a single two-dimensional image (ii). B, Thy1-YFP;hSOD1G93A and Thy1-YFP;hSOD1WT mice (hemizygous for both the Thy1-YFP and the hSOD1G93A genes) were generated by serial breeding Thy1-YFP mice and hSOD1G93A or hSOD1WT mice, respectively. Thy1-YFP;WT mice were used as controls. C–E, Insets (Ci, Di, Ei, Ciii, Diii, Eiii) show higher-magnification views of the dorsal funiculi, indicating CST axons at both the cervical (i) and lumbar (iii) segments of the spinal cord. C, At P120, the CST did not show any sign of degeneration in Thy1-YFP;hSOD1WT mice. The CST is normal in the cervical (i), thoracic (ii), and lumbar spinal cord (iii). D, E, In Thy1-YFP;hSOD1G93A mice, CST degeneration is most dramatic between P60 and P120. There is substantial loss of cervical (Di) and thoracic (Dii) CST axons by P120, and innervation of the lumbar spinal cord is drastically reduced to absent in Thy1-YFP; hSOD1G93A mice at P120 (Diii).
Discussion
Together, using a combination of anatomic, cellular, transgenic labeling, and newly available neuronal subtype-specific molecular analyses of both laminar and neuronal subtype identity, these results demonstrate that early and neuron type-specific CSMN/SCPN (both motor and nonmotor) neuronal degeneration is faithfully recapitulated in hSOD1G93A transgenic ALS mice. Other neuron populations in other layers of the neocortex, other neuron populations within layer V, and interspersed cortical interneurons do not undergo such degeneration. Thus, hSOD1G93A mice develop specific CSMN/SCPN degeneration that mirrors the cortical pathology found in human ALS, and nonmotor SCPN degeneration might explain some or much of the nonmotor cortical pathology identified in ALS. This neuron type-specific degeneration in hSOD1G93A mice begins exceptionally early, in the presymptomatic stage, coincident with the earliest degeneration of spinal motor neurons (Gurney et al., 1994; Tu et al., 1996; Hall et al., 1998; Cleveland and Rothstein, 2001; Bruijn et al., 2004; Wengenack et al., 2004; Hegedus et al., 2007). These results are consistent with longstanding clinical evidence and more recent human magnetic resonance imaging studies that clearly indicate that CSMN in motor cortex are affected at early stages of ALS (Brown, 2005; Graham et al., 2004; Stewart et al., 2006). The data reveal early events of CSMN degeneration by P30, with progressive CSMN degeneration occurring via apoptosis. Neocortical neuron degeneration is specific to CSMN/SCPN in the cerebral cortex of hSOD1G93A mice. In contrast, other cortical neuron populations do not undergo such degeneration—other cortical projection neuron populations in other layers of the cortex, other projection neuron populations within cortical layer V, or interneurons. CSMN undergo true cellular degeneration, not simply defects in retrograde axonal transport, shown to be separable issues by multistage retrograde labeling experiments. CSMN-specific degeneration is accompanied by striking corticospinal tract degeneration later in disease progression, revealed by transgenic labeling of corticospinal tract axons in Thy1-YFP;hSOD1G93A mice. These experiments and findings provide a foundation for detailed investigation of CSMN/SCPN biology in ALS, and of potentially shared biology between CSMN and SMN upper and lower motor neurons, respectively.
It is quite notable that CSMN degeneration is accompanied by degeneration of the related broader category of SCPN. Types of SCPN other than CSMN are molecularly and developmentally closely related to CSMN, and their coincident degeneration might be in part responsible for the broader cortical pathology in cognitive, association, and even sensory systems in many ALS cases. This further links the findings in hSOD1G93A mice to this emergingly important biology in humans with ALS. To further our understanding of CSMN/SCPN degeneration in human ALS, neuron type-specific analysis coupled with cellular and genetic labeling now enables deeper investigation regarding the cortical component of pathology in hSOD1G93A mice. Such investigation can now build on both basic anatomical analysis of the cortex/corticospinal tract, and on new molecular knowledge about subtype-specific differentiation and markers (Arlotta et al., 2005; Molyneaux et al., 2005, 2007, 2009; Tomassy et al., 2010). Using a combination of anatomical, cellular, transgenic labeling, and newly available neuronal subtype-specific molecular analyses to identify, quantify, and assess progressive and neuron type-specific degeneration of CSMN and related SCPN in hSOD1G93A transgenic ALS mice, these experiments closely link the mouse and human corticospinal and broader cortical biology of ALS. These analyses can now be extended by other investigators to other mammalian models of ALS to more deeply and rigorously elucidate the extent and mechanisms of cortical pathology. Such investigations will likely also elucidate mechanisms of CSMN/SCPN degeneration in other upper motor neuron neurodegenerative diseases, such as hereditary spastic paraplegia and primary lateral sclerosis.
CSMN and SMN degenerate independently, but their degeneration is potentially mechanistically related. CSMN and SMN are located at distant locations in the CNS, have very different developmental histories, and are positioned in distinct but potentially related microenvironments (with related glutamatergic synapses, transmission, and functions of surrounding astroglia, and potentially some shared channel properties). However, their progressive and specific degeneration indicates the relevance and importance of potentially common biology between these upper and lower motor neurons in ALS.
Sufficient information is developing to begin to elucidate common and/or unique biology between CSMN and SMN during development, and in ALS and related neurodegenerative diseases. There has been deep and important work on the differentiation, development, and maturation of spinal motor neurons (Tanabe and Jessell, 1996; Ericson et al., 1997; Arber et al., 1999; Briscoe et al., 1999; Pierani et al., 2001; Wichterle et al., 2002; William et al., 2003; Dasen et al., 2003, 2005; Kania and Jessell, 2003; Lanuza et al., 2004; Fox et al., 2007; Azim et al., 2009a,b; Molyneaux et al., 2009). In addition, cellular and molecular controls over differentiation, development, and maturation of CSMN/SCPN and other populations of neocortical projection neurons are beginning to emerge (Arlotta et al., 2005; B. Chen et al., 2005; J. G. Chen et al., 2005; Molyneaux et al., 2005, 2007, 2009; Ozdinler and Macklis, 2006; Joshi et al., 2008; Lai et al., 2008; Azim et al., 2009a,b; Tomassy et al., 2010). Moreover, there have been multiple recent studies investigating early defects in corticospinal motor neuron circuitry in the cerebral cortex of ALS patients and the progressive cortical pathology with respect to spinal motor neuron degeneration (Vucic et al., 2008; Sivák et al., 2010; van der Graaff et al., 2010; Mohammadi et al., 2011). Emerging evidence also indicates the relevance of SOD1 pathology in at least some sporadic ALS cases and extends the importance of understanding SOD1-mediated pathology well beyond familial ALS (Bosco et al., 2010). These studies, together with increasing understanding regarding the biology of both upper and lower motor neurons, might contribute toward development of future diagnostic and therapeutic approaches.
Understanding cellular and molecular mechanisms of cell type-specific degeneration in neurodegenerative diseases is of major and important interest toward both prevention and therapy. Together, our investigations establish and clarify the central, early, specific, and independent involvement of CSMN/SCPN in hSOD1G93A mice, and develop neuron type-specific approaches for analysis of the upper motor neuron component of neurodegeneration and its potential prevention or therapy in hSOD1G93A mice and other rodent models of ALS.
Footnotes
This work was supported by grants from the National Institutes of Health (NS49553; additional infrastructure provided by NS41590 and NS45523), the ALS Association, and the Harvard Stem Cell Institute to J.D.M., and the Les Turner ALS Foundation to P.H.O. P.H.O. was partially supported by a fellowship from the Harvard NeuroDiscovery Center, and by a program grant from the Harvard Stem Cell Institute to J.D.M. We thank K. Billmers, A. Palmer, A. Sanders, M. Jonathan for technical assistance; members of the Macklis and Brown laboratories for important input and suggestions; and E. Azim, S. Sohur, and J. Emsley for critical reading of an earlier version of this manuscript.
References
- Arai M, Sasaki A, Saito N, Nakazato Y. Immunohistochemical analysis of cleaved caspase-3 detects high level of apoptosis frequently in diffuse large B-cell lymphomas of the central nervous system. Pathol Int. 2005;55:122–129. doi: 10.1111/j.1440-1827.2005.01808.x. [DOI] [PubMed] [Google Scholar]
- Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/s0896-6273(01)80026-x. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Azim E, Jabaudon D, Fame RM, Macklis JD. SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat Neurosci. 2009a;12:1238–1247. doi: 10.1038/nn.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azim E, Shnider SJ, Cederquist GY, Sohur US, Macklis JD. Lmo4 and Clim1 progressively delineate cortical projection neuron subtypes during development. Cereb Cortex. 2009b;19(Suppl 1):i62–69. doi: 10.1093/cercor/bhp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat Med. 2005;11:1355–1360. doi: 10.1038/nm1331. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Carrì MT. Lessons from models of SOD1-linked familial ALS. Trends Mol Med. 2004;10:393–400. doi: 10.1016/j.molmed.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Boillée S, Cleveland DW. Gene therapy for ALS delivers. Trends Neurosci. 2004;27:235–238. doi: 10.1016/j.tins.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH., Jr Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Sussel L, Serup P, Hartigan-O'Connor D, Jessell TM, Rubenstein JL, Ericson J. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature. 1999;398:622–627. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- Brown RH, Jr, Robberecht W. Amyotrophic lateral sclerosis: pathogenesis. Semin Neurol. 2001;21:131–139. doi: 10.1055/s-2001-15260. [DOI] [PubMed] [Google Scholar]
- Brown RH., Jr . Amyotrophic lateral sclerosis. In: Hauser S.L., Kasper D.L., Braunwald E., Fauci A.S., Jameson L., Isselbacher K.J., editors. Harrison's textbook of internal medicine. Ed 16 2005. [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Chen B, Schaevitz LR, McConnell SK. Fezl regulates the differentiation and axon targeting of layer 5 subcortical projection neurons in cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17184–17189. doi: 10.1073/pnas.0508732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Rasin MR, Kwan KY, Sestan N. Zfp312 is required for subcortical axonal projections and dendritic morphology of deep-layer pyramidal neurons of the cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17792–17797. doi: 10.1073/pnas.0509032102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Dasen JS, Liu JP, Jessell TM. Motor neuron columnar fate imposed by sequential phases of Hox-c activity. Nature. 2003;425:926–933. doi: 10.1038/nature02051. [DOI] [PubMed] [Google Scholar]
- Dasen JS, Tice BC, Brenner-Morton S, Jessell TM. A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell. 2005;123:477–491. doi: 10.1016/j.cell.2005.09.009. [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JC, Casey GP, Aleem S, Park WM, Corriveau RA. NMDA receptors promote survival in somatosensory relay nuclei by inhibiting Bax-dependent developmental cell death. Proc Natl Acad Sci U S A. 2006;103:16971–16976. doi: 10.1073/pnas.0608068103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia: the pace quickens. Ann Neurol. 2002;51:669–672. doi: 10.1002/ana.10258. [DOI] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- Fox MA, Sanes JR, Borza DB, Eswarakumar VP, Fassler R, Hudson BG, John SW, Ninomiya Y, Pedchenko V, Pfaff SL, Rheault MN, Sado Y, Segal Y, Werle MJ, Umemori H. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- Gavrilina TO, McGovern VL, Workman E, Crawford TO, Gogliotti RG, Didonato CJ, Monani UR, Morris GE, Burghes HM. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle specific SMN expression has no phenotypic effect. Hum Mol Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JM, Papadakis N, Evans J, Widjaja E, Romanowski CA, Paley MN, Wallis LI, Wilkinson ID, Shaw PJ, Griffiths PD. Diffusion tensor imaging for the assessment of upper motor neuron integrity in ALS. Neurology. 2004;63:2111–2119. doi: 10.1212/01.wnl.0000145766.03057.e7. [DOI] [PubMed] [Google Scholar]
- Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci. 1997;152(Suppl 1):S67–S73. doi: 10.1016/s0022-510x(97)00247-5. [DOI] [PubMed] [Google Scholar]
- Gurney ME. What transgenic mice tell us about neurodegenerative disease. Bioessays. 2000;22:297–304. doi: 10.1002/(SICI)1521-1878(200003)22:3<297::AID-BIES12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Cheng W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Joshi PS, Molyneaux BJ, Feng L, Xie X, Macklis JD, Gan L. Bhlhb5 regulates the postmitotic acquisition of area identities in layers II-V of the developing neocortex. Neuron. 2008;60:258–272. doi: 10.1016/j.neuron.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania A, Jessell TM. Topographic motor projections in the limb imposed by LIM homeodomain protein regulation of ephrin-A:EphA interactions. Neuron. 2003;38:581–596. doi: 10.1016/s0896-6273(03)00292-7. [DOI] [PubMed] [Google Scholar]
- Kunst CB, Mezey E, Brownstein MJ, Patterson D. Mutations in SOD1 associated with amyotrophic lateral sclerosis cause novel protein interactions. Nat Genet. 1997;15:91–94. doi: 10.1038/ng0197-91. [DOI] [PubMed] [Google Scholar]
- Lai T, Jabaudon D, Molyneaux BJ, Azim E, Arlotta P, Menezes JR, Macklis JD. SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron. 2008;57:232–247. doi: 10.1016/j.neuron.2007.12.023. [DOI] [PubMed] [Google Scholar]
- Lanuza GM, Gosgnach S, Pierani A, Jessell TM, Goulding M. Genetic identification of spinal interneurons that coordinate left-right locomotor activity necessary for walking movements. Neuron. 2004;42:375–386. doi: 10.1016/s0896-6273(04)00249-1. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Frézal J, Munnich A, Melki J. The role of the SMN gene in proximal spinal muscular atrophy. Hum Mol Genet. 1998;7:1531–1536. doi: 10.1093/hmg/7.10.1531. [DOI] [PubMed] [Google Scholar]
- Li M, Ona VO, Guégan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Garcia ML, Ward CM, Cleveland DW. Altered axonal architecture by removal of the heavily phosphorylated neurofilament tail domains strongly slows superoxide dismutase 1 mutant-mediated ALS. Proc Natl Acad Sci U S A. 2005;102:10351–10356. doi: 10.1073/pnas.0503862102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60:1094–1097. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- Minciacchi D, Kassa RM, Del Tongo C, Mariotti R, Bentivoglio M. Voronoi-based spatial analysis reveals selective interneuron changes in the cortex of FALS mice. Exp Neurol. 2009;215:77–86. doi: 10.1016/j.expneurol.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Mohammadi B, Kollewe K, Samii A, Dengler R, Munte TF. Functional neuroimaging at different disease stages reveals distinct phases of neuroplastic changes in amyotrophic lateral sclerosis. Hum Brain Mapp. 2011 doi: 10.1002/hbm.21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Fame RM, MacDonald JL, MacQuarrie KL, Macklis JD. Novel subtype-specific genes identify distinct subpopulations of callosal projection neurons. J Neurosci. 2009;29:12343–12354. doi: 10.1523/JNEUROSCI.6108-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen HG, Angenstein F, Sander K, Kunz WS, Teuchert M, Ludolph AC, Heinze HJ, Scheich H, Vielhaber S. In vivo quantification of spinal and bulbar motor neuron degeneration in the G93A-SOD1 transgenic mouse model of ALS by T2 relaxation time and apparent diffusion coefficient. Exp Neurol. 2006;201:293–300. doi: 10.1016/j.expneurol.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pierani A, Moran-Rivard L, Sunshine MJ, Littman DR, Goulding M, Jessell TM. Control of interneuron fate in the developing spinal cord by the progenitor homeodomain protein Dbx1. Neuron. 2001;29:367–384. doi: 10.1016/s0896-6273(01)00212-4. [DOI] [PubMed] [Google Scholar]
- Przedborski S. Programmed cell death in amyotrophic lateral sclerosis: a mechanism of pathogenic and therapeutic importance. Neurologist. 2004;10:1–7. doi: 10.1097/01.nrl.0000106920.84668.37. [DOI] [PubMed] [Google Scholar]
- Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6) Am J Hum Genet. 2003;73:967–971. doi: 10.1086/378817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, Wong LF, Bilsland LG, Greensmith L, Kingsman SM, Mitrophanous KA, Mazarakis ND, Azzouz M. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- Rösler KM, Truffert A, Hess CW, Magistris MR. Quantification of upper motor neuron loss in amyotrophic lateral sclerosis. Clin Neurophysiol. 2000;111:2208–2218. doi: 10.1016/s1388-2457(00)00481-8. [DOI] [PubMed] [Google Scholar]
- Sivák S, Bittšanský M, Kurča E, Turčanová-Koprušáková M, Grofik M, Nosál′ V, Poláček, Dobrota D. Proton magnetic resonance spectroscopy in patients with early stages of amyotrophic lateral sclerosis. Neuroradiology. 2010;52:1079–1085. doi: 10.1007/s00234-010-0685-6. [DOI] [PubMed] [Google Scholar]
- Stewart HG, Andersen PM, Eisen A, Weber M. Corticomotoneuronal dysfunction in ALS patients with different SOD1 mutations. Clin Neurophysiol. 2006;117:1850–1861. doi: 10.1016/j.clinph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Strong MJ. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9:323–338. doi: 10.1080/17482960802372371. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science. 1996;274:1115–1123. doi: 10.1126/science.274.5290.1115. [DOI] [PubMed] [Google Scholar]
- Tomassy GS, De Leonibus E, Jabaudon D, Lodato S, Alfano C, Mele A, Macklis JD, Studer M. Area-specific temporal control of corticospinal motor neuron differentiation by COUP-TFI. Proc Natl Acad Sci U S A. 2010;107:3576–3581. doi: 10.1073/pnas.0911792107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci U S A. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- van der Graaff MM, Lavini C, Akkerman EM, Majoie Ch B, Nederveen AJ, Zwinderman AH, Brugman F, van den Berg LH, de Jong JM, de Visser M. MR Spectroscopy Findings in Early Stages of Motor Neuron Disease. AJNR Am J Neuroradiol. 2010;31:1799–1806. doi: 10.3174/ajnr.A2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540–1550. doi: 10.1093/brain/awn071. [DOI] [PubMed] [Google Scholar]
- Wengenack TM, Holasek SS, Montano CM, Gregor D, Curran GL, Poduslo JF. Activation of programmed cell death markers in ventral horn motor neurons during early presymptomatic stages of amyotrophic lateral sclerosis in a transgenic mouse model. Brain Res. 2004;1027:73–86. doi: 10.1016/j.brainres.2004.08.054. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- William CM, Tanabe Y, Jessell TM. Regulation of motor neuron subtype identity by repressor activity of Mnx class homeodomain proteins. Development. 2003;130:1523–1536. doi: 10.1242/dev.00358. [DOI] [PubMed] [Google Scholar]
- Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci. 2002;5:633–639. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Miller TM, McAlonis-Downes M, Chun SJ, Cleveland DW. Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann Neurol. 2006;60:95–104. doi: 10.1002/ana.20888. [DOI] [PubMed] [Google Scholar]
- Yang F, Sun X, Beech W, Teter B, Wu S, Sigel J, Vinters HV, Frautschy SA, Cole GM. Antibody to caspase-cleaved actin detects apoptosis in differentiated neuroblastoma and plaque-associated neurons and microglia in Alzheimer's disease. Am J Pathol. 1998;152:379–389. [PMC free article] [PubMed] [Google Scholar]
- Zanette G, Tamburin S, Manganotti P, Refatti N, Forgione A, Rizzuto N. Changes in motor cortex inhibition over time in patients with amyotrophic lateral sclerosis. J Neurol. 2002;249:1723–1728. doi: 10.1007/s00415-002-0926-7. [DOI] [PubMed] [Google Scholar]
- Zang DW, Cheema SS. Degeneration of corticospinal and bulbospinal systems in the superoxide dismutase 1(G93A G1H) transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2002;332:99–102. doi: 10.1016/s0304-3940(02)00944-8. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Winter M, Reimers CD, Reimers K, Tergau F, Paulus W. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis. Evidence from paired transcranial magnetic stimulation. Neurology. 1997;49:1292–1298. doi: 10.1212/wnl.49.5.1292. [DOI] [PubMed] [Google Scholar]