


Abstract
Alternative mRNA splicing is a mechanism to regulate protein isoform expression and is regulated by alternative splicing factors. The alternative splicing factor 45 (SPF45) is overexpressed in cancer, although few biological effects of SPF45 are known, and few splicing targets have been identified. We previously showed that Extracellular Regulated Kinase 2 (ERK2) phosphorylation of SPF45 regulates cell proliferation and adhesion to fibronectin. In this work, we show that Cdc2-like kinase 1 (Clk1) phosphorylates SPF45 on eight serine residues. Clk1 expression enhanced, whereas Clk1 inhibition reduced, SPF45-induced exon 6 exclusion from Fas mRNA. Mutational analysis of the Clk1 phosphorylation sites on SPF45 showed both positive and negative regulation of splicing, with a net effect of inhibiting SPF45-induced exon 6 exclusion, correlating with reduced Fas mRNA binding. However, Clk1 enhanced SPF45 protein expression, but not mRNA expression, whereas inhibition of Clk1 increased SPF45 degradation through a proteasome-dependent pathway. Overexpression of SPF45 or a phospho-mimetic mutant, but not a phospho-inhibitory mutant, stimulated ovarian cancer cell migration and invasion, correlating with increased fibronectin expression, ERK activation and enhanced splicing and phosphorylation of full-length cortactin. Our results demonstrate for the first time that SPF45 overexpression enhances cell migration and invasion, dependent on biochemical regulation by Clk1.
INTRODUCTION
Alternative pre-mRNA splicing is an important molecular mechanism for stimulating proteomic diversity. It has been shown by combining mRNA-Seq and EST–cDNA sequence data that alternative splicing occurs in ∼95% of all human genes with multiple exons (1). Mutations occurring at sites of pre-mRNA splicing significantly contribute to the number of somatic mutations that are known to occur in cancer and other genetic diseases (2–4). Splicing is carried out by the spliceosome, a large complex consisting of both small ribonuclear proteins and other associated proteins (5). Both constitutive and alternative splicing are regulated by the serine/arginine-rich (SR) protein family and the heterogeneous ribonucleoprotein particles family of proteins, which have antagonistic effects on splice site utilization (6–9), while other splicing factors fall outside of these protein families. Splicing factors are characterized by RNA recognition motifs (RRMs), protein–protein interaction domains and in the case of SR proteins, Arg-Ser (RS) rich motifs that can become heavily phosphorylated on serine residues (10). Alternative splicing site utilization by these RNA binding proteins is dependent on their relative concentrations, with increased expression enhancing their ability to increase alternative pre-mRNA splicing (11). Differential expression of alternative splicing factors has been observed in cancer with the potential to profoundly regulate protein diversity (12,13). Phosphorylation can regulate protein–protein interactions within the spliceosome as well as alternative splice site utilization (14–17), and several kinases and phosphatases have been identified that regulate phosphorylation of mRNA splicing factors (18–21). Cdc2-like kinase 1 (Clk1) is a nuclear kinase that has been shown to to be a major regulator of several splicing factors, phosphorylating them on multiple serine residues and regulating their intranuclear localization and splice site utilization on pre-mRNA (19,20,22–24). Once the splicesome machinery is assembled, dephosphorylation of splicing proteins can be required for the catalytic process of splicing (25–27).
Splicing factor 45 (SPF45) was first identified in mammalian cells as a member of the spliceosome complex (28). The SPF45 protein consists of an unstructured N-terminal domain, followed by an α-helical G-patch motif (29) involved in protein–protein (30) and protein–nucleic acid interactions (31,32), and a C-terminal RRM domain required for mRNA splicing (33). In mammalian cells, SPF45 regulates splicing of Fas exon 6, which encodes the transmembrane domain of this death receptor (33), and exon 6 exclusion generates a secreted dominant-negative Fas protein (34). SPF45 overexpression induces inclusion of the extra domain A (EDA) region into mature fibronectin transcripts, regulating cell adhesion to fibronectin (21). SPF45 expression is low in normal tissues, but is overexpressed in several forms of cancer, including breast, ovarian and prostate (35). Stable overexpression of SPF45 in HeLa cervical cancer cells and in A2780 ovarian cancer cells was reported to induce multidrug resistance (35,36).
We recently reported that SPF45 is a substrate for the ERK, Jun N-terminal Kinase (JNK) and p38 MAP kinases in response to extracellular stimulation, regulating SPF45 splice site utilization, ovarian cancer cell proliferation and cell adhesion to fibronectin (21). In this study, we investigated whether the SR protein kinase Clk1 plays a role in the regulation of human SPF45. We demonstrate that Clk1 directly phosphorylated SPF45 in vitro on eight serine residues, stabilized SPF45 protein levels and regulated SPF45-induced exon 6 skipping in Fas pre-mRNA. Moreover, we found that SPF45 overexpression induced cell migration and invasion in ovarian cancer cells, fibronectin expression and splicing and phosphorylation of the actin regulatory protein cortactin, all of which were dependent on the identified Clk1 phosphorylation sites. These data identify novel biochemical and biological functions of SPF45 that are governed by Clk1 phosphorylation.
MATERIALS AND METHODS
Plamids and siRNA
SPF45 and Clk1 mutants were generated using the GeneTailor™ site-directed mutagenesis kit (Invitrogen, Carlsbad, CA). The ΔFas minigene was generated using genomic DNA from immortalized ovarian surface epithelial (IOSE) cells as described previously (14). Clk1 plasmid was purchased from Addgene (Cambridge, MA). Clk1 siRNA, control siRNA and SPF45 siRNA were from Invitrogen.
Cell culture and transfection
COS-1, HeLa and SKOV-3 cells (American Type Culture Collection, Manassas, VA) were grown in Dulbecco’s Modified Eagle medium (Thermo Scientific) (COS-1 and HeLa) or McCoy’s 5A medium (SKOV-3 and ES-2) (Sigma, St. Louis, MO). A2780, OV2008 and OVCAR5 cells were grown in RPMI1640 medium (Sigma). All media was supplemented with 10% fetal bovine serum (FBS) (PAA, Dartmouth, MA), and cells were grown at 37°C with 5% CO2. Transient plasmid transfections were performed using Lipofectamine 2000 and siRNA transfections with Lipofectamine RNAiMAX (Invitrogen). SKOV-3-Myc-SPF45 and OV2008-Myc-SPF45 stable cells were generated by retroviral transduction as described previously (21). Cellular populations expressing Myc-SPF45 were selected with 1.5 μg/ml puromycin for 2 weeks and maintained in 0.75 μg/ml.
RT-PCR
Total RNA was extracted from cells using TRIzol (Invitrogen). The cDNA was transcribed from 2 μg of total RNA using a high capacity cDNA reverse transcription kit (Applied Biosystems, Foster, CA). PCR was performed using a Mycycler thermal cycler (Bio-Rad, Hurcules, CA) and analysed on agarose gels. Primer sequences are in Supplementary Table S1. Spliced products were quantified using Gel-Pro Analyzer 3.1. Results were expressed as a relative ratio, which was calculated by comparing the ratios of the lower bands to the upper bands in other groups with the ratio in the indicated group, which was set to 1. Values are means ± standard error (SE) from three independent experiments performed in duplicate. Quantitative real-time PCR was carried out using either Taqman Gene Expression Assays: Hs00269734_m1 for Clk1 and Hs99999901_sl for the housekeeping gene of 18S RNA on a 7300 Real-Time PCR System (Applied Biosystems) or Bio-Rad’s SsoAdvanced™ SYBR® Green Supermix on an Eppendorf (Hauppage, NY) Mastercycler Realplex 2. Quantification was determined by Relative Quantification Software (Applied Biosystems). Statistical comparisons of exon exclusion were performed using analysis of variance (ANOVA) followed by Fisher’s Protected Least Significance Difference (PLSD). Values of all parameters were considered significantly different at a value of P < 0.05.
Immunoprecipitations and western blotting
Cells were lysed in M2 lysis buffer (37), sonicated and protein concentrations determined using the BCA kit (Pierce, Rockford, IL). Typically, 25 µg of protein was run on an SDS–PAGE gel and transferred to nitrocellulose. For immunoprecipitations (IP), transfected cells were lysed, and equal amounts of protein were immunoprecipitated with anti-Myc antibody (Sigma) conjugated to protein G agarose (Roche), washed and the immunoprecipitates run on SDS–PAGE. Nitrocellulose membranes were blocked in 5% bovine serum albumin and were probed with antibodies overnight: anti-Myc, actin and fibronectin antibodies (Sigma); anti-Clk1, SF1, SF3b155 and U2AF65 (Abcam, Cambridge, MA); anti-p-ERK and cortactin (Cell Signaling); anti-cortactin (clone 4F11) (38), cortactin p405 and cortactin p418 (39); anti-ERK (Michael Weber, University of Virginia); and anti-phospho-serine (Millipore). Polyclonal anti-SPF45 antibody was generated to recombinant His-SPF45 protein in rabbits (Pacific Immunology, San Diego, CA). Secondary antibodies conjugated to horseradish peroxidase were followed by enhanced chemiluminescence (Pierce). Results were confirmed by at least three independent experiments.
In vitro kinase assay
Recombinant SPF45 proteins were prepared as described previously (21). Recombinant, active Clk1 (Sigma) was incubated with His-SPF45 or a His-SPF45 phosphorylation site mutant in 5 mM MOPS, 2.5 mM beta-glycerophosphate, 1 mM EGTA, 0.25 mM EDTA, 5 mM MgCl2, 0.5 mM dithiothreitol and 100 μM ATP. Radioactive experiments contained 10 μCi [γ-32 P]-ATP per reaction. The samples were incubated at 30°C for 30 min before the reaction was stopped with 2× Laemmli sample buffer and run on SDS–PAGE. Radioactive samples were transferred to nitrocellulose and exposed for autoradiography. Gels containing samples for mass spectrometry analysis were stained with Coomassie stain, and SPF45 was excised from the gel. After trypsin digest, samples were analysed via liquid chromatography (LC)-electrospray ionization-tandem mass spectrometry (MS/MS) on a linear ion trap mass spectrometer (LTQ XL, Thermo Finnigan) coupled to a Dionex Ultimate 3000 nano LC system. Phosphorylation sites were confirmed by manual inspection of the data.
RNA immunoprecipitation
RNA immunoprecipitation (RIP) was performed as described previously (40). Briefly, COS-1 cells were cotransfected with ΔFas and either empty vector, Myc-SPF45, Myc-SPF45-8 A or Myc-SPF45-8D. Protein G agarose beads pre-coated with anti-Myc monoclonal antibody (Sigma) or isotype control IgG (Santa Cruz Biotechnology, Santa Cruz, CA) in NET2 buffer [50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.05% Nonidet P-40, 10 mM dithiothreitol (DTT), 150 mM EDTA, RNase OUT™ and Superase IN™] were incubated briefly (5 s) with cell lysates. The beads were quickly pelleted, an aliquot of lysate was removed as input control and the beads were reintroduced for immunoprecipitation of Myc-SPF45 at 4°C. To recover RNA from the RIP complexes, the washed beads were re-suspended in 100 µl of NET2 and 100 µl of proteinase K (0.5 mg/ml) buffer for 30 min at 55°C. RNA was extracted with phenol-chloroform-isoamyl alcohol and precipitated in the presence of glycogen. For Fas RNA expression analysis, the RNA isolated from the IP was subjected to quantitative RT-PCR (qRT-PCR). Values were means ± SD from six experiments. Statistical analysis were performed by using ANOVA followed by Fisher’s PLSD, where P < 0.05 was considered significant.
Cell migration and invasion
For wound-healing assays, confluent cultures of stable SKOV-3 and OV2008 cell lines were scratched using a 10 µl of pipette tip. The culture medium and cell debris were removed and replaced with fresh culture medium containing 5 µg/ml mitomycin C. Cells were photographed at the time of the scratch (0 h) and at 20 or 24 h, and the percentage of wound closure was calculated using ImageJ software. Values are means ± SE from six experiments. Statistical analysis were performed by using ANOVA followed by Fisher’s PLSD, where P < 0.05 was considered significant. For transwell migration, stable SKOV-3 cells (2 × 104 cells) or OV2008 cells (5 × 104 cells) in 200 µl of medium containing 0.1% FBS were added to the upper chamber, and 400 µl of culture medium with 10% FBS was added to the lower chamber of a transwell dish (BD Biocoat™, BD Biosciences, Franklin Lakes, NJ). After 24 h, the non-migrated cells were removed from the upper surface, and cells on the underside of the membrane were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Migrated cells from 10 fields were counted and photographed. Values were means ± SE from six experiments. Statistical analysis were performed by using ANOVA followed by Fisher’s PLSD, where P < 0.05 was considered significant. For the cell invasion assays, 2 × 104 SKOV-3 cells or 5 × 104 OV2008 cells in 0.5 ml of culture medium containing 0.1% FBS were introduced to the upper chamber of BD Biocoat™ GFR Matrigel Invasion chamber (BD Biosciences, Franklin Lakes, NJ). The lower chamber was filled with 750 µl of culture medium with 10% FBS. Analysis of invading cells was carried out as aforementioned.
RESULTS
Clk1 promotes SPF45-induced exon 6 exclusion
SPF45 enhances exon 6 exclusion from Fas pre-mRNA in a cellular minigene assay (21,33). To determine whether endogenous SPF45 has a similar effect on splicing of endogenous Fas pre-mRNA, we transfected SKOV-3 ovarian cancer cells with siRNA against SPF45. Seventy-two hour after transfection, RNA was harvested, and the endogenous Fas mRNA isoforms containing or excluding exon 6 were analysed by RT-PCR using primers flanking exon 6. SPF45 knockdown inhibited the splicing of the short Fas mRNA isoform (lacking exon 6) from Fas mRNA (Figure 1A). No other bands were detected on the gel. Immunoblotting of parallel cultures with an anti-SPF45 polyclonal antibody confirmed SPF45 knockdown (Figure 1B). This polyclonal antibody generated to recombinant SPF45 protein recognizes endogenous SPF45 from several ovarian cancer cell lines and HeLa cells, with specificity determined by comparison with lysate from COS-1 cells transfected with untagged SPF45 (COS-1 +con) and SPF45 shRNA knockdown in OV2008 cells (Figure 1C). Consistent with the above-splicing results, transient transfection of SPF45 into COS-1 cells increased splicing of the short isoform of endogenous Fas mRNA, demonstrating enhanced exon 6 exclusion (Figure 1D). Clk1 is an SR protein kinase that phosphorylates splicing factors to regulate their localization and splice site utilization (19,20,22–24). To determine whether Clk1-regulated SPF45-dependent exon 6 exclusion from endogenous Fas mRNA, COS-1 cells were cotransfected with Clk1 and SPF45. Overexpression of Clk1 with SPF45 significantly increased exon 6 exclusion from endogenous Fas mRNA compared with transfection of SPF45 and empty vector (Figure 1D). We used our previously described ΔFas minigene (14,21), comprised of exon 5 through exon 7 of genomic Fas, including introns (Figure 1E), to study the effect of Clk1 on SPF45 splice site utilization. Cotransfection of COS-1 cells with ΔFas and increasing amounts of Myc-SPF45 plasmid caused a dose-dependent increase in exon 6 exclusion, shown as an increase in the lower band, demonstrating that our ΔFas minigene worked as a target of SPF45 in cells (Figure 1F) (21). COS-1 cells were chosen for minigene splicing assays owing to their low expression of endogenous SPF45 (Figure 1C), which allowed us to better determine SPF45-specific effects on alternative splicing of ΔFas mRNA. Clk1 was co-transfected into cells with SPF45 and the ΔFas minigene and exon 6 exclusion of ΔFas was determined. Clk1 enhanced SPF45-induced exon 6 excluion of ΔFas by >50% (Figure 1G), suggesting that SPF45 may be a cellular target of Clk1. Interestingly, Clk1 overexpression also enhanced Myc-SPF45 protein expression (Figure 1H). As SPF45 concentration directly affects splice site utilization (Figure 1F), the enhancement of SPF45 expression by Clk1 could be a possible mechanism for the enhanced exon 6 exclusion and was investigated futher.
Figure 1.

Clk1 enhances SPF45-induced exon 6 exclusion from Fas mRNA. (A) SKOV3 cells were transfected with siRNA against SPF45 (siSPF45) or scrambled control siRNA (scr) for 72 h, and RNA was isolated. Endogenous Fas spliced isoforms were analysed by RT-PCR using primers flanking exon 6. PCR products representing mRNA including exon 6 [Fas (L)] and excluding exon 6 [Fas (S)] are shown and are quantified in the graph, with the ratio Fas (S) to Fas (L) set to one in the control siRNA group. The resuslts are from three independent experiments performed in duplicate and were statistically significant. (B) Parallel cultures to those in (A) were lysed for western blotting using antibodies to SPF45 and actin. (C) Cell lysates from the indicated cell lines were immunoblotted for SPF45 using a polyclonal antibody to recombinant His-SPF45. COS-1 cells transfected with untagged SPF45 (COS-1, +con) and OV2008 cells expressing control or SPF45-specific shRNA served as positive controls. The SPF45 band (SPF45) and an SPF45 degradation product (degr.) in the positive control lane are labelled. (D) COS-1 cells were cotransfected with Myc-SPF45 (0.6 μg) and Clk1 (0.8 μg) and endogenous Fas spliced products were analysed by RT-PCR 24 h after transfection. A representative gel is shown. The means and SE for the relative ratio of exon 6 exclusion from three experiments done in duplicate are shown under the gel images. (E) Schematic of the ΔFas minigene splicing reporter used in transfection assays and the different mRNA isoforms derived from it, representing inclusion or exclusion of exon 6. (F) SPF45 expression induced exon 6 exclusion in a dose-dependent manner. COS-1 cells were co-transfected with ΔFas and increasing amounts of Myc-SPF45. Total RNA was extracted at 24 h and analysed by RT-PCR using ΔFas-specific primers. A representaive gel from at least three independent experiments is shown, and the ratio of the lower band to the upper band is shown below the gel. The lower panel represents a western blot of Myc-SPF45 and actin protein expression from one experiment. (G) Clk1 overexpression promotes SPF45 alternative splicing activity. COS-1 cells were cotransfected with ΔFas (0.3 μg), Myc-SPF45 (0.6 μg) and Clk1 (0.8 μg), and spliced products were analysed by RT-PCR 24 h after transfection. A representative gel is shown. The means and SE for the relative ratios of exon 6 exclusion are shown under the gel images. Results were derived from three independent experiments done in duplicate. Statistical significance (P < 0.01) is indicated in the graph. (H) Clk1 overexpression enhanced SPF45 protein levels. Protein lysates were prepared from cells transfected as in (G) and were subjected to western blotting using antibodies to Myc, Clk1 and actin.
Clk1 inhibition prevents exon 6 exclusion by SPF45
To further determine the role of Clk1 kinase activity in regulating SPF45 exon 6 exclusion of Fas pre-mRNA, we generated a kinase-dead Clk1, Clk1-K233R, and co-transfected COS-1 cells with plasmids for SPF45 and ΔFas. Clk1-K233R decreased both SPF45-induced exon 6 exclusion (Figure 2A) and SPF45 protein levels (Figure 2B) compared with wild-type Clk1. To confirm this result, a selective Clk1 inhibitor, TG003 (41), was used to block endogenous Clk1 activity. TG003 not only inhibited the increase of exon 6 exclusion induced by SPF45 (Figure 2C) but also decreased SPF45 protein expression (Figure 2D), similar to what was seen with kinase-dead Clk1 expression. Next, we used siRNA to knock down endogenous Clk1 expression. Transfection of cells with any of three different siRNA-targeting Clk1 led to a signifcant decrease in endogenous Clk1 mRNA and protein levels after 48 h, as determined by real-time PCR and western blot analysis, respectively (Figure 3A and B). The siRNA-mediated knockdown of Clk1 in COS-1 cells significantly decreased exon 6 exclusion in an SPF45-dependent manner (Figure 3C and D) and greatly inhibited Myc-SPF45 protein expression (Figure 3E), most likely contributing to the reduction in exon 6 exclusion.
Figure 2.

Inhibition of Clk1 decreases both SPF45 protein levels and exon 6 exclusion by SPF45. (A) COS-1 cells were cotransfected with ΔFas (0.3 μg), Myc-SPF45 (0.6 μg) and Clk1 (0.8 μg) or Clk1-K233R (0.8 μg), and spliced products were analysed by RT-PCR 24 h after transfection. A representative gel is shown. The graph under the gel image represents the quantification of the corresponding bands from three experiments done in duplicate. (B) COS-1 cells were cotransfected as in (A), and whole-cell protein lysates were immunoblotted with anti-Myc and anti-actin antibodies. (C) COS-1 cells were pretreated with the Clk inhibitor TG003 (10 μM) for 1 h and then cotransfected with ΔFas and Myc-SPF45 or empty vector. Twenty-four hour post-transfection, spliced products were analysed by RT-PCR. A representative gel is shown. A graph under the gel image showed the relative ratio of the lower band to the upper band. The results were derived from three independent experiments done in duplicate and statistical significance (P < 0.01) is indicated. (D) COS-1 cells were pretreated with TG003 for 1 h and then cotransfected as in (C). Whole-cell protein lysates were immunoblotted as in (B).
Figure 3.

Knockdown of Clk1 decreases both SPF45 protein levels and SPF45-induced exon 6 exclusion. (A) COS-1 cells were transfected with three siRNA against Clk1 or scrambled control siRNA (scr). Total RNA was extracted at 48 h post-transfection and was subjected to real-time PCR analysis using primers specific to Clk1. (B) COS-1 cells were transfected as in (A), and whole-cell protein lysates were immunoblotted with anti-Clk1 and anti-actin antibodies. (C) COS-1 cells were transfected with siRNA against Clk1 (siClk1-1, siClk1-2 and siClk1-3) or scrambled control siRNA for 48 h and then cotransfected with plasmids for ΔFas and SPF45 or empty vector. Twenty-four hour after plasmid transfection, mRNA was collected, and ΔFas spliced products were analysed by RT-PCR. (D) Graph showing the relative ratio of splicing products (the lower band to the upper band) after quantification of the corresponding lanes in panel (C). The ratio of splicing products for the scrambled siRNA in the presence of transfected SPF45 was set to 1. The results are from three independent experiments done in duplicate and statistical significance (P < 0.01) is indicated. (E) COS-1 cells were transfected as in (C), and whole-cell protein lysates were immunoblotted with anti-Myc and anti-actin antibodies.
Inhibition of Clk1 decreases the half-life of SPF45 through a proteasome-dependent pathway
We suspected that the enhancement or inhibition of exon 6 exclusion by SPF45 with Clk1 activation or inhibition, respectively, was partially due to regulation of SPF45 protein levels, as SPF45 expression directly impacts exon 6 exclusion (Figure 1F). To determine whether Clk1 regulated the half-life of SPF45, A2780 ovarian cancer cells, which express high levels of endogenous SPF45 (Figure 1C), were treated with cycloheximide in the absence or presence of TG003 for 24 h (Figure 4A). TG003 significantly decreased the half-life of endogenous SPF45 from 15 to 8 h (Figure 4B). In addition, SKOV-3 ovarian cancer cells stably expressing Myc-SPF45 were treated with DMSO or TG003 in the presence of cycloheximide. Consistent with the aforementioned results, TG003 decreased the half-life of Myc-SPF45 from 16 to 7 h (Figure 4C and D). Moreover, knockdown of endogenous Clk1 with siRNA decreased endogenous SPF45 protein levels in both SKOV-3 and HeLa cells (Figure 4E). To determine whether the decrease in SPF45 half-life in the presence of TG003 was dependent on the proteasome, A2780 cells were treated with the proteasome inhibitor MG132 in the absence or presence of TG003 (Figure 4F). MG132 co-treatment with TG003 prevented the downregulation of SPF45 protein levels that was induced by TG003 treatment, indicating that Clk1 inhibition regulated SPF45 degradation through a proteasome-dependent pathway.
Figure 4.

Clk1 inhibition decreases the half-life of SPF45 through a proteasome-dependent pathway. (A) A2780 cells were treated with cycloheximide (50 μg/ml) and TG003 (10 μM) or DMSO for the indicated times, followed by western blot analysis with anti-SPF45 and anti-actin antibodies to detect endogenous proteins. (B) Graph of SPF45 protein levels relative to the actin loading control from (A). (C) SKOV-3-Myc-SPF45 stable cells were treated with cycloheximide and TG003 or DMSO for the indicated times followed by western blot analysis with anti-Myc and anti-actin antibodies. (D) Graph of Myc-SPF45 protein levels relative to actin from (C). (E) Knockdown of Clk1 inhibits expression of endogenous SPF45. SKOV-3 cells and HeLa cells were transfected with siRNA to Clk1 or control siRNA (scr) for 48 h. Cell lysates were immunoblotted for endogenous Clk1, SPF45 and actin. (F) The increased degradation of SPF45 with Clk1 inhibition is proteasome-dependent. A2780 cells were treated with MG132 (10 μM) and TG003 or DMSO for the indicated times, followed by western blot analysis with anti-SPF45 and anti-actin antibodies. Quantitation of SFP45 expression relative to actin is below each lane, with the DMSO control lane set to 1.
Clk1 phosphorylates SPF45
The aforementioned data and the ability of Clk1 to regulate splicing factors suggest that Clk1 may regulate SPF45 through direct phosphorylation. To test this, we performed an in vitro kinase assay with recombinant Clk1, bacterially expressed histidine-tagged SPF45 (His-SPF45) and [γ-32 P]-ATP and observed a robust phosphorylation of SPF45 by Clk1 (Figure 5A). To determine the site(s) of phosphorylation, parallel reactions were performed using unlabelled ATP, and the His-SPF45 was excised from the subsequent Coomassie stained gel. Eight serine phosphorylation sites were identified by (LC)-electrospray ionization-tandem mass spectrometry (MS/MS): serines 48, 62, 202, 204, 222, 266, 288 and 291 (Figure 5B). We generated single alanine mutants at S202 or S204 and observed reduced phosphorylation of each mutant by Clk1 in vitro using [γ-32P]-ATP (Figure 5C). Mutation of six residues (serines 48, 62, 222, 266, 288 and 291; SPF45-6A) showed greatly reduced phosphorylation by Clk1 in a radioactive in vitro kinase assay (Figure 5C), and mutation of all eight sites to alanine (SPF45-8A) blocked phosphorylated by Clk1 in vitro, demonstrating that there were no additional Clk1 phosphorylation sites on SPF45. When transfected into COS-1 cells, Myc-SPF45-8A showed greatly reduced serine phosphorylation compared with wild-type Myc-SPF45, demonstrating that these sites are phosphorylated in cells (Figure 5D).
Figure 5.

Clk1 phosphorylates eight serines in SPF45. (A) His-SPF45 was incubated in the absence or presence of recombinant CLK1 and [γ-32 P]-ATP in an in vitro kinase assay. The reactions were run on a gel, transferred to nitrocellulose and exposed for autoradiography, followed by western blotting for SPF45. (B) Recombinant Clk1 was used to phosphorylate His-SPF45 from bacteria in vitro, and the phosphorylated protein was run on a gel and processed for mass spectrometry. Eight phosphorylated serine residues in SPF45 were identified. (C) Recombinant Clk1 was used to phosphorylate His-SPF45, His-SPF45-S202A, His-SPF45-S204A, His-SPF45-6 A (S48/62/222/266/288/291 A) and His-SPF45-8 A (S48/62/202/204/222/266/288/291 A) in vitro using [γ-32 P]-ATP. The reactions were run on a gel, transferred to nitrocellulose, exposed for autoradiography and immunoblotted with anti-SPF45 antibody. (D) COS-1 cells were transfected with empty vector, Myc-SPF45 or Myc-SPF45-8 A. Anti-Myc immunoprecipitates were immunoblotted with anti-phospho-serine and anti-Myc antibodies.
Effects of SPF45 phosphorylation site mutation on Fas exon 6 exclusion
To determine the collective effect of Clk1 phosphorylation on SPF45, we compared exon 6 exclusion by SPF45, SPF45-8A or a phospho-mimetic SPF45-8D in the ΔFas minigene assay. Surprisingly, Myc-SPF45-8A demonstrated a significant 25% increase in exon 6 exclusion compared with wild-type SPF45 in the absence of exogenous Clk1, suggesting that phosphorylation of these sites in total inhibited SPF45-induced exon 6 exclusion (Figure 6A and B). Similarly, mutation of these eight residues to aspartate inhibited exon 6 exclusion by a comparable amount. When Clk1 was co-expressed, wild-type Myc-SPF45 induction of exon 6 was enhanced, as aforementioned, but the significant difference between wild-type Myc-SPF45 and Myc-SPF45-8A was lost, suggesting that Clk1 had little effect on SPF45 exon 6 exclusion when the 8 phosphorylation sites were mutated to alanine. Similarly, Clk1 expression had little effect on Myc-SPF45-8D-induced exon 6 exclusion. SPF45 mRNA expression was unaffected by Clk1 expression (Figure 6C), whereas expression of the mutant proteins was similar to wild-type (Figure 6D).
Figure 6.

Mutation of Clk1 phosphorylation sites on SPF45 regulates SPF45 splice site utilization. (A) COS-1 cells were transfected for ΔFas splicing assays as above using either Myc-SPF45, Myc-SPF45-8 A or Myc-SPF45-8D in the absence or presence of either empty vector or Clk1. Twenty-four hour post-transfection, spliced products were analysed by RT-PCR. A representative gel is shown. (B) The means and SE for the relative ratios of exon 6 exclusion from (A) are shown in the graph. Results were derived from three independent experiments in duplicate and statistical significance (P < 0.01) is indicated. (C) The same as in (A), but total RNA was subjected to RT-PCR analysis using primers specific to Myc-SPF45 and GAPDH. (D) Protein lysates from cells transfected in parallel were immunoblotted with anti-Myc and anti-actin antibodies.
Clk1 phosphorylation sites in SPF45 differentially affect exon 6 exclusion
To further determine whether each of the identified Clk1 phosphorylation sites on SPF45 exert the same effect on SPF45 splice site utilization in Fas pre-mRNA, we generated different combinations of alanine mutants and tested their ability to induce exon 6 exclusion from ΔFas. Both Myc-SPF45-S48/222/266A and Myc-SPF45-S48/62/266A significantly increased exon 6 exclusion compared with wild-type Myc-SPF45 (Figure 7A), whereas Myc-SPF45-S48/202/204/266A, Myc-SPF45-S48/202/204/266/288/291A and Myc-SPF45-S202/204/288/291A significantly decreased exon 6 exclusion compared with wild-type Myc-SPF45, suggesting that phosphorylation of serines 48, 222 and 266 by Clk1 inhibited exon 6 exclusion and phosphorylation of serines 202 and 204 promoted exon 6 exclusion. To confirm this result, we made single mutants of serines 48A, 62A, 222A and 266A and double mutants of serine 202/204A and S288/291A. COS-1 cells were transiently transfected with these mutants and Myc-SPF45. The results showed that Myc-SPF45-S48A, Myc-SPF45-S222A and Myc-SPF45-S266A slightly, but significantly, increased exon 6 exclusion (Figure 7B), whereas Myc-SPF45-S202/204A decreased exon 6 exclusion, which were consistent with the aforementioned results. Collectively, these data indicate that Clk1 differentially regulates SPF45 alternative splicing activity through phosphorylation of different serines.
Figure 7.

Mutation of Clk1 phosphorylation sites in SPF45 differentially affects SPF45-indcuced exon 6 exclusion. (A and B) COS-1 cells were transfected with plasmids for ΔFas (0.3 μg), wild-type (wt)-SPF45 (0.6 μg) or an SPF45 mutant (0.6 μg) as indicated. Twenty-four hour post-transfection, spliced products were analysed by RT-PCR. The graph shows the relative ratios (short form to long form) of exon 6 exclusion. The table under the graph indicates the means and SE for each SPF45 mutant. Results were derived from three independent experiments done in duplicate. *P < 0.05 versus wt group, **P < 0.01 versus wt group. The bottom panels show western blotting of protein lyasates using anti-Myc and anti-actin antibodies.
Mutation of the Clk1 phosphorylation sites on SPF45 affects Fas mRNA binding, but not association with other splicing factors
SPF45 has been shown to associate with the splicing factors SF3b155, U2AF65 and SF1 through residues in its RRM domain (33,42). To determine whether SPF45 phosphorylation on the Clk1 sites altered binding to these proteins, COS-1 cells were transfected with either empty vector, Myc-SPF45, Myc-SPF45-8A or Myc-SPF45-8D. Anti-Myc immunoprecipitates were immunoblotted for endogenous splicing factors. All three splicing factors bound equally well to Myc-SPF45 and either of the SPF45 mutants (Figure 8A), suggesting that Clk1 phosphorylation of SPF45 does not significantly affect association with these proteins. To determine whether phosphorylation affected SPF45 binding to ΔFas RNA, we performed RIP analysis on COS-1 cells, which were cotransfected with ΔFas and wild-type or mutant Myc-SPF45. The ΔFas mRNA detected in anti-Myc immunoprecipitates was analysed by qRT-PCR and was shown to specifically interact with Myc-SPF45 (Figure 8B). Myc-SPF45-8A showed enhanced binding to ΔFas mRNA, whereas Myc-SPF45-8D showed reduced binding, with both reflecting the relative changes in exon 6 exclusion observed in the minigene splicing assays. These data suggest that SPF45 phosphorylation affects Fas mRNA binding.
Figure 8.

Mutation of the Clk1 phosphorylation sites in SPF45 affects ΔFas mRNA binding, but not binding to other splicing factors. (A) COS-1 cells were transfected with either empty vector, Myc-SPF45, Myc-SPF45-8 A or Myc-SPF45-8D. After 24 h, the cells were lysed, and anti-Myc immunoprecipitates were run on a gel and immunoblotted for Myc and co-immunoprecipitating endogenous SF3b155, U2AF65 and SF1. Cell lysates (Lys.) were immunoblotted as indicated. (B) COS-1 cells were co-transfected with ΔFas and either empty vector, Myc-SPF45, Myc-SPF45-8 A or Myc-SPF45-8D. Twenty-four hour after transfection, the cells were subjected to RNA IP using anti-Myc antibody followed by RT-qPCR analysis to detect the binding of ΔFas mRNA to Myc-SPF45. Relative binding to the control IP from six experiments are shown as mean ± SE. *P < 0.05 versus vector group, #P < 0.05 versus wt-SPF45 group.
SPF45 overexpression enhances cell migration and invasion in a phosphorylation-dependent manner
We previously showed that SPF45 overexpression affects cell proliferation and cell-matrix adhesion (21). To investigate the effect of SPF45 on cell motility, scratch wound healing assays were performed on SKOV-3 cells stably expressing Myc-SPF45, Myc-SPF45-8A, Myc-SPF45-8D or empty vector. Equal expression of Myc-SPF45 proteins was observed in the stable cell lines (Supplementary Figure S1). SPF45 and SPF45-8D significantly increased cell migration compared with vector control, whereas SPF45-8A did not (Figure 9A and B). Similar results were observed in OV2008 stable cell lines (Figure 9C and D). To confirm these results, we performed transwell cell migration assays towards a 10% serum chemoattractant. In SKOV-3 cells, SPF45 and SPF45-8D overexpression significantly increased serum-stimulated migration 2.1-fold and 2.2-fold, respectively, compared with vector control cells, whereas SPF45-8A inhibited cell migration by 41% (Figure 9E). In OV2008 cells, SPF45 and SPF45-8D enhanced migration by 3.2-fold and 3.4-fold, respectively, compared with vector control cells, whereas SPF45-8A had no effect (Figure 9F).
Figure 9.

SPF45 overexpression enhances ovarian cancer cell migration in a phosphorylation site-specific manner. (A) SKOV-3 cell lines stably expressing Myc-SPF45, Myc-SPF45-8A, Myc-SPF45-8D or empty vector were grown to confluence. Cells were pre-treated with mitomycin-C for 3 h before being scratched and wound closure was recorded at 20 h by phase contrast microscopy. Representative images of six experiments are shown. (B) Wound closure in (A) was calculated using Image J software and expressed as a percentage of the initial scratched area. Results shown are mean ± SE. *P < 0.05 versus vector group, #P < 0.05 versus wt-SPF45 group. (C and D) Scratch assays in OV2008 stable cells. The methods were the same as in (A and B). (E and F) Transwell migration assays. Stable (E) SKOV-3 cells (2 × 104) or (F) OV2008 cells (5 × 104 cells) in 0.1% FBS were added to the upper chamber, and 400 µl of 10% FBS was added to the lower chamber of a transwell dish. After 24 h, non-migrating cells were removed from the upper surface of the membrane, and the migrating cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Migrating cells were photographed and counted. Results from six experiments are shown as mean ± SE. *P < 0.05 versus vector group, #P < 0.05 versus wt-SPF45 group.
In addition, we determined the effect of SPF45 overexpression on cell invasion through matrigel in a transwell chamber. In SKOV-3 cells, SPF45 and SPF45-8D significantly increased cell invasion by 1.9-fold and 2.1-fold, respectively, compared with vector control cells, whereas 8A-SPF45 inhibited cell invasion by 45% (Figure 10A and C). In OV2008 cells, SPF45 and SPF45-8D enhanced invasion by 3.2-fold and 3.5-fold, respectively, whereas SPF45-8A did not significantly affect invasion (Figure 10B and D). Collectively, these results show that SPF45 overexpression promoted cell migration and invasion in a phosphorylation-dependent manner.
Figure 10.

SPF45 overexpression enhances ovarian cancer cell invasion in a phosphorylation site-specific manner. (A) Stable SKOV-3 cells (2 × 104) and (B) OV2008 (5 × 104) cells in 0.5 ml of 0.1% FBS were added to the top of a matrigel invasion chamber, with 0.75 ml of 10% FBS in the lower chamber. After 48 h, the non-invasive cells were removed from the upper surface of the membrane, and the invading cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Representative pictures of invading cells are shown. (C and D) Quantification of cell invasion from (C) SKOV-3 and (D) OV2008 stable cell lines in (A and B) from six experiments are shown as mean ± SE. *P < 0.05 versus vec group, #P < 0.05 versus wt group.
SPF45 regulates fibronectin and cortactin in a phosphorylation-dependent manner
Fibronectin enhances cell migration (43–46), and we previously showed that SPF45 overexpression enhanced fibronectin expression in SKOV-3 cells in a manner independent of SPF45 phosphorylation by MAP kinases (21). Stable expression of SPF45 or SPF45-8D in SKOV-3 cells increased fibronectin mRNA and protein expression, whereas SPF45-8A expression completely inhibited fibronectin expression (Figure 11A and B), indicating that SPF45 regulates fibronectin expression in a Clk1 phosphorylation-dependent manner. In OV2008 cells, SPF45 and SPF45-8D overexpression also increased fibronectin protein levels compared with vector cell and cells expressing SPF45-8A (Figure 11C).
Figure 11.
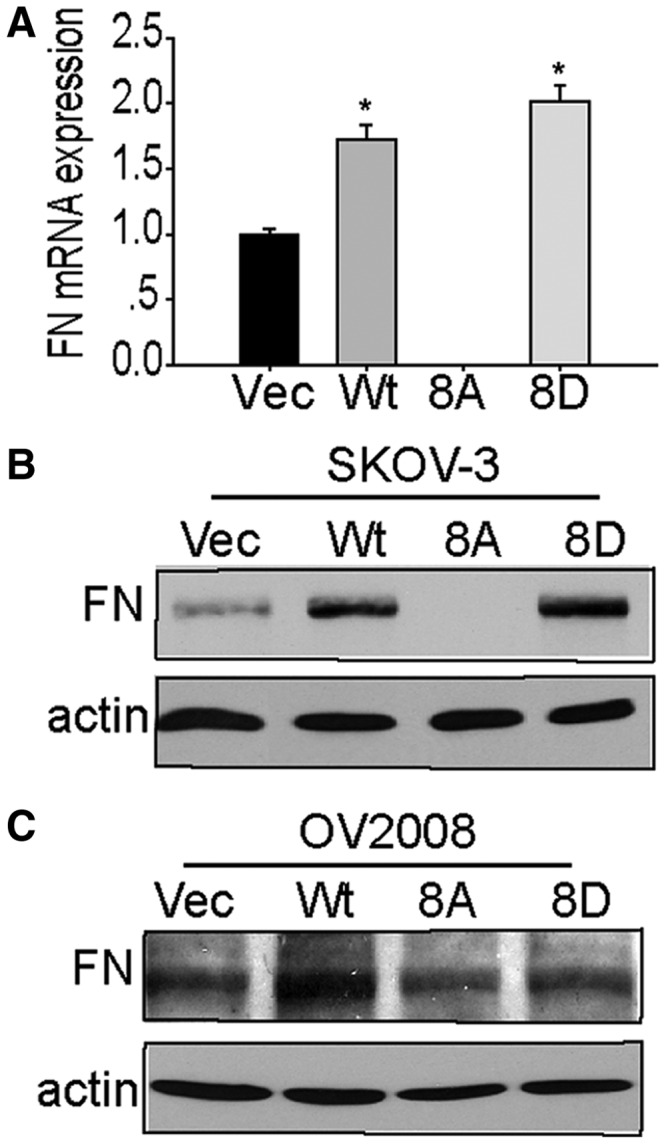
SPF45 overexpression enhances fibronectin expression in a phosphorylation site-specific manner. (A) Fibronectin mRNA expression was measured in stable SKOV-3 cells by qRT-PCR. Results from six experiments are shown as mean ± SE. *P < 0.05 versus vec group, #P < 0.05 versus wt group. (B) Protein lysates from cells in (A) were immunoblotted with anti-fibronectin (FN) and anti-actin antibodies. (C) Fibronectin protein levels in OV2008 stable cell lines were determined by immunoblotting as in (B).
Analysis of alternative splicing in SKOV-3-Myc-SPF45 cells identified the filamentous actin-binding protein cortactin as a potential splicing target of SPF45 (Al-Ayoubi and Eblen, unpublished data). Cortactin overexpression enhances cell migration and invasion (47), whereas cortactin splice variant (SV) 1 induces less migration and SV2 inhibits migration when overexpressed (48). The qRT-PCR using primers specific to wt-cortactin or total cortactin (wt + SV1 + SV2) (Figure 12A) in the SKOV-3 stable cell lines showed that SPF45 and SPF45-8D significantly increased the ratio of wt:total cortactin compared with vector control cells by 64 and 72%, respectively, whereas SPF45-8A did not show a significant increase (Figure 12B). These differences were also evident at the protein level (Figure 12C), where the wt-cortactin doublet and faster migrating SV1, but not SV2, were observed. In addition, expression of SPF45 and SPF45-8D enhanced ERK activation and phosphorylation of Ser405 and Ser418 on cortactin (Figure 12C), two ERK-dependent sites that enhance cortactin function (39,49,50). Similar results were obtained in OV2008 stable cell lines (Figure 12D and E).
Figure 12.

SPF45 overexpression inhibits alternative splicing of cortactin and enhanced phosphorylation of cortactin and ERK in a phosphorylation site-dependent manner. (A) Schematic of cortactin alternative splicing, showing exon inclusion in wt cortactin and the two known SVs, representing inclusion or exclusion of exons 10 and 11 (48). Arrows indicate the position of primers to detect total cortactin and wild-type (wt) cortactin. (B) SPF45 and SPF45-8D enhance splicing of wild-type cortactin. Quantitative real-time PCR analysis of wild-type cortactin and total cortactin (wt + SV1 + SV2) from SKOV-3 cell lines stably overexpressing wt-SPF45, SPF45-8A, SPF45-8D or empty vector. The results, expressed as the ratio of wt-cortactin to total cortactin, are from three experiments done in duplicate and are shown as mean ± SE. *P < 0.05 versus vec group, #P < 0.05 versus wt-SPF45 group. (C) SPF45 and SPF45-8D overexpression enhance ERK activation and cortactin phosphorylation. Protein lysates from SKOV-3 stable cell lines were immunoblotted for phosphorylated and total ERK and cortactin. S and L signify short and long exposure. (D) The ratio of wild-type to total cortactin was measured in OV2008 stable cell lines as in (B). (E) Total and phosphorylated ERK and cortactin protein were measured by immunoblotting OV2008 cell lysates as in (C). All immunoblots are representative images from three to four independent experiments.
DISCUSSION
We recently showed that ERK, JNK and p38 MAP kinases phosphorylate SPF45 on a serine and threonine residues, inhibiting ΔFas exon 6 exlusion and regulaing the splicing and expression of fibronectin in a phosphorylation-dependent and -independent manner, respectively (21). In the present work, we show that Clk1 phosphorylates SPF45 in vitro on eight serine residues, all of which are N-terminal to the RRM domain required for splicing (33). Ser266 is in the G patch domain, and Ser288 and 291 are just C-terminal to this domain. All other identified phosphorylation sites lie in the unstructured N-terminal domain of unknown function (33). Clk1 phosphorylation of ASF/SF2, a physiological target, occurs on serines in the arginine/serine dipeptide rich region of the protein (22,51). Clk1 prefers to phosphorylate serine residues with a basic residue at the −3 and +3 position (52), and many of the residues that we identified loosely fit this consensus, with positively charged residues within three residues of each side of the site of phosphorylation.
Clk1 expression enhanced SPF45 exon 6 exclusion from Fas mRNA through multiple antagonistic mechanisms involving both positive and negative regulation via direct phosphorylation, negative regulation of RNA binding and positive regulation of protein half-life (Figure 13). Mutagenesis of all eight phosphorylation sites at once showed that Clk1 phosphorylation inhibits SFP45-induced exon 6 exclusion and Fas pre-mRNA binding, whereas mutatagenesis of select groups of phosphorylation sites demonstrated positive or negative regulation of splicing by individual sites; however, the effect of Clk1 phosphorylation on SPF45 may vary when only certain sites are phosphoryalted and may vary by RNA target, as it is possible that only certain sites are phosphorylated at a given time. In addition, it is possible that other kinases also regulate phosphorylation of one or more of these sites, as we have shown that MAP kinases also phosphorylate SPF45 on Ser222 (21).
Figure 13.

Clk1 regulates SPF45-induced exon 6 exclusion from fas mRNA through antagonistic mechanisms. The model shows that Clk1 regulates SPF45 splice site utilization through multiple mechanisms involving increases in SPF45 stability, phosphorylation and regulation of mRNA binding. SPF45 overexpression causes enhanced migration and invasion, dependent on the identified phosphorylation sites and regulation of fibronectin and cortactin.
Clk1 expression enhanced the expression of co-transfected SPF45, whereas inhibition of Clk1 through various means inhibited SPF45 expression and exon 6 exclusion. The relative concentration of splicing factors within the cell controls splice site utilization (11), and our data showing that SPF45 induced exon 6 exclusion in a dose-dependent manner support a role for enhanced splicing of Fas mRNA partially occurring through stabilization of SPF45 protein levels. Mutational analysis of the Clk1 phosphorylation sites did not identify a single or group of phosphorylation sites that regulate SPF45 stability, suggesting that the mechanism of protein stabilization is not through direct phosphorylation of SPF45. Another study has shown that Clk1 overexpression promoted SRp55 degradation via a proteasome dependent pathway (53), whereas our data demonstrate that Clk1 inhibits SPF45 proteosomal-mediated degradation.
Phosphorylation of the RS domains of proteins regulates their interactions with other proteins and with RNA (15–17,54). We found that mutation of the Clk1 phosphorylation sites in SPF45 did not affect binding to splicing factors SF1, SF3b155 and U2AF, which are known binding partners (33,42). Two regions of SPF45 that interact with these proteins are an RYF sequence starting at R375 and charge interactions at N319 and E325 (33), all of which are C-terminal to our identified sites. Mutations at the binding sites not only disrupt these interactions but also SPF45 splicing of Fas pre-mRNA (33). Our data showing that the SPF45 phosphorylation mutants still regulate splicing of Fas further support that these interactions are not affected by the identified phosphorylation sites. This suggests that these phosphorylation sites regulate binding to other splicing proteins or may regulate binding to RNA, and our data showed that mutation of these phosphorylation sites affected binding to Fas mRNA. A previous report showed that SPF45 binds to Sxl pre-mRNA in Drosophila (42); however, whether SPF45 is capable of directly RNA binding was questioned by Corsini et al. (33) who could not show binding of SPF45 to an RNA oligonucleotide from the β-thalassemic mutant β110 of the β-globin gene, a known splicing target of SPF45 (42), and by virtue of the use of a crosslinking reagent in the Sxl mRNA binding experiment. In our study, we did not use a crosslinking agent. Our data support a role for SPF45 phosphorylation in RNA association, either direct or indirect, and correlate with our results on alternative splicing of Fas, suggesting this as a potential mechanism for regulation of SPF45 splicing by phosphorylation. If association with RNA occurs through interactions with an unidentified binding partner, it is likely that phosphorylation regulates association with this protein.
Little is known about the biological function of SPF45 and how it is regulated by phosphorylation. SPF45 is overexpressed in several human cancers (35) and was correlated with a multidrug resistant phenotype (36). We recently showed that SPF45 overexpression inhibits cell proliferation and adhesion to fibronectin in a MAP kinase phosphorylation-dependent manner (21). Our current data show that SPF45 overexpression induces cell migration and invasion in a manner dependent on the identified Clk1 phosphorylation sites. In both assays, overexpression of a phospho-inhibitory mutant of SPF45 (SPF45-8A) acted as a null, with no increase in migration and invasion compared with cells expressing empty vector, whereas cells expressing SPF45-8D behaved similar to wild-type SPF45. These data suggest a role for SPF45 in cancer metastasis and that phosphorylation of these sites control SPF45 biological functions. We identified two potential mechanisms by which SPF45 may regulate these processes: overexpression of fibronectin and splicing and phosphorylation of cortactin. Fibronectin and its integrin receptors increase cell migration and invasion in several cell systems by various mechanisms (43–46). We have previously shown that SPF45 induces fibronectin expression independent of phosphorylation by MAP kinases, but now show that cells expressing SPF45-8A do not induce fibronectin expression, suggesting that phosphorylation of these sites regulates SPF45-induced fibronectin expression, and that this may contribute to a lack of induction of migration and invasion. SPF45 likely regulates a transcription factor or pathway that enhances fibronectin induction.
In addition, cortactin overexpression promotes cell migration and invasion through the regulation of actin polymerization in both lamellipodia and invadopodia (55). There are three SVs of cortactin: wild-type, full-length cortactin; SV1, which lacks the sixth cortactin repeat; and SV2, which lacks the fifth and sixth repeats (48). Overexpression of wild-type cortactin and, to a much lesser extent, SV1, induce migration, whereas SV2 inhibits migration (48). We show that cells overexpressing SPF45 or SPF45-8D have enhanced splicing of wild-type cortactin relative to total cortactin (wt + SV1 + SV2) compared with cells expressing SPF45-8A or empty vector, suggesting that SFP45 regulates cortactin splicing, dependent on these phosphorylation sites. Our data show increased ERK activation in cells expressing SPF45 or SPF45-8D and a corresponding increase in cortactin phosphorylation on serines 405 and 418. ERK phosphorylation of cortactin at these sites (49) increases cortactin-mediated actin polymerization via association with N-WASP and activation of the Arp2/3 complex (50) and increases cortactin localization to sites of dynamic actin regulation, promoting increased duration of lamellipodia formation (39).
In summary, our data establish novel roles for SPF45 in the regulation of ovarian cancer cell migration and invasion, two processes that are important for cancer metastasis. In addition, we have demonstrated that these roles are regulated by SPF45 phosphorylation by Clk1 and showed how these phosphorylations regulate SPF45 biochemical and biological functions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figure 1.
FUNDING
National Institutes of Health [1R01CA131200 to S.T.E.]. Funding for open access charge: 1R01CA131200 from the National Cancer Institute/National Institutes of Health.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Michael Weber for ERK antibody, Thomas Hamilton for OVCAR5 cells, Andrew Godwin for A2780 cells, Nelly Auersperg for IOSE cells and Runzhao Li for OV2008 and ES-2 cells.
REFERENCES
- 1.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 2.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 3.Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 4.Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? Int. J. Biochem. Cell Biol. 2007;39:1432–1449. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 5.Rino J, Carmo-Fonseca M. The spliceosome: a self-organized macromolecular machine in the nucleus? Trends Cell Biol. 2009;19:375–384. doi: 10.1016/j.tcb.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Mayeda A, Krainer AR. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell. 1992;68:365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- 7.Caputi M, Mayeda A, Krainer AR, Zahler AM. hnRNP A/B proteins are required for inhibition of HIV-1 pre-mRNA splicing. EMBO J. 1999;18:4060–4067. doi: 10.1093/emboj/18.14.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothrock CR, House AE, Lynch KW. HnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J. 2005;24:2792–2802. doi: 10.1038/sj.emboj.7600745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Expert-Bezancon A, Sureau A, Durosay P, Salesse R, Groeneveld H, Lecaer JP, Marie J. hnRNP A1 and the SR proteins ASF/SF2 and SC35 have antagonistic functions in splicing of beta-tropomyosin exon 6B. J. Biol. Chem. 2004;279:38249–38259. doi: 10.1074/jbc.M405377200. [DOI] [PubMed] [Google Scholar]
- 10.Shepard PJ, Hertel KJ. The SR protein family. Genome Biol. 2009;10:242. doi: 10.1186/gb-2009-10-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanamura A, Caceres JF, Mayeda A, Franza BR, Jr, Krainer AR. Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA. 1998;4:430–444. [PMC free article] [PubMed] [Google Scholar]
- 12.Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO Rep. 2008;9:1087–1093. doi: 10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grosso AR, Gomes AQ, Barbosa-Morais NL, Caldeira S, Thorne NP, Grech G, von Lindern M, Carmo-Fonseca M. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 2008;36:4823–4832. doi: 10.1093/nar/gkn463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigo R, Bilbao D, Valcarcel J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 15.Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J. Biol. Chem. 2008;283:1223–1227. doi: 10.1074/jbc.R700034200. [DOI] [PubMed] [Google Scholar]
- 16.Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997;11:334–344. doi: 10.1101/gad.11.3.334. [DOI] [PubMed] [Google Scholar]
- 17.Shin C, Feng Y, Manley JL. Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature. 2004;427:553–558. doi: 10.1038/nature02288. [DOI] [PubMed] [Google Scholar]
- 18.Ma CT, Ghosh G, Fu XD, Adams JA. Mechanism of dephosphorylation of the SR protein ASF/SF2 by protein phosphatase 1. J. Mol. Biol. 2010;403:386–404. doi: 10.1016/j.jmb.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duncan PI, Stojdl DF, Marius RM, Bell JC. In vivo regulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Mol. Cell. Biol. 1997;17:5996–6001. doi: 10.1128/mcb.17.10.5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duncan PI, Stojdl DF, Marius RM, Scheit KH, Bell JC. The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Exp. Cell Res. 1998;241:300–308. doi: 10.1006/excr.1998.4083. [DOI] [PubMed] [Google Scholar]
- 21.Al-Ayoubi AM, Zheng H, Liu Y, Bai T, Eblen ST. Mitogen-activated protein kinase phosphorylation of splicing factor 45 (SPF45) regulates SPF45 alternative splicing site utilization, proliferation, and cell adhesion. Mol. Cell. Biol. 2012;32:2880–2893. doi: 10.1128/MCB.06327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- 23.Misteli T, Caceres JF, Clement JQ, Krainer AR, Wilkinson MF, Spector DL. Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J. Cell Biol. 1998;143:297–307. doi: 10.1083/jcb.143.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yomoda J, Muraki M, Kataoka N, Hosoya T, Suzuki M, Hagiwara M, Kimura H. Combination of Clk family kinase and SRp75 modulates alternative splicing of Adenovirus E1A. Genes Cells. 2008;13:233–244. doi: 10.1111/j.1365-2443.2008.01163.x. [DOI] [PubMed] [Google Scholar]
- 25.Mermoud JE, Cohen P, Lamond AI. Ser/Thr-specific protein phosphatases are required for both catalytic steps of pre-mRNA splicing. Nucleic Acids Res. 1992;20:5263–5269. doi: 10.1093/nar/20.20.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tazi J, Daugeron MC, Cathala G, Brunel C, Jeanteur P. Adenosine phosphorothioates (ATP alpha S and ATP tau S) differentially affect the two steps of mammalian pre-mRNA splicing. J. Biol. Chem. 1992;267:4322–4326. [PubMed] [Google Scholar]
- 27.Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA. 1997;3:1456–1467. [PMC free article] [PubMed] [Google Scholar]
- 28.Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, Mann M. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat. Genet. 1998;20:46–50. doi: 10.1038/1700. [DOI] [PubMed] [Google Scholar]
- 29.Aravind L, Koonin EV. G-patch: a new conserved domain in eukaryotic RNA-processing proteins and type D retroviral polyproteins. Trends Biochem. Sci. 1999;24:342–344. doi: 10.1016/s0968-0004(99)01437-1. [DOI] [PubMed] [Google Scholar]
- 30.Silverman EJ, Maeda A, Wei J, Smith P, Beggs JD, Lin RJ. Interaction between a G-patch protein and a spliceosomal DEXD/H-box ATPase that is critical for splicing. Mol. Cell. Biol. 2004;24:10101–10110. doi: 10.1128/MCB.24.23.10101-10110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Callebaut I, Wecker K, Prochnicka-Chalufour A, Dendouga N, Zinn-Justin S, Delepierre M, Tomavo S, Wolff N. Structural and functional characterization of the TgDRE multidomain protein, a DNA repair enzyme from Toxoplasma gondii. Biochemistry. 2006;45:4867–4874. doi: 10.1021/bi051948e. [DOI] [PubMed] [Google Scholar]
- 32.Svec M, Bauerova H, Pichova I, Konvalinka J, Strisovsky K. Proteinases of betaretroviruses bind single-stranded nucleic acids through a novel interaction module, the G-patch. FEBS Lett. 2004;576:271–276. doi: 10.1016/j.febslet.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Corsini L, Bonna S, Basquin J, Hothorn M, Scheffzek K, Valcarcel J, Sattler M. U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat. Struct. Mol. Biol. 2007;14:620–629. doi: 10.1038/nsmb1260. [DOI] [PubMed] [Google Scholar]
- 34.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 35.Sampath J, Long PR, Shepard RL, Xia X, Devanarayan V, Sandusky GE, Perry WL, 3rd, Dantzig AH, Williamson M, Rolfe M, et al. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am. J. Pathol. 2003;163:1781–1790. doi: 10.1016/S0002-9440(10)63538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perry WL, 3rd, Shepard RL, Sampath J, Yaden B, Chin WW, Iversen PW, Jin S, Lesoon A, O'Brien KA, Peek VL, et al. Human splicing factor SPF45 (RBM17) confers broad multidrug resistance to anticancer drugs when overexpressed–a phenotype partially reversed by selective estrogen receptor modulators. Cancer Res. 2005;65:6593–6600. doi: 10.1158/0008-5472.CAN-03-3675. [DOI] [PubMed] [Google Scholar]
- 37.Eblen ST, Catling AD, Assanah MC, Weber MJ. Biochemical and biological functions of the N-terminal, noncatalytic domain of extracellular signal-regulated kinase 2. Mol. Cell. Biol. 2001;21:249–259. doi: 10.1128/MCB.21.1.249-259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Reynolds AB, Kanner SB, Vines RR, Parsons JT. Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol. Cell. Biol. 1991;11:5113–5124. doi: 10.1128/mcb.11.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelley LC, Hayes KE, Ammer AG, Martin KH, Weed SA. Cortactin phosphorylated by ERK1/2 localizes to sites of dynamic actin regulation and is required for carcinoma lamellipodia persistence. PLoS One. 2010;5:e13847. doi: 10.1371/journal.pone.0013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talwar S, Jin J, Carroll B, Liu A, Gillespie MB, Palanisamy V. Caspase-mediated cleavage of RNA-binding protein HuR regulates c-Myc protein expression after hypoxic stress. J. Biol. Chem. 2011;286:32333–32343. doi: 10.1074/jbc.M111.255927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, Sumi K, Yomoda J, Murray MV, Kimura H, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- 42.Lallena MJ, Chalmers KJ, Llamazares S, Lamond AI, Valcarcel J. Splicing regulation at the second catalytic step by Sex-lethal involves 3' splice site recognition by SPF45. Cell. 2002;109:285–296. doi: 10.1016/s0092-8674(02)00730-4. [DOI] [PubMed] [Google Scholar]
- 43.Zou L, Cao S, Kang N, Huebert RC, Shah VH. Fibronectin induces endothelial cell migration through beta1 integrin and Src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J. Biol. Chem. 2012;287:7190–7202. doi: 10.1074/jbc.M111.304972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitra AK, Sawada K, Tiwari P, Mui K, Gwin K, Lengyel E. Ligand-independent activation of c-Met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene. 2011;30:1566–1576. doi: 10.1038/onc.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caswell PT, Spence HJ, Parsons M, White DP, Clark K, Cheng KW, Mills GB, Humphries MJ, Messent AJ, Anderson KI, et al. Rab25 associates with alpha5beta1 integrin to promote invasive migration in 3D microenvironments. Dev. Cell. 2007;13:496–510. doi: 10.1016/j.devcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 46.Nishida T, Nakagawa S, Awata T, Ohashi Y, Watanabe K, Manabe R. Fibronectin promotes epithelial migration of cultured rabbit cornea in situ. J. Cell Biol. 1983;97:1653–1657. doi: 10.1083/jcb.97.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirkbride KC, Sung BH, Sinha S, Weaver AM. Cortactin: a multifunctional regulator of cellular invasiveness. Cell Adh. Migr. 2011;5:187–198. doi: 10.4161/cam.5.2.14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Rossum AG, de Graaf JH, Schuuring-Scholtes E, Kluin PM, Fan YX, Zhan X, Moolenaar WH, Schuuring E. Alternative splicing of the actin binding domain of human cortactin affects cell migration. J. Biol. Chem. 2003;278:45672–45679. doi: 10.1074/jbc.M306688200. [DOI] [PubMed] [Google Scholar]
- 49.Campbell DH, Sutherland RL, Daly RJ. Signaling pathways and structural domains required for phosphorylation of EMS1/cortactin. Cancer Res. 1999;59:5376–5385. [PubMed] [Google Scholar]
- 50.Martinez-Quiles N, Ho HY, Kirschner MW, Ramesh N, Geha RS. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol. Cell. Biol. 2004;24:5269–5280. doi: 10.1128/MCB.24.12.5269-5280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, Fu XD. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J. Biol. Chem. 1996;271:24569–24575. doi: 10.1074/jbc.271.40.24569. [DOI] [PubMed] [Google Scholar]
- 52.Menegay HJ, Myers MP, Moeslein FM, Landreth GE. Biochemical characterization and localization of the dual specificity kinase CLK1. J. Cell Sci. 2000;113(Pt.18):3241–3253. doi: 10.1242/jcs.113.18.3241. [DOI] [PubMed] [Google Scholar]
- 53.Lai MC, Kuo HW, Chang WC, Tarn WY. A novel splicing regulator shares a nuclear import pathway with SR proteins. EMBO J. 2003;22:1359–1369. doi: 10.1093/emboj/cdg126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc. Natl Acad. Sci. USA. 2004;101:9666–9670. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacGrath SM, Koleske AJ. Cortactin in cell migration and cancer at a glance. J. Cell Sci. 2012;125:1621–1626. doi: 10.1242/jcs.093781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.