


Abstract
Nucleotide excision DNA repair is mechanistically conserved across all kingdoms of life. In prokaryotes, this multi-enzyme process requires six proteins: UvrA–D, DNA polymerase I and DNA ligase. To examine how UvrC locates the UvrB–DNA pre-incision complex at a site of damage, we have labeled UvrB and UvrC with different colored quantum dots and quantitatively observed their interactions with DNA tightropes under a variety of solution conditions using oblique angle fluorescence imaging. Alone, UvrC predominantly interacts statically with DNA at low salt. Surprisingly, however, UvrC and UvrB together in solution bind to form the previously unseen UvrBC complex on duplex DNA. This UvrBC complex is highly motile and engages in unbiased one-dimensional diffusion. To test whether UvrB makes direct contact with the DNA in the UvrBC–DNA complex, we investigated three UvrB mutants: Y96A, a β-hairpin deletion and D338N. These mutants affected the motile properties of the UvrBC complex, indicating that UvrB is in intimate contact with the DNA when bound to UvrC. Given the in vivo excess of UvrB and the abundance of UvrBC in our experiments, this newly identified complex is likely to be the predominant form of UvrC in the cell.
INTRODUCTION
The genomes of all living organisms are under constant assault from DNA-damaging agents ranging from reactive oxygen species produced during normal cellular respiration to UV irradiation from sunlight. A number of enzymatic repair pathways have evolved to deal with the huge variety of resulting DNA lesions. Nucleotide excision repair (NER) corrects a diverse range of chemically and structurally unrelated lesions from UV-induced photoproducts to carcinogen DNA adducts (1–4). NER is a highly conserved process from bacteria to mammals (2,5). In bacteria, this pathway is initiated by three key proteins: UvrA, UvrB and UvrC. UvrA and UvrB together scan DNA in search for lesions (3,5–7). On encountering a DNA lesion, the UvrA dimer passes the lesion to UvrB and dissociates resulting in a tight UvrB–DNA pre-incision complex (8–11). This pre-incision complex marks the DNA for cleavage by UvrC, which incises the DNA on both sides of the damage, on the same DNA strand (12–14). In the final steps of NER, DNA polymerase I and UvrD (helicase II) remove the post-incision complex and synthesize the repair patch, which is sealed by DNA ligase (15,16). Expression of both UvrA and UvrB is upregulated by an approximate order of magnitude during the SOS response initiated when damage is detected (1). Although UvrD is also upregulated, this is not essential for its function during NER (17). UvrC is not upregulated in response to damage and remains at ∼10 molecules per cell (1,13). Given the large amount of DNA that UvrC would need to search for the UvrB–DNA pre-incision complex, this task is likely the rate-determining step of the NER system.
UvrC is a dual-functional molecule that incises damaged DNA 3′ and 5′ to the lesion (18). The N-terminal domain of UvrC possesses the nuclease activity that incises three to four nucleotides 3′ of the lesion (12,14,19). However, alone the purified N-terminal domain does not bind DNA and is incapable of incision (20). UvrC’s 5′ incision activity, which incises at the seventh nucleotide 5′ to the lesion, is found in its C-terminal domain. The C-terminal region of the molecule contains two tandem helix–hairpin–helix motifs (HhH)2 and is connected via a flexible hinge to the C-terminal endonuclease domain (21). The flexible hinge allows the (HhH)2 region to reach around and potentially clamp the opposite face of the DNA to the endonuclease domain.
Formation of a UvrB–DNA pre-incision complex occurs after UvrA is ejected, which may be facilitated by the wrapping of DNA around UvrB in some bacterial species (22). UvrA acts to relieve UvrB’s auto-inhibitory C-terminal domain, allowing UvrB’s β-hairpin to insert between DNA’s strands to detect damage (23). A critical residue located at the base of this hairpin is tyrosine 96, which is 100% conserved among all bacterial species (24,25). This aromatic side chain is essential for damage verification, as alanine substitution renders UvrB incapable of accepting the damaged DNA from UvrA, causing arrest at the UvrAB–DNA complex (8,25). When UvrB forms a pre-incision complex, non-specific electrostatic interactions are thought to precede unwinding and strand separation, which exposes nucleotide bases thus permitting more specific interactions to form (24,26). These specific interactions include hydrogen bonds and stacking interactions that may cause the bending of the DNA backbone and the stabilization of the unwound DNA (27,28). It is not clear how UvrB signals UvrC to bind and incise DNA; however, stacking interactions may be weakened in the presence of a lesion causing a conformational change in UvrB, which in turn promotes UvrC binding (28). Alternatively, the ATPase and limited helicase activity of UvrB promote conformational changes that expose single-stranded DNA to the nuclease domains of UvrC (25).
Single-molecule studies have been useful in studying a number of systems that track along DNA to find their target binding sites (6,29–35). These studies have probed both the mechanisms of enzyme action and target search. Several modes of motion have been proposed for how proteins efficiently locate their target sites among an excess of similar but non-cognate DNA including (36) (i) jumping, (ii) hopping, (iii) sliding, (iv) intersegmental transfer and (v) active translocation [also reviewed in (29,33,37)]. In reality, it is likely that proteins use more than one of these mechanisms. How UvrA and the UvrAB complex search DNA for lesions was recently determined to be an efficient combination of three-dimensional (3D) and one-dimensional (1D) DNA sampling, distinct from UvrA alone which only uses 3D sampling (6).
Here, we investigate the DNA interactions of UvrC alone and in combination with UvrB using real-time single-molecule imaging on DNA tightropes (6,30). UvrB and UvrC were differentially labeled with quantum dots (Qdots) and imaged using oblique angle fluorescence (OAF) microscopy (6). We have found that UvrC is capable of binding and then, in a salt-dependent manner, diffusing one-dimensionally on DNA. Surprisingly, however, we found that UvrC when pre-mixed with UvrB formed abundant UvrBC–DNA complexes that displayed striking differences in search behavior compared with UvrC alone. Both the interaction lifetimes and the diffusion constants were found to be affected by salt concentration, consistent with a hopping search mechanism. At low salt, more UvrBC molecules were capable of diffusing, but the diffusion constant was an order of magnitude slower than UvrC alone. To determine whether UvrB contacts DNA within the UvrBC–DNA complex, we compared the motile properties of three UvrB mutants in complex with UvrC and found their behavior to be consistent with UvrB making a significant contact with DNA. Together with our observation that UvrB in solution cannot be captured by preformed UvrC–DNA complexes, these data indicate a novel interaction between UvrB and UvrC in solution, which activates a DNA-binding activity of UvrB.
MATERIALS AND METHODS
Standard conditions
Unless otherwise stated, all experimental procedures were performed at room temperature in ABC buffer [50 mM Tris–HCl (pH 7.5), 50 mM KCl and 10 mM MgCl2].
Protein purification and labeling
To conjugate UvrC to Qdots, the biotin ligase recognition sequence GLNDIFEAQKIEWHEGGG (AviTag™) was added to the C-terminus of Bacillus caldotenax UvrC. First, amplification of nucleotides 2211–2859 of uvrC gene on pTYB1-wt uvrCbca vector was achieved using the forward primer (5′-CAT CCG CTC GAG TTT GAA CGG GCG AAA GAA TAC-3′) and reverse primer (5′-CAT TGG TAC CCT TGG CAA AGC ATT CGT GCC ATT CGA TTT TCT GAG CCT CGA AGA TGT CGT TCA GAC CAC CGC CAC CTT CAT GCA GTT TTT CAT AGA TTT TCT CCG CCA C-3′). The PCR products were digested with XhoI and KpnI, gel purified and cloned into the pTYB1-Wt uvrCbca vector that was also digested with same restriction enzymes. Insertion of the AviTag into the vector (pTYB1-uvrC-avi) was confirmed by DNA sequencing. For the expression of in vivo biotinylated UvrC, pTYB1-uvrC-avi plasmid (ampicillin resistance) and a pACYC184 plasmid with an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible birA gene (chloramphenicol resistance) were co-transformed into the BL21-CodonPlus® (DE3)-RIL cells (Invitrogen). Biotin (50 µM) was added at the time of IPTG (10 mM) induction. Biotinylated UvrC was purified using the IMPACT™ system (intein-mediated purification with an affinity chitin-binding tag; New England Biolabs) (23). Proteins were quantified using Image-Quant® software after the staining of SDS–PAGE protein gel with SimplyBlue SafeStain (Invitrogen). WT UvrB (HA-tagged), Y96A, Δhairpin and D338N UvrB mutants were purified following the procedures described previously (25,38). All expressed Uvr proteins were heated at 65°C for 10 min to denature the endogenous Escherichia coli Uvr proteins thus avoiding the possibility of contamination. To test UvrC-avi for its interaction with UvrB and incision activity, a 50-bp 32P-radiolabeled DNA (F50/NDB) containing a modified thymine at position 26 was incised by various UvrC preparations in the presence of UvrA and UvrB (Supplementary Figure S1). The avi-tagged UvrC retains a similar incision activity as compared with WT protein (Supplementary Figure S1, compare lanes 2 and 3).
Assessment of the oligomeric state of UvrC on DNA was achieved by pre-conjugating UvrC separately with two differently colored Qdots (655 and 565 nm), mixing and then examining them on DNA tightropes. Approximately 10% of complexes showed dual color labeling (Supplementary Figure S2); thus, although we cannot rule out that Qdot conjugation interferes with oligomerization, the majority of the complexes consisted of singly labeled presumably monomeric UvrC.
DNA substrates
All single-molecule fluorescence experiments used undamaged bacteriophage λ-DNA (New England Bio Labs). For atomic force microscopy (AFM) imaging, a 458-bp linear DNA substrate was made by TaqI digestion of pUC18 followed by separation over a 1% agarose gel and purification using the Illustra GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare).
Single-molecule fluorescence imaging
UvrC–Qdot conjugates were prepared by incubating biotinylated UvrC-avi (100 nM) with 400 nM 655 or 565 nm streptavidin-conjugated Qdots (Invitrogen) in ABC buffer for 30 min before dilution to ∼1 nM for imaging. A 4:1 excess of Qdot to protein was used to increase the probability of generating singly labeled Qdots (39). To prevent label cross-talk, UvrB expressed with an N-terminal hemagglutinin tag (HA) was conjugated to Qdots using an antibody sandwich as described previously (39). During imaging, 100 mM dithiothreitol (DTT) was added to ABC buffer to decrease Qdot blinking (40) and to reduce photobleaching of YOYO-1. When used, ATP was supplemented to a final concentration of 1 mM. Likewise, elevated ionic strengths were achieved by altering the final [KCl] in ABC buffer to 100 or 150 mM KCl.
To visualize protein interactions with DNA, λ-DNA tightropes were suspended between 5 µm surface adhered beads to create DNA tightropes as described previously (6,30). Briefly, the surface of a custom-built flow cell was blocked using mPEG5000 N-succinimidyl propionate (Sigma-Aldrich). Poly-l-lysine (Sigma-Aldrich)–coated 5-µm diameter silica spheres (Polysciences Inc.) were flushed into the flow cell and left to randomly adhere to the surface before washing with ABC buffer. DNA was passed through the flow cell at an appropriate rate to unravel it for adherence to the coated 5 µm silica spheres, thus forming DNA tightropes. YOYO-1 (Invitrogen) at a ratio of ∼1:4000 to DNA base pairs (base pairs in excess) was flowed into the chamber to check for tightropes by fluorescence microscopy before being washed out with ABC + DTT buffer. Qdot-tagged proteins in the imaging buffer were subsequently added and visualized. Imaging was performed using OAF microscopy on a custom-built imaging platform (6) and recorded using a DU897 EMCCD camera (Andor, Belfast, UK).
Atomic force microscopy imaging
DNA substrates were heated at 65°C for 15 min before slow cooling to room temperature. UvrC–Qdot conjugates were prepared by incubating 5 µl of biotinylated UvrC-avi (15 nM) with 5 µl of streptavidin-conjugated Qdot (655 nm, Invitrogen, 75 nM) in ABC buffer for 30 min at room temperature. To form protein–DNA complexes, 1 µl of the UvrC–Qdot conjugates were mixed with 1.5 µl of 458-bp DNA (50 nM, diluted in ABC buffer) and 0.5 µl of ABC buffer followed by incubation for 10 min.
UvrB–Qdot conjugates were prepared by first incubating 2 µl of UvrB–HA [300 nM; (39)] with 2 µl of 300 nM mouse monoclonal hemagglutinin antibody (HA-Ab, Covance) for 30 min. Two microliters of goat anti-mouse IgG 565 Qdot conjugates (75 nM, Invitrogen) was incubated with 2 µl of the UvrB–HA:HA-Ab solution diluted in ABC to 15 nM for 30 min. To 2 µl of this UvrB–Qdot conjugate, we added 1 µl of UvrC-avi (15 nM) and incubated for an additional 10 min. The 458-bp DNA fragments (3 µl, 50 nM) were then added to the Qdot–UvrB and UvrC solution and incubated for an additional 10 min.
All reactions were diluted 1:7 into AFM deposition buffer [25 mM NaOAc, 25 mM HEPES–KOH (pH 7.5) and 10 mM Mg(OAc)2] before deposition onto freshly cleaved mica, rinsed with deionized water and dried under a gentle stream of nitrogen gas. Images were collected using a MultiModeV microscope (Bruker Corporation) using an E scanner in tapping mode. Pointprobe® plus non-contact/tapping mode silicon probes (PPP-NCL, Agilent) with spring constants of ∼50 N/m and resonance frequencies of ∼190 kHz were used. Images were captured at a scan size of 1 × 1 μm, scan rate of ∼3 Hz, target amplitude of 0.30–0.35 V and resolution of 512 × 512 pixels.
Data analysis
Kymographic time streaks of the Qdot-conjugated proteins interacting with DNA were created using ImageJ (NIH, USA). Multiple binding events could be observed in each visual field; however, only DNA with a protein occupancy of six molecules or less was chosen to be analyzed, to avoid interference from adjacent proteins during data analysis. Furthermore, any complexes that were excessively bright indicating the presence of multiple Qdots were ignored during analysis. A protein was considered motile if its kymograph showed movement of three pixels over three frames from the previous position. The length of the streak corresponds to the lifetime of attachment, only streaks that began and ended in a movie were analyzed which may lead to a small systematic underestimation of the attached lifetime. These data were plotted as cumulative frequency histograms and fitted to:
| (1) |
where N is the number of observed points, t the bin, tmax the maximum bin size and k the reciprocal of the dwell time.
Streak analysis permitted rapid inspection of the time-lapse recordings for examining whether and how a protein is moving. For unbiased data analysis, a custom-written automated fitting routine was used for each time streak (see Supplementary Information for more detail). This routine used the Gaussian distribution approximation of the point spread function for single fluorophores to provide positional accuracy beyond the limit of diffraction, which was determined as 8.7 nm (41).
To quantify the motion of individual molecules their mean-squared displacements (MSDs) were determined using:
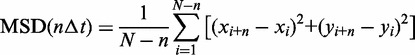 |
(2) |
where N is the total number of frames in the kymograph, n the frame, xi and yi the position of the protein and Δt the time window.
MSDs were fitted to a straight line; however, it was not possible to fit the entire MSD, as non-linearities appear even for simulated random walkers (data not shown). Therefore, the r2 value of the linear regression was used to determine the quantity of data to be fitted. When the r2 value of the fit dropped <0.7, no more data were used; this corresponded to approximately the first quarter of the data set. The slope of this linear plot provides the diffusion constant. To determine the mechanism of motion, the MSD was re-plotted on log–log axes, and the slope of this plot defined ‘α’, the diffusive exponent. When α is 1, unbiased diffusion is indicated, 2 indicates directed motion and <1 suggests sub-diffusion (42). For each condition, we collected enough data such that 30–100 determinations of D and α could be made. In the tables, we report mean values (±SE) of these values; however, in cases where sub-populations may exist, the distributions may not be normally distributed; therefore, these means and standard error values are less accurate. Supplementary Figure S4 shows an attempt to fit certain of these data using a Gaussian mixture model, which suggests there is indeed a distinct sub-population of D and α. All P-values quoted herein are derived from a Student’s t-test.
RESULTS
Characterization of UvrC–Qdot conjugates
To study the dynamic interaction of UvrC with DNA in real time at the single-molecule level, we conjugated UvrC to Qdots through a biotin ligase recognition sequence (avi-tag), which was cloned onto the C-terminus of UvrC (UvrC-avi). UvrC-avi was biotinylated in vivo through co-expression with BirA biotin ligase (see ‘Materials and Methods’ section).
AFM imaging was used to assess the conjugation of biotinylated UvrC to streptavidin-coated Qdots and the ability of UvrC–Qdot conjugates to bind dsDNA. This approach revealed that UvrC–Qdots could bind to 458-bp dsDNA fragment (Figure 1A).
Figure 1.

UvrC–Qdots interact with DNA tightropes. (A) AFM image of a UvrC–Qdot conjugate bound to DNA. (B) An illustration of DNA tightrope showing three Qdot-labeled protein complexes bound to a single tightrope of DNA suspended between two 5 -µm silica beads. (C) UvrC–Qdots (purple) bound to individual λ-DNA strands labeled with YOYO-1 (green).
Dynamic interaction between UvrC–Qdot and DNA
The interaction between UvrC–Qdot conjugates and DNA was examined using the DNA tightrope assay (Figure 1B). This assay involves suspending elongated DNA (∼90% contour length) between surface adhered poly-l-lysine-coated microspheres using hydrodynamic flow within custom-built microfluidic cells (6,30). This DNA tightrope assay provides the advantages of isolating DNA from the surface, reducing background signal and avoiding the need for continuous buffer flow. Imaging was performed using OAF microscopy, which reduces background by selectively illuminating the sample plane. Together this approach has permitted the direct observation of the movements of Qdot-labeled DNA repair proteins UvrA, UvrB and glycosylase proteins (6,30). To visualize UvrC–Qdots on DNA, we conjugated biotinylated UvrC with streptavidin-coated Qdots, introduced the complexes into flow chambers with DNA tightropes and monitored the UvrC–Qdot DNA binding using OAF. We observed numerous UvrC–Qdots bound to tightropes throughout the visual field (Figure 1C and Supplementary Movie S1). Streptavidin-coated Qdots alone or in the presence of un-tagged WT UvrC did not bind to the DNA (data not shown), indicating that the binding of UvrC–Qdot to DNA is specific. The motion of bound complexes on DNA was studied using kymographic streak analysis (6); this approach allowed quick assessment of diffusion constants and lifetimes.
ATP was found to have no effect on UvrC’s DNA lifetime bound to DNA; therefore, results from experiments with and without ATP were combined, generating lifetime measurements of 35.3 (±2.0), 14.6 (±0.9) and 21.7 (±0.8) s at 50, 100 and 150 mM KCl, respectively (Table 1).
Table 1.
Motile properties of UvrC–Qdot on DNA
| UvrC | 50 mM KCl | 100 mM KCl | 150 mM KCl |
|---|---|---|---|
| Lifetime (±SE) (s) | 35.3 (±2.0) | 14.6 (±0.9) | 21.7 (±0.8) |
| n = 145 | n = 136 | n = 157 | |
| D (±SE) ×10−3 µm2 s−1 | 36.0 (±6.7) | 7.0 (±1.6) | 9.3 (±1.4) |
| n = 91 | n = 98 | n = 91 | |
| Diffusive exponent (±SE) | 0.53 (±0.05) | 0.64 (±0.03) | 0.80 (±0.04) |
| n = 91 | n = 98 | n = 91 |
All errors quoted are standard errors.
At 50 mM KCl, UvrC was observed to have a low incidence (∼15%) of motility. However, at elevated KCl concentrations, a significant increase in the number of motile UvrC molecules was observed, from 15 to 30% at 100 mM KCl to 43% at 150 mM KCl (P < 0.01) (Figure 2A and Supplementary Movie S2). As expected, 1 mM ATP induced no significant difference in protein motility at any KCl concentrations (data not shown). Of the moving proteins (n = 1949), ∼90% moved continuously without visibly pausing during the recording. Approximately 95% of these molecules were fit to Equation (2) for diffusional or sub-diffusional motion; the small remainder could not be confidently characterized. The diffusion constant of UvrC decreased from 36.0 (±6.7) ×10−3 µm2 s−1 at 50 mM KCl to 7.0 (±1.6) ×10−3 µm2 s−1 at 100 mM KCl and 9.3 (±1.4) ×10−3 µm2 s−1 at 150 mM KCl (Table 1 and Figure 2B). Further insight into how UvrC diffuses on the DNA was provided by characterization of its diffusive exponents. For all salt concentrations studied, the average exponent value was <1; such values suggest a stop–start motion, which indicates searching followed by interrogation of the DNA (30,42). However, as the salt concentration was raised, a clear trend toward a value of 1 was seen (Table 1 and Figure 2B), suggesting reduced sub-diffusive behavior (see ‘Materials and Methods’ section for explanation of α values).
Figure 2.

The motile characteristics of UvrC. (A) Percentage of moving UvrC versus salt concentration. Values for mean percentage (±SE, where n refers to experiments repeated on different days) moving were 14.9% (±3.7, n = 13), 29.8% (±5.8, n = 10) and 43.3% (±3.5, n = 10) for 50, 100 and 150 mM KCl, respectively. Cumulative number of molecules examined was 1949; the number of molecules examined per condition is shown in the figure. (B) 3D density plots of the diffusion constant versus the α factor (slope of log–log MSD versus time plot) of UvrC under different salt concentrations. The coloring is a percentage scale relative to the maximum bin size. N was 44, 44 and 45 in increasing order of the respective salt condition. See Supplementary Figure S5 for original data with errors and representative kymographs.
Imaging of UvrB and UvrC on DNA
Previous studies have only been able to suggest the potential presence of UvrBC complexes on a specific Y-substrate DNA (18,43). To investigate the possible formation of UvrBC complexes on normal dsDNA, we differentially labeled UvrB and UvrC for direct fluorescence imaging. We used UvrB–Qdot conjugates, which were formed using HA-tagged UvrB, primary HA antibody and secondary antibody-coated Qdots (39), in combination with avi-tagged UvrC. UvrB and UvrC were pre-incubated at a 2:1 ratio (UvrB:UvrC) before introduction to DNA tightropes in the flow chamber. We observed clear and abundant binding of UvrB–Qdot on DNA only in the presence of UvrC, under the conditions where only the UvrB was labeled or when both proteins were differentially color labeled with Qdots (Figure 3; Supplementary Movies S3 and S4). No difference in diffusion constant, diffusive exponent and percentage of motile complexes was seen between labeling strategies (Figure 3 and Supplementary Table S2). To confirm our imaging data, AFM was used to demonstrate that UvrB only bound to dsDNA in the presence of UvrC (Figure 3). Together, these data represent the first direct demonstration of UvrBC complexes on dsDNA in the absence of UvrA or a pre-formed bubble or DNA flap (18,43). Importantly, when UvrC–Qdots were pre-loaded onto DNA before addition of UvrB–Qdots (with a wash in between to remove free UvrC and UvrC–Qdots in solution), no loading of UvrB–Qdots on DNA was observed. This result is consistent with previous observations that UvrB does not independently bind to dsDNA (4,6) and implies that the UvrB-interacting domain on UvrC was inaccessible for UvrB once UvrC was loaded onto DNA.
Figure 3.

UvrBC binding to DNA. (A) Imaging of dual color Qdot-labeled UvrBC shows the presence of both UvrB (green Qdot) and UvrC (purple Qdot) on DNA tightropes (unlabeled). The bright object on the lower right is the corner of a silica pedestal bead. (B) UvrBC diffusion is shown as movement on an unlabeled DNA tightrope over successive frames temporally separated by 1.03 s. (C) The percentage of motile complexes are shown to be unaffected by labeling strategy. Mean (±SE) values were plotted: 34.3% (±5.9, n = 58 molecules), 33.6% (±4.3, n = 130 molecules) and 32.2% (±5.8, n = 45 molecules) for dual labeled, labeled B or labeled C, respectively. AFM images of 458-bp dsDNA incubated with UvrB–Qdot conjugates (D) in the absence and (E) presence of in vivo biotinylated UvrC-avi. The image size is at 1 × 1 µm, and the Z scale is at 2 nm. (F) and (G) are zoomed 3D displays of the marked areas in (D) and (E).
In the absence of nucleotide cofactors, ∼20% of all the motile UvrBC proteins observed were of sufficient duration and clarity to be analyzed for MSD (total observed = 1256, MSD analyzed = 287). The percentage of motile molecules decreased as the salt concentration was raised (Figure 4A). There was a small increase in diffusion constant as the concentration of KCl was increased from 100 to 150 mM (Table 2 and Figure 4B) that is not predicted for a sliding molecule (37). It may be possible that some of the electrostatic contacts with DNA are impaired either through direct shielding of protein–DNA contacts or through a salt-induced conformational change in the protein. The salt dependency of the diffusive exponent for UvrBC (+ATP or −ATP) was similar to that observed with UvrC alone; at low salt, sub-diffusion was observed [α = 0.57 (+ATP) at 50 mM and 0.69 (+ATP) at 100 mM KCl]; at 150 mM KCl, the diffusive exponent increased [α = 0.87 (+ATP)], indicating free diffusion along a smoother diffusive landscape.
Figure 4.

The motile characteristics of UvrBC. (A) Mean percentage (±SE, where n refers to experiments repeated on different days) of moving UvrBC in the presence of ATP (light gray bars). Values were 47.9% (±4.8, n = 7), 46.9% (±4.5, n = 5), 49.9% (±2.9, n = 5) at 50, 100 and 150 mM KCl, respectively. In the absence of ATP (dark gray bars), values were 64.1% (±4.2, n = 7), 58.5% (±1.2, n = 6) and 42.7% (±4.5, n = 5) at 50, 100 and 150 mM KCl, respectively. Cumulative number of molecules examined was 1256; the number of molecules examined per condition is shown in the figure. (B) 3D density plots of the diffusion constant versus the α factor (slope of log–log MSD versus time plot) of UvrBC versus salt concentration in the presence of ATP. The coloring is a percentage scale relative to the maximum bin size. N was 49, 48 and 72 in increasing order of salt concentration. See Supplementary Figure S6 for original data with errors and representative kymographs.
Table 2.
Motile properties of UvrBC–Qdot on DNA
| UvrBC | 50 mM KCl | 100 mM KCl | 150 mM KCl |
|---|---|---|---|
| Lifetime (±SE) (s) −ATP | 32.1 (±7.3) | 18.6 (±0.5) | 13.3 (±0.6) |
| n = 74 | n = 94 | n = 73 | |
| D (±SE) ×10−3 µm2 s−1 −ATP | 3.7 (±0.8) | 2.6 (±0.9) | 10.0 (±0.9) |
| n = 64 | n = 43 | n = 43 | |
| Diffusive exponent (±SE) –ATP | 0.66 (±0.05) | 0.44 (±0.04) | 0.88 (±0.04) |
| n = 64 | n = 43 | n = 43 | |
| Lifetime (±SE) (s) +ATP | 47.6 (±3.4) | 25.1 (±1.0) | 10.8 (±0.4) |
| n = 63 | n = 70 | n = 80 | |
| D (±SE) ×10−3 µm2 s−1 +ATP | 2.8 (±0.4) | 6.0 (±1.3) | 15.0 (±2.0) |
| n = 49 | n = 48 | n = 72 | |
| Diffusive exponent (±SE) +ATP | 0.57 (±0.06) | 0.69 (±0.04) | 0.87 (±0.03) |
| n = 49 | n = 48 | n = 72 |
All errors quoted are standard errors.
Domains 1a and 3 of UvrB bind ATP at the inter-domain interface (27). To investigate whether ATP plays a role in influencing the diffusional properties of UvrBC complexes, we carried out experiments in the presence of ATP. In the presence of ATP, no significant change in the percentage of motile molecules was observed as the salt concentration was raised (Figure 4). At 50 mM KCl, ATP was found to have a slight effect on the movement of UvrBC, resulting in a reduction of the percentage of motile complexes from 64 to 48% (Figure 4A). However, ATP increased the diffusion constant of the UvrBC complex from 2.6 × 10−3 (±0.9) to 6.0 × 10−3 (±1.3) µm2 s−1 at 100 mM and from 10.0 × 10−3 (±0.9) to 15.0 × 10−3 (±2.0) µm2 s−1 at 150 mM KCl (Table 2). Importantly, the salt dependency of the diffusive exponent for UvrBC in the presence of ATP was similar to that observed in the absence of ATP, with increased diffusive exponent at 150 mM KCl.
We also studied UvrBC (using labeled UvrB and unlabeled UvrC) motion in the presence of ADP. Experiments at 50 and 150 mM KCl showed no significant difference (P > 0.05) in lifetime and diffusion constant for UvrBC with ADP (Supplementary Table S1) compared with data in the presence or absence of ATP (Table 2). However, a loss of the low-diffusion low-α population was observed when ADP was present (Supplementary Figure S3), consistent with a loss of pauses in the data (30). Examination of data traces confirmed no visible pauses; however, the α value is still low and, therefore, may indicate the presence of short pauses not visible in the data.
Structure/function analysis of the UvrBC–DNA complex
Our observation of UvrC-dependent binding of UvrB to dsDNA raised the question of whether the UvrB molecule within the UvrBC–DNA complex makes direct contact with DNA. We had observed that at 50 mM KCl, the majority of UvrBC complexes (57%, combining +ATP and −ATP data) were motile, unlike UvrC alone where only 15% of molecules were seen to move (Figures 2 and 4). These results suggest that UvrBC possesses different DNA contacts compared with UvrC.
To address this question directly, we studied three UvrB mutants. The first two UvrB variants, Y96A (8) and the β-hairpin deletion (8,44), affect DNA binding. The final UvrB mutant, D338N, is deficient in ATP hydrolysis (45). All of the UvrB mutants were pre-complexed with UvrC before addition to the DNA tightropes, and their motion studied in low-salt conditions where the greatest differences in behavior would be expected. The first mutant, UvrBY96A, was designed to remove a crucial residue located at the base of the β-hairpin that inserts into the DNA to detect damage (8,25,27,44). At 50 mM KCl, this mutant possessed a statistically different lifetime (36.8 s: Table 3, P < 0.001) compared with wild-type UvrBC [47.6 s (+ATP): Table 2]. Furthermore as shown in Figure 5A, the number of motile complexes for UvrBY96A was considerably higher (83%) compared with WT + ATP (48%). Additionally, UvrBY96A mutant’s diffusion constant (12.1 × 10−3 (±3.0) µm2 s−1) was significantly (P < 0.001) greater than WT + ATP [2.8 × 10−3 (±0.4) µm2 s−1] at low salt (Table 3). The diffusive exponent (α = 0.75) was also higher, suggesting free diffusion, and is almost identical to WT at 150 mM KCl (α = 0.87). All of these results suggest that UvrBY96A has affected the motion of the UvrBC complex by reducing the incidence of sub-diffusion along the DNA. Given the UvrBY96A mutant showed such a substantial change from WT, we made the more severe mutation of deleting the entire β-hairpin implicated in DNA damage recognition (27,44). Removing this β-hairpin should eliminate a large interaction between UvrB and DNA, but not affect its ability to interact with UvrC. At low salt, this deletion mutant possessed a lifetime (14.0 s) and diffusive exponent (α = 0.87) significantly different from those of the WT protein at 50 mM KCl, and in fact, more closely resembled WT UvrBC data at 150 mM KCl (Table 3 and Figure 5). In addition, the β-hairpin deletion mutant’s diffusion constant was approximately twice that of wild-type at high-salt concentrations, suggesting that UvrBΔhairpinC complex was moving unencumbered along the DNA. Importantly, these results indicate that alterations in the DNA interacting region of UvrB affect UvrBC motility.
Table 3.
Motile properties of mutant UvrBC–Qdot on DNA
| Mutant | UvrBY96AC + ATPa | UvrBΔhairpinC + ATPa | UvrBD338NC + ATPa |
|---|---|---|---|
| Lifetime (±SE) (s) | 36.8 (±1.1) | 14.0 (±0.7) | 43.6 (±1.1) |
| n = 95 | n = 30 | n = 72 | |
| D (±SE) ×10−3 µm2 s−1 | 12.1 (±3.0) | 31 (±9.0) | 2.6 (±1.0) |
| n = 61 | n = 31 | n = 48 | |
| Diffusive exponent (±SE) | 0.75 (±0.03) | 0.87 (±0.05) | 0.71 (±0.03) |
| n = 61 | n = 31 | n = 48 |
All errors quoted are standard errors.
aData gathered at 50 mM KCl.
Figure 5.

The effect of UvrB mutation on the motile characteristics of UvrBC at 50 mM KCl and in the presence of ATP. (A) Percentage of moving UvrBC for WT, UvrBY96AC, UvrBΔhairpinC and UvrBD338NC complexes. Mean (±SE, where n refers to experiments repeated on different days) values were 47.9% (±4.8, n = 7), 84.1% (±2.3, n = 4), 48.4% (±9.9, n = 6) and 60.1% (±5.9, n = 3), respectively. (B) 3D density plots of the diffusion constant versus the α factor for each UvrBC mutant complex. The coloring is a percentage scale relative to the maximum bin size. N (in same order as aforementioned) was 49, 61, 41 and 48. See Supplementary Figure S7 for original data with errors and representative kymographs.
To further understand how ATP influences the diffusional properties of the UvrBC complex on DNA, we also studied UvrBD338N. This mutation in the helicase II motif results in an impaired ATPase, non-functional incision and the inability to form a pre-incision complex (45). For this mutant, only the α value factor was significantly different between the WT and mutant UvrBC complexes (0.71 versus 0.57: P < 0.01). All other motile parameters remained unchanged (Table 3 and Figure 5). These data suggest that binding of ATP and lack of hydrolysis cause the diffusive behavior of the UvrBC complex to be altered.
DISCUSSION
NER of DNA-distorting lesions is a highly coordinated multi-enzyme process that must proceed efficiently and accurately. The occurrence of spurious repair activity would be detrimental to the integrity of the genome by tying up valuable repair complexes and endangering the genome through gratuitous incision activity (46). At the cost of ATP, kinetic proofreading mechanisms have evolved to ensure high fidelity preventing unwanted entry into repair; however, it is unclear how the regulation of incision is achieved. The rate-limiting step in DNA repair is probably the rate at which UvrC finds pre-incision complexes given its low abundance of ∼10 copies in the cell (1,13). Therefore, understanding the physical mechanism of the search process is imperative. Here, we have used single-molecule approaches to understand the behavior of UvrC on DNA. Using direct single-molecule fluorescence imaging on a DNA tightrope, we robustly detect UvrC binding to DNA in the absence of any other factors. We also observe that salt changes UvrC’s motility on DNA. Given that UvrB is in excess over UvrC in the cell, we investigated their interaction and have directly observed, for the first time, the formation of a UvrBC complex on normal dsDNA. This UvrBC complex is highly abundant in our studies and, therefore, is likely the steady-state complex in the cell. Compared with UvrC alone at low salt, UvrBC behaves differently on DNA. We also found that mutations in the DNA binding or ATPase domain of UvrB affected the DNA interaction of the UvrBC complex. These data suggest that the UvrBC complex makes direct contact with DNA at least partly through the DNA-binding domain of UvrB. In addition, we also infer that at 50 mM KCl, ATP hydrolysis by UvrB in the UvrBC complex may cause the complex to pause during its diffusive motion.
UvrBC forms complexes on DNA
Mixing UvrB and UvrC before incubation with DNA tightropes resulted in the formation of DNA bound complexes containing UvrB. These complexes showed labeling consistent with the presence of UvrBC bound to the DNA. This surprising observation indicates that UvrB, which cannot bind dsDNA alone, is brought to DNA by UvrC. Previously, all biochemical studies have indicated that UvrB can only be brought to dsDNA by UvrA (4), now it seems a second enzyme is capable of bringing UvrB to dsDNA. The existence of a UvrBC complex has only been suggested before in solution (47) and when bound to specific DNA substrates designed to expose a double-strand–single-strand DNA junction (48). In our assays, the formation of a UvrBC–DNA complex was not rare, suggesting this is a real complex and likely exists within cells. Indeed, given the relative cellular concentrations of UvrB and UvrC and that we observe UvrBC formation in the nanomolar concentration range, it is possible that all of the available UvrC is bound to UvrB. Therefore, what is the significance of the UvrBC complex? To date there are no data suggesting that in the absence of UvrA, UvrB can form a pre-incision complex and excise an oligonucleotide containing damage in normal dsDNA (48). This indicates UvrBC is not searching for damage and the NER process cannot occur without UvrA. Therefore, another possibility could be that UvrC is using UvrB to help in its search for a pre-incision complex.
UvrC is present at a very low-copy number of ∼10 molecules in the cell (13) and is not induced by the SOS response. This raises the possibility that recruitment of UvrC to the pre-incision complex is a bottleneck in the process of DNA repair. To test this hypothesis, we studied the search mechanism of UvrBC versus UvrC. At low salt, there was a strong distinction between UvrBC and UvrC alone; a large number of UvrBC complexes diffuse on the DNA, whereas UvrC was largely static. Why approximately one in six molecules of UvrC slide on the DNA is not clear, perhaps UvrC exists in different conformations in solution, one capable of sliding and one not. UvrC is believed to make contact with DNA through its two C-terminal (HhH)2 domains (49). Crystal structure analysis of this region indicates that it is highly flexible and exists in multiple conformations (21). On binding to DNA, the free energy of interaction may trap UvrC in one of these conformations; therefore, this assay gives an approximate equilibrium picture of the conformational states of UvrC. This statement is supported by our observation that none of the sliding complexes would become static and equally once a complex binds statically it would not initiate sliding (acquisitions timescales were up to 720 s). If we assume that the motile fraction search the DNA at the diffusion constant observed in low salt, which is significantly (P < 0.001) higher for UvrC alone than for UvrBC, we can estimate the expected search times for the E. coli genome (see Supplementary Information). The predicted search time for UvrC is 42 min; however, despite the greater percentage of moving UvrBC complexes, its search time was not improved (76 min) because of its considerably slower diffusion constant (Supplementary Table S3). With no correlation between the diffusion constant of singly or doubly labeled complexes, the viscous drag of the Qdots could not have generated this difference (Supplementary Table S2).
At higher (physiological) salt concentrations, we found the diffusional properties of UvrC and UvrBC on DNA to converge. From the diffusion constants we determined the ‘roughness’ of the energy landscape to motion (see Supplementary Information); at low salt, there was considerable difference in roughness where UvrBC experienced an average ∼2.5 κT energy barrier and UvrC experienced none. At high salt, this roughness was reduced and similar for UvrBC and UvrC (0.8–1.4 κT), resulting in UvrBC and UvrC taking similar times to scan the genome of 15 and 22 min, respectively (Supplementary Table S3). This scan time is now within the expected time for bacterial cell division; further aided by the SOS response triggered arrest of the cell division cycle once damage is detected (50). Therefore, UvrBC does not apparently serve to deliver UvrC faster to the pre-incision complex.
The 1D diffusion properties of UvrC and UvrBC
Although we have established that UvrBC does not enhance the pre-incision complex search time for UvrC, we can use our observations to better understand how DNA is searched by the UvrBC complex. To investigate the mode of motion, we studied attachment times and diffusion constants across salt concentrations. Salt acts to impede electrostatic contacts between the protein and DNA; therefore, as the amount of salt is raised, the number of bound proteins should decrease (37), but what happens to the protein–DNA interface? On binding to DNA, a number of counterions are liberated from the DNA, which are energetically balanced by the contacts made with the protein. To slide, the protein must break an electrostatic contact with DNA at one end and make a new one at the other. This results in a displacement of a DNA-counterion from the leading edge of the motile protein and generation of a counterion-free interaction at the rear end. Therefore, assuming a rapid equilibrium of ions, the released counterion is balanced by another one binding, and there is no net change in binding energy (36). Across salt concentrations, therefore, there should be no effect on the diffusion coefficient if the protein complex is sliding. For UvrBC, we observe a drop in attached lifetime as the salt concentration is raised. Together with an increase in the diffusion constant this suggests hopping (36). However, because the precise UvrBC–DNA contacts are not well understood, further experiments are necessary to confirm the occurrence of hopping. It is also possible that some electrostatic contacts between the protein complex and the DNA may not lie on the backbone and, therefore, when broken are not re-made immediately impeding rather than facilitate sliding. Such contacts could be shielded by the increase in salt concentration, which would result in a decrease in the energy barrier for movement. In such a scenario, at higher salt, this contact is broken weakening the protein–DNA interaction both accounting for the reduced lifetime of attachment and the increased diffusion constant. UvrB’s β-hairpin domain may facilitate this type of interaction (see later in the text).
A comparison of the diffusion versus α plots for UvrC (Figure 2) and UvrBC (Figure 4) shows a distinctly different pattern. As salt is raised, UvrC’s α value rises toward 1, which indicates free diffusion. However, the observed diffusion versus α values is homogenously distributed about the peak value. This behavior is unlike that of UvrBC which shows a second component of the distribution at lower α, and as salt is raised, this second component disappears. One explanation for this phenomenon, made previously for DNA glycosylase fpg, is a stop and start DNA interrogation process (30). The response of UvrBC to salt suggests a more complex picture than a simple electrostatic interface as proposed for the Lac repressor (36). A better understanding of the protein–DNA interface of this UvrBC complex is necessary and may be provided by X-ray crystallography in the future. Together these data indicate that UvrC molecules pause less as salt is raised, and UvrBC behaves similarly. However, there is a sub-population of UvrBC molecules that are involved in a much more static interaction with DNA, which is reversed by salt. UvrBC’s diffusion constant increases with increasing salt concentrations, which is characteristic of hopping on DNA (35,36) (Figure 4, Table 2). This result raises the presently untested possibility that UvrB may help UvrC to overcome static obstacles on the DNA.
Protein–DNA contacts in the UvrBC complex
An important observation made in this study is that to form a UvrBC–DNA complex, pre-incubation of UvrB with UvrC is required before the introduction of DNA. If UvrC is pre-bound to DNA, the introduction of UvrB does not lead to the formation of UvrBC–DNA complex. This suggests that once bound to DNA, the UvrC domain responsible for binding UvrB (47,51) is hidden either because of a conformational change or because access to the binding site is sterically blocked. The latter could occur if this region is close to or involved with DNA binding (20). To shed light on whether UvrB is involved in contacting the DNA in the UvrBC–DNA complex, we studied a number of UvrB mutants at low salt, as the largest difference in behavior between UvrC and UvrBC was seen in these conditions. Two mutations were chosen around the DNA-binding site, Y96A and Δβ-hairpin. The hydrophobic tip of β-hairpin of UvrB is believed to insert between the strands of the DNA in the pre-incision UvrB–DNA complex and potentially during damage location as part of the UvrAB complex (24,46). In doing so the hairpin is thought to verify DNA damage, mediated in particular by a hydrophobic residue at the base of the hairpin, Tyr96, the substitution of which for alanine results in a protein incapable of discerning damage (8). In this study, we have observed that alteration of this residue affects the interaction of UvrBC with DNA, thus providing a clear indication that UvrB is contacting the DNA in the UvrBC complex. However, as UvrA is absolutely required for incision at damaged sites, it is unlikely that UvrB, as part of the UvrBC complex, is searching for damage on double-stranded DNA. It is important to note that UvrBC complex can act on a Y-substrate and that the presence of damage has been previously observed to increase incision by the UvrBC complex (18,43). The third mutant studied here was D338N UvrB; this residue lies in the helicase II motif and is indirectly involved in coordination of ATP binding (24). D338N reduces the ATPase of UvrB and traps the NER complex UvrAB–DNA preventing the formation of a pre-incision complex. This mutant provided an additional control, as this was not a direct DNA-binding site mutant and allowed us to study the role of UvrB’s ATPase. Only the α factor showed any significant difference (P < 0.05) between this mutant and WT. This difference in α factor is notable because of its trend: UvrBCWT + ATP < UvrBCWT + ADP < UvrBCWT − ATP < UvrBD338NC + ATP (only significantly different between UvrBD338N and UvrBWT). In addition, the low α component in the diffusion versus α plots (Figure 5B) is lost with the D338N UvrB mutant and wild-type UvrB in the presence of ADP or absence of nucleotide altogether (Supplementary Table S1 and Supplementary Figure S3). These observations strongly suggest that ATP hydrolysis is associated with a conformational change in UvrB that effects pausing and underlies sub-diffusive behavior.
CONCLUSIONS
In this study, we have shown that UvrC can independently form a stable DNA-bound complex. Remarkably, however, we have also observed that UvrC facilitates UvrB binding to DNA. This was previously thought to only occur through UvrB’s interaction with UvrA in a forward reaction at the beginning of NER. We have no evidence to suggest that UvrC is performing the same function as UvrA; therefore, UvrBC is not capable of detecting lesions on unaltered dsDNA. However, we have found that this complex is abundant, suggesting that given the excess of UvrB over UvrC in the cell, all UvrC are present as a UvrBC complex in vivo. UvrBC can one-dimensionally diffuse along DNA; however, this mode of motion does not accelerate the search process relative to UvrC alone, as it is limited by the number of available UvrC, and the diffusion constants for UvrC or UvrBC are not significantly different. The diffusive behavior of UvrBC is complex with respect to salt, suggesting that the protein–DNA interaction is modified in a more complex way than can be explained by Debye shielding effects. The increase in diffusion constant with increasing salt suggests DNA hopping. Thus, UvrB might facilitate UvrC movement along DNA that is littered with other DNA-binding proteins, which would otherwise prevent sliding. We have used a number of mutants to also show that UvrBC binds to DNA through UvrB’s DNA-binding domain, perhaps explaining the origin of UvrBC’s idiosyncratic behavior. Perhaps one function of the sliding/hopping UvrBC complex is to bring UvrC to a pre-incision UvrB–DNA complex and to hand over the UvrC to perform its function. Furthermore, UvrC’s endonuclease activity if left unchecked would certainly be deleterious to the genome positing, another plausible function of the UvrBC complex—to chaperone UvrC’s nuclease function. Given the significance and abundance of this newly determined complex, we hope our work will lead to further studies into the role of this complex and the determination of its high-resolution structure.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3, Supplementary Figures 1–7, Supplementary Methods, Supplementary Movies 1–4 and Supplementary References [52–56].
FUNDING
BBSRC [BB/I003460/1 to N.M.K. and M.S.]; National Institutes of Health [1R01ES019566 to B.V.H. and 4R00ES016758 to H.W]. Funding for open access charge: BBSRC.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Robert Keller for his help in the early part of this project and Merle Nazareth for her technical assistance. They also thank the whole of the Kad laboratory for useful discussions.
REFERENCES
- 1.Van Houten B. Nucleotide excision repair in Escherichia coli. Microbiol. Rev. 1990;54:18–51. doi: 10.1128/mr.54.1.18-51.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sancar A. DNA excision repair. Annu. Rev. Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 3.Truglio JJ, Croteau DL, Van Houten B, Kisker C. Prokaryotic nucleotide excision repair: the UvrABC system. Chem. Rev. 2006;106:233–252. doi: 10.1021/cr040471u. [DOI] [PubMed] [Google Scholar]
- 4.Van Houten B, Croteau DL, DellaVecchia MJ, Wang H, Kisker C. ‘Close-fitting sleeves': DNA damage recognition by the UvrABC nuclease system. Mutat. Res. 2005;577:92–117. doi: 10.1016/j.mrfmmm.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Peng Y, Wang H, Santana-Santos L, Kisker C, Van Houten B. In: Chemical Carcinogenesis. Penning TM, editor. New York: Springer Science + Business Media; 2011. pp. 267–296. [Google Scholar]
- 6.Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Mol. Cell. 2010;37:702–713. doi: 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goosen N, Moolenaar GF. Repair of UV damage in bacteria. DNA Repair. 2008;7:353–379. doi: 10.1016/j.dnarep.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 8.DellaVecchia MJ, Croteau DL, Skorvaga M, Dezhurov SV, Lavrik OI, Van Houten B. Analyzing the handoff of DNA from UvrA to UvrB utilizing DNA-protein photoaffinity labeling. J. Biol. Chem. 2004;279:45245–45256. doi: 10.1074/jbc.M408659200. [DOI] [PubMed] [Google Scholar]
- 9.Visse R, King A, Moolenaar GF, Goosen N, van de Putte P. Protein-DNA interactions and alterations in the DNA structure upon UvrB-DNA preincision complex formation during nucleotide excision repair in Escherichia coli. Biochemistry. 1994;33:9881–9888. doi: 10.1021/bi00199a009. [DOI] [PubMed] [Google Scholar]
- 10.Orren DK, Sancar A. The (A)BC excinuclease of Escherichia coli has only the UvrB and UvrC subunits in the incision complex. Proc. Natl Acad. Sci. USA. 1989;86:5237–5241. doi: 10.1073/pnas.86.14.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orren DK, Sancar A. Formation and enzymatic properties of the UvrB.DNA complex. J. Biol. Chem. 1990;265:15796–15803. [PubMed] [Google Scholar]
- 12.Verhoeven EE, van Kesteren M, Moolenaar GF, Visse R, Goosen N. Catalytic sites for 3′ and 5′ incision of Escherichia coli nucleotide excision repair are both located in UvrC. J. Biol. Chem. 2000;275:5120–5123. doi: 10.1074/jbc.275.7.5120. [DOI] [PubMed] [Google Scholar]
- 13.Yoakum GH, Grossman L. Identification of E. coli uvrC protein. Nature. 1981;292:171–173. doi: 10.1038/292171a0. [DOI] [PubMed] [Google Scholar]
- 14.Sancar A, Rupp WD. A novel repair enzyme: UVRABC excision nuclease of Escherichia coli cuts a DNA strand on both sides of the damaged region. Cell. 1983;33:249–260. doi: 10.1016/0092-8674(83)90354-9. [DOI] [PubMed] [Google Scholar]
- 15.Caron PR, Kushner SR, Grossman L. Involvement of helicase II (uvrD gene product) and DNA polymerase I in excision mediated by the uvrABC protein complex. Proc. Natl Acad. Sci. USA. 1985;82:4925–4929. doi: 10.1073/pnas.82.15.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husain I, Van Houten B, Thomas DC, Abdel-Monem M, Sancar A. Effect of DNA polymerase I and DNA helicase II on the turnover rate of UvrABC excision nuclease. Proc. Natl Acad. Sci. USA. 1985;82:6774–6778. doi: 10.1073/pnas.82.20.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowley DJ, Hanawalt PC. The SOS-dependent upregulation of uvrD is not required for efficient nucleotide excision repair of ultraviolet light induced DNA photoproducts in Escherichia coli. Mutat. Res. 2001;485:319–329. doi: 10.1016/s0921-8777(01)00068-4. [DOI] [PubMed] [Google Scholar]
- 18.Moolenaar GF, Bazuine M, van Knippenberg IC, Visse R, Goosen N. Characterization of the Escherichia coli damage-independent UvrBC endonuclease activity. J. Biol. Chem. 1998;273:34896–34903. doi: 10.1074/jbc.273.52.34896. [DOI] [PubMed] [Google Scholar]
- 19.Lin JJ, Sancar A. Active site of (A)BC excinuclease. I. Evidence for 5' incision by UvrC through a catalytic site involving Asp399, Asp438, Asp466, and His538 residues. J. Biol. Chem. 1992;267:17688–17692. [PubMed] [Google Scholar]
- 20.Truglio JJ, Rhau B, Croteau DL, Wang L, Skorvaga M, Karakas E, DellaVecchia MJ, Wang H, Van Houten B, Kisker C. Structural insights into the first incision reaction during nucleotide excision repair. EMBO J. 2005;24:885–894. doi: 10.1038/sj.emboj.7600568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karakas E, Truglio JJ, Croteau D, Rhau B, Wang L, Van Houten B, Kisker C. Structure of the C-terminal half of UvrC reveals an RNase H endonuclease domain with an Argonaute-like catalytic triad. EMBO J. 2007;26:613–622. doi: 10.1038/sj.emboj.7601497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Lu M, Tang MS, Van Houten B, Ross JB, Weinfeld M, Le XC. DNA wrapping is required for DNA damage recognition in the Escherichia coli DNA nucleotide excision repair pathway. Proc. Natl Acad. Sci. USA. 2009;106:12849–12854. doi: 10.1073/pnas.0902281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, DellaVecchia MJ, Skorvaga M, Croteau DL, Erie DA, Van Houten B. UvrB domain 4, an autoinhibitory gate for regulation of DNA binding and ATPase activity. J. Biol. Chem. 2006;281:15227–15237. doi: 10.1074/jbc.M601476200. [DOI] [PubMed] [Google Scholar]
- 24.Theis K, Chen PJ, Skorvaga M, Van Houten B, Kisker C. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999;18:6899–6907. doi: 10.1093/emboj/18.24.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skorvaga M, DellaVecchia MJ, Croteau DL, Theis K, Truglio JJ, Mandavilli BS, Kisker C, Van Houten B. Identification of residues within UvrB that are important for efficient DNA binding and damage processing. J. Biol. Chem. 2004;279:51574–51580. doi: 10.1074/jbc.M409266200. [DOI] [PubMed] [Google Scholar]
- 26.Machius M, Henry L, Palnitkar M, Deisenhofer J. Crystal structure of the DNA nucleotide excision repair enzyme UvrB from Thermus thermophilus. Proc. Natl Acad. Sci. USA. 1999;96:11717–11722. doi: 10.1073/pnas.96.21.11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Truglio JJ, Karakas E, Rhau B, Wang H, DellaVecchia MJ, Van Houten B, Kisker C. Structural basis for DNA recognition and processing by UvrB. Nat. Struct. Mol. Biol. 2006;13:360–364. doi: 10.1038/nsmb1072. [DOI] [PubMed] [Google Scholar]
- 28.Verhoeven EE, Wyman C, Moolenaar GF, Hoeijmakers JH, Goosen N. Architecture of nucleotide excision repair complexes: DNA is wrapped by UvrB before and after damage recognition. EMBO J. 2001;20:601–611. doi: 10.1093/emboj/20.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kad NM, Van Houten B. Dynamics of lesion processing by bacterial nucleotide excision repair proteins. Prog. Mol. Biol. Transl. Sci. 2012;110:1–24. doi: 10.1016/B978-0-12-387665-2.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn AR, Kad NM, Nelson SR, Warshaw DM, Wallace SS. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011;39:7487–7498. doi: 10.1093/nar/gkr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl Acad. Sci. USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harada Y, Funatsu T, Murakami K, Nonoyama Y, Ishihama A, Yanagida T. Single-molecule imaging of RNA polymerase-DNA interactions in real time. Biophys. J. 1999;76:709–715. doi: 10.1016/S0006-3495(99)77237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorman J, Plys AJ, Visnapuu ML, Alani E, Greene EC. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat. Struct. Mol. Biol. 2010;17:932–938. doi: 10.1038/nsmb.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finkelstein IJ, Visnapuu ML, Greene EC. Single-molecule imaging reveals mechanisms of protein disruption by a DNA translocase. Nature. 2010;468:983–987. doi: 10.1038/nature09561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tafvizi A, Huang F, Leith JS, Fersht AR, Mirny LA, van Oijen AM. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys. J. 2008;95:L01–L03. doi: 10.1529/biophysj.108.134122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 37.von Hippel PH, Berg OG. Facilitated target location in biological systems. J. Biol. Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 38.Croteau DL, DellaVecchia MJ, Wang H, Bienstock RJ, Melton MA, Van Houten B. The C-terminal zinc finger of UvrA does not bind DNA directly but regulates damage-specific DNA binding. J. Biol. Chem. 2006;281:26370–26381. doi: 10.1074/jbc.M603093200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Tessmer I, Croteau DL, Erie DA, Van Houten B. Functional characterization and atomic force microscopy of a DNA repair protein conjugated to a quantum dot. Nano Letts. 2008;8:1631–1637. doi: 10.1021/nl080316l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hohng S, Ha T. Near-complete suppression of quantum dot blinking in ambient conditions. J. Am. Chem. Soc. 2004;126:1324–1325. doi: 10.1021/ja039686w. [DOI] [PubMed] [Google Scholar]
- 41.Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saxton MJ. Anomalous subdiffusion in fluorescence photobleaching recovery: a Monte Carlo study. Biophys. J. 2001;81:2226–2240. doi: 10.1016/S0006-3495(01)75870-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou Y, Walker R, Bassett H, Geacintov NE, Van Houten B. Formation of DNA repair intermediates and incision by the ATP-dependent UvrB-UvrC endonuclease. J. Biol. Chem. 1997;272:4820–4827. doi: 10.1074/jbc.272.8.4820. [DOI] [PubMed] [Google Scholar]
- 44.Skorvaga M, Theis K, Mandavilli BS, Kisker C, Van Houten B. The beta -hairpin motif of UvrB is essential for DNA binding, damage processing, and UvrC-mediated incisions. J. Biol. Chem. 2002;277:1553–1559. doi: 10.1074/jbc.M108847200. [DOI] [PubMed] [Google Scholar]
- 45.Bienstock RJ, Skorvaga M, Mandavilli BS, Van Houten B. Structural and functional characterization of the human DNA repair helicase XPD by comparative molecular modeling and site-directed mutagenesis of the bacterial repair protein UvrB. J. Biol. Chem. 2003;278:5309–5316. doi: 10.1074/jbc.M210159200. [DOI] [PubMed] [Google Scholar]
- 46.Moolenaar GF, Hoglund L, Goosen N. Clue to damage recognition by UvrB: residues in the beta-hairpin structure prevent binding to non-damaged DNA. EMBO J. 2001;20:6140–6149. doi: 10.1093/emboj/20.21.6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu DS, Kim ST, Sun Q, Sancar A. Structure and function of the UvrB protein. J. Biol. Chem. 1995;270:8319–8327. doi: 10.1074/jbc.270.14.8319. [DOI] [PubMed] [Google Scholar]
- 48.Zou Y, Van Houten B. Strand opening by the UvrA(2)B complex allows dynamic recognition of DNA damage. EMBO J. 1999;18:4889–4901. doi: 10.1093/emboj/18.17.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh S, Folkers GE, Bonvin AM, Boelens R, Wechselberger R, Niztayev A, Kaptein R. Solution structure and DNA-binding properties of the C-terminal domain of UvrC from E.coli. EMBO J. 2002;21:6257–6266. doi: 10.1093/emboj/cdf627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huisman O, D'Ari R, Gottesman S. Cell-division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation. Proc. Natl Acad. Sci. USA. 1984;81:4490–4494. doi: 10.1073/pnas.81.14.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yildiz A, Selvin PR. Fluorescence imaging with one nanometer accuracy: application to molecular motors. Acc. Chem. Res. 2005;38:574–582. doi: 10.1021/ar040136s. [DOI] [PubMed] [Google Scholar]
- 53.Hughes BD. Random Walks and Random Environment. Oxford: Clarendon Press; 1995. [Google Scholar]
- 54.Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schurr JM. The one-dimensional diffusion coefficient of proteins absorbed on DNA. Hydrodynamic considerations. Biophys. Chem. 1979;9:413–414. [PubMed] [Google Scholar]
- 56.Bagchi B, Blainey PC, Xie XS. Diffusion constant of a nonspecifically bound protein undergoing curvilinear motion along DNA. J. Phys. Chem. B. 2008;112:6282–6284. doi: 10.1021/jp077568f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.