

Abstract
The formation of reactive oxygen species by the cytochrome P450 monoxygenase system is thought to be due to autooxidation of NADPH-cytochrome P450 reductase and the non-productive decay of oxygen-bound cytochrome P450 intermediates. To characterize this process in recombinant microsomal enzymes, we used a highly sensitive hydrogen peroxide assay based on Amplex-Red oxidation. This assay is 20 times more sensitive (LLD = 5.0 pmoles/assay, and LLQ = 30 pmoles/assay) than the standard ferrous thiocyanate assay for detection of hydrogen peroxide. We found low, but detectable spontaneous generation of hydrogen peroxide by recombinant human NADPH-cytochrome P450 reductase complexes (0.034 nmoles hydrogen peroxide/min/100 Units of NADPH-cytochrome P450 reductase). Significantly higher rates of hydrogen peroxide production were observed when recombinant cytochrome P450 enzymes were coexpressed with NADPH-cytochrome P450 reductase (0.31 nmoles of hydrogen peroxide/min/100 Units of NADPH-cytochrome P450 reductase). This was independent of the addition of any exogenous cytochrome P450 substrates. These data demonstrate that cytochrome P450’s are a major source of hydrogen peroxide in the recombinant cytochrome P450 monooxygenase system. Moreover, substrate binding is not required for the cytochrome P450’s to generate reactive oxygen species.
Keywords: Cytochrome P450, NADPH cytochrome P450 reductase, hydrogen peroxide
Introduction
Cytochrome P450-dependent monooxygenases are localized in the endoplasmic reticulum, and are the primary intracellular enzymes mediating the metabolism of xenobiotics, as well as physiologically important endogenous compounds [1,2]. This system consists of NADPH-cytochrome P450 reductase (EC 1.6.2.4), and various forms of cytochrome P450, an important class of heme containing enzymes [3]. Metabolism involves electron transfer from NADPH-cytochrome P450 reductase to the P450’s leading to the formation of reduced P450’s which then bind oxygen and, in a multi-step process, generate iron oxygen complexes or “active” oxygen species [4,5]. One of the unique attributes of this oxygen activation process is a low efficiency, “uncoupling”, due to the non-productive decay of some oxygen-bound cytochrome P450 intermediates [6,7]. During this process, superoxide anion and/or H2O2 are formed as products of microsomal electron transport chain activity. It is generally thought that the microsomal enzymes are one of largest sources of intracellular H2O2 [7–9]; however, the mechanisms or factors and relative contribution of the different members of the P450 system leading to “uncoupling” and H2O2 production are still poorly understood.
The cytochrome P450 system is often studied using recombinant forms of the enzymes expressed in insect cells [10, 11]. However, little is known about the ability of these preparations to undergo “uncoupling” and generate H2O2. In the present studies we used an ultra sensitive Amplex Red/Horseradish peroxidase (AR/HRP)-based fluorescence assay to compare H2O2 formation by native microsomal preparations and microsomal preparations containing recombinant cytochrome P450 isoenzymes and NADPH-cytochrome P450 reductase. Using this system, we found that in the absence of detectable cytochrome P450’s, human NADPH-cytochrome P450 reductase was capable of generating H2O2, albeit at very low rates. This activity increased significantly in mixtures of human cytochrome P450’s coexpressed with NADPH-cytochrome P450 reductase. These data demonstrate that cytochrome P450’s are a significant source of intracellular H2O2. Further studies using this system will be useful for determining the relative activities and contributions of different microsomal enzymes to intracellular H2O2 production.
Materials and Methods
Chemicals and reagents
Type 1 HRP (cat. no. P-8125), catalase (from Aspergillus niger as an ammonium sulfate suspension, cat. No. C3515), superoxide dismutase (SOD) (from bovine liver, cat no S2515), NADPH, sodium azide, dimethyl sulfoxide, diethylenetriaminepentaacetic acid (DETAPAC), and 30% H2O2 were from Sigma-Aldrich (St. Louis, MO). AR was from Molecular Probes (Eugene, OR) and was prepared in oxygen-free dimethyl sulfoxide as a 10 mM stock solution. Human liver microsomes and liver microsomes from male SD rats were obtained from BD Gentest (Woburn, MA) and Xenotech (Lenexa, KS). Recombinant human microsomal enzymes (Supersomes™) as microsomal fractions of insect cells infected with baculovirus including control (cat no 456201), human NADPH-cytochrome P450 reductase coexpressed with cytochrome b5 (cat. no. 456244), and Supermix™ (cat no 456406), which consists of human NADPH-cytochrome P450 reductase and cytochrome b5 coexpressed with human CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 were obtained from BD Gentest (Woburn, MA). Activities of the components in the Supermix™ were similar to their counterparts in human liver microsomes. The storage, preparation of stock suspensions, handling, and general characteristics of the enzyme preparations were provided by the manufacturer. All other chemicals and water used in this study were of the highest available purity and were used without additional treatments.
AR/HRP method for analyzing H2O2
In this assay, HRP interacts with H2O2 forming Compound I. Oxidation of AR by Compound I results in the formation of Compound II and an AR radical intermediate. Subsequent reaction of this radical with Compound II results in the formation of HRP and resorufin, a highly fluorescent product that can readily be quantified using a fluorometer [12, 13]. Under conditions where the molar ratio of AR to H2O2 is greater than five, resorufin is generated in a 1:1 stoichiometry with H2O2 [12–14]. Since the determination of H2O2 by the AR/HRP method is not direct, relative fluorescence units (RFU) need to be extrapolated to amounts of H2O2 produced using appropriate calibration standards. To prepare these standards, H2O2 was diluted with water to a concentration of 1.6 mM. This solution was further diluted to a 16 μM H2O2 solution with potassium phosphate buffer (50 mM, pH 7.7, containing 0.5 mM diethylenetriaminepentaacetic acid). Serial two-fold dilutions of this solution were used to prepare calibration standards. Samples without H2O2 were used as blanks. Fifty μl of blank and each calibration standard were mixed with 50 μl of the phosphate buffer supplemented with components required to maximize the enzymatic generation of H2O2 including 50 mM potassium phosphate buffer, pH 7.7, 0.1 mM NADPH, and an NADPH regenerating system consisting of 10 mM glucose-6-phosphate and 0.5 Units/ml glucose-6-phosphate dehydrogenase, 0.5 mM diethylenetriaminepentaacetic acid, 1 mM sodium azide, and 500 Units/ml SOD in a final reaction volume of 100 μl. The H2O2 concentrations in the calibration standards prior to the addition of the AR/HRP mix were 0 μM, 0.0625 μM, 0.125 μM, 0.25 μM, 0.5 μM, 1 μM, 2 μM, 4 μM, and 8 μM. The samples were preincubated at 37°C for 5 min in black, flat bottom 96 well plates. Fifty μl of the AR/HRP mix (25 μM AR, final concentration, and 0.1 units of HRP, in 50 mM potassium phosphate buffer, pH 7.7) were then added to each well using a multichannel pipetter. Fluorescence of resorufin (ex 530 nm/em 587 nm) was recorded after approximately 45 sec and then every 1 min using a Molecular Devices SpectraMax M5/M5e microplate reader controlled by PC SoftMax Pro 5 software (Sunnyvale, CA). Under these conditions, the appearance of resorufin fluorescence related to H2O2 formation is very rapid; maximal fluorescence was obtained in less than 45 sec at all standard concentrations of H2O2 tested (Fig. 1A).
Figure 1. Fluorescence analysis of H2O2 standards using the AR/HRP assay.

Panel A. Reactions were initiated by the addition of AR/HRP (arrow). Fluorescence of resorufin was recorded ≈45 sec later. In this calibration experiment, each point represents the mean ± SD (n = 3). The lowest curve represents background and temporal fluorescence changes of blank samples (in the absence of H2O2). In most cases, the error bars were less than 5% and fell within the symbols.
Panel B. Comparison of the AR/HRP assay and ferrous-thiocyanate assay for analyzing H2O2 standards. Upper panels. Analysis of H2O2 using the AR/HRP assay. The curve represents the best fit of the average of relative fluorescence units (RFU) for each H2O2 standard obtained from three replicates assayed on 6 different days. Each point and error bar on the curve represent the mean ± SD (n = 18). The inset shows the calibration curve at low H2O2 concentrations (from 0.0 to 100 pmoles/well). The dashed line on the Y-axis indicates the mean +10 SD’s of blank samples and the dashed line on the x-axis indicates the LLQ value which is 12 pmoles of H2O2/well. Lower panels. Analysis of H2O2 in the ferrous thiocyanate assay. Each point on the curve represents the mean ± SD of H2O2 standards obtained with three replicates assayed on 4 days. OD values were adjusted automatically to 1 cm on the microplate reader using the path check option. The inset shows the calibration curve at low H2O2 concentrations (from 0.0 to 1500 pmoles/well). The dashed line on the Y-axis indicates the mean +10 SD’s of blank samples and the dashed line on the x-axis indicates the LLQ value which is 280 pmoles of H2O2/well.
With the exception of NADPH, none of the components required for optimal activity interfered with the H2O2 assay. NADPH increased background fluorescence, recorded as a low, but detectable, time-dependent increase in fluorescence with no enzyme additions in reaction mixes. This enzyme-independent oxidation of AR was not prevented by SOD, but was abolished by the addition of exogenous catalase (not shown). Therefore, for extrapolation of RFU into absolute amounts of H2O2, the NADPH-dependent background fluorescence was subtracted.
It should be noted that microsomal preparations may also contain P450-related peroxidase or monooxygenase activities which could potentially mediate the metabolism of AR into resorufin in the absence of HRP. However, if HRP was omitted from reaction mixes, we found only background fluorescence over a 30 min incubation period.
Ferrous-thiocyanate method for analyzing H2O2
In the ferrous thiocyanate assay, H2O2 oxidizes ferrous ion to ferric ion. Subsequent reaction of ferric ion with thiocyanate ion under strongly acidic conditions results in a ferric iron-thiocyanate red-colored complex which can be quantified spectrophotometrically [15, 16]. The ferrous thiocyanate assay for H2O2 was conducted as described by Hildebrandt et al. [16] and modified for use in clear flat bottom 96 well plates. H2O2 standards in the range of 1.0 μM to 100 μM were prepared as described above. After loading of 100 μl of H2O2 standards and blank samples into each well of the plate, 50 μl of concentrated HCl was added. This was followed by the additions of 20 μl ferrous ammonium sulfate (5 mM final concentration) and 30 μl potassium thiocyanate (200 mM final concentration). After 10 min incubation at room temperature, plates were analyzed using a microplate reader by monitoring changes in absorption at 483 nm. Since ferrous ammonium sulfate in solution was found to be highly unstable, it was routinely prepared fresh ≈ 10 min prior to use.
Comparison of methods for assaying H2O2
In initial studies, we compared the AR/HRP and ferrous-thiocyanate methods for detection of H2O2. In both assays, H2O2 could readily be detected (Fig. 1B–E). Components of incubation mixes including diethylenetriaminepentaacetic acid, an iron chelator, sodium azide, an inhibitor of catalase, and SOD, did not interfere with the assays. In contrast, NADPH increased background fluorescence by approximately 2–5% in the AR/HRP assay, but not in the ferrous thiocyanate assay. However, this effect did not alter analysis of the calibration standards. The AR/HRP assay was found to be significantly more sensitive for detecting H2O2. The LLD (mean of background values + 3 SD’s) of H2O2 with AR/HRP was estimated to be 5 pmoles/assay, while the LLQ (mean of background values + 10 SD’s) was 30 pmoles/assay. By comparison, the LLD and LLQ values using the ferrous thiocyanate method were 80 and 600 pmoles of H2O2/assay, respectively.
H2O2 generated by enzymatic reactions
As indicated above, the incubation buffer for microsomal enzyme activities contained components required to maximize the enzymatic generation of H2O2. In this assay, 90 μl of this incubation mix was loaded into individual wells of a 96 well plate which was then preheated at 37°C for 3 min. The H2O2 generating reaction was initiated by the addition of 10 μl microsomal proteins (0.5–20 μg/well). Changes in fluorescence were recorded in either a “continuous” mode where AR/HRP was added to in the incubation mix prior to the microsomes, or a “fixed” time point mode where AR/HRP was added at selected time points after the addition of microsomes (see details in Figure legends). Only linear phases of the reactions (typically between 4 and 15 min after the initiation of the reaction) were used to calculate the absolute rates of H2O2 production.
Data collection and treatment
RFU or OD values for the H2O2 calibration standards were presented as the means ± SD. Standard curves were fitted to a line by regression analysis using GraphPad Prism, Version 5.0 software (GraphPad, La Jolla, CA). For both the AR/HRP and the ferrous thiocyanate assays, the lowest limit of detection (LLD) and lowest limit of quantification (LLQ) were calculated as the amount of H2O2 (in pmoles/sample) equal to the mean + 3 SD’s and the mean + 10 SD’s of background values, respectively (n=18) [17]. For the AR/HRP assay, linearity was observed at concentrations of H2O2 ranging from 0.0 to 4.0 μM (at r2 = 0.995). For the ferrous thiocyanate assay, changes in OD were linear for the entire range of H2O2 concentrations tested.
Results
Quantification of H2O2 generation by microsomal enzymes using the AR/HRP assay
The enzymatic generation of H2O2 in native rat and human liver microsomal enzymes was quantified using the AR/HRP assay. In these studies, H2O2 generation was monitored by recording changes in resorufin fluorescence continuously and at fixed time points after the initiation of the assay (Fig. 2). The reaction kinetics of H2O2 formation was linear for at least 10 min in both assay modes, and the rates of H2O2 generation were proportional to amounts of microsomal protein. Moreover, as little as 2 μg/well of microsomal protein generated H2O2 at a rate that was more than 10-fold above background (i.e., with no enzyme present). Absolute rates of H2O2 generation calculated for microsomal preparations from rat and human liver microsomes are shown in Table 1. These values are generally similar to those previously reported [15, 19, 20]. It should be noted that the generation of H2O2 by the liver microsomal preparations was inhibited by catalase (1000 Units/ml), and by DPI (20 μM), an inhibitor of flavin-containing enzymes (not shown).
Figure 2. Kinetics of H2O2 generation by native microsomes.
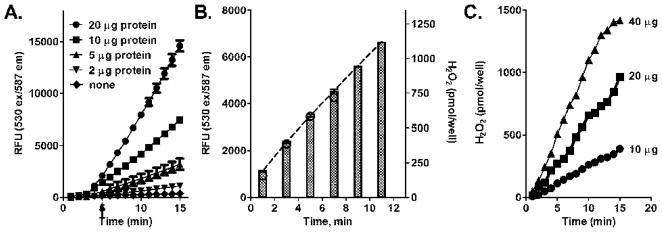
Panels A and C. Continuous recordings of H2O2 generation by rat and human liver microsomes. Each well on a 96 well plate was loaded with 90 μl of incubation media containing: potassium phosphate buffer, 50 mM, pH 7.7, 1 mM DETAPAC, 1 mM azide, 500 Units/ml SOD, 0.1 mM NADPH, and an NADPH regenerating system, (10.0 mM glucose-6-phosphate and 0.5 Units/ml glucose-6- phosphate dehydrogenase). The plate was preincubated at 37°C for 5 min in the plate reader chamber. The wells were then loaded with 50 μl of AR/HRP. The reactions were started by the addition of 10 μl microsomal preparations using a multichannel pipette (as indicated by arrow). The lines represent time-dependent changes of fluorescence generated with different amounts of microsomal protein (as indicated). The data for rats are the mean ± SD of two experiments and humans, are the mean ± SD of three experiments. Each microsomal protein concentration was done in triplicate. Panel B. H2O2 generation by rat liver microsomes assayed at fixed time points. Mixes were prepared as indicated above; reactions were initiated by the addition of 10 μg of microsomal protein. In this mode of assay, the 50 μl of AR/HRP mixture were added to the wells at the indicated time points. The bars reflect the fluorescence intensity recorded at those time points. The dashed line connecting the top of the bars was generated by GraphPad Prism and used for calculating the absolute rates of H2O2 production.
Table 1.
Rates of H2O2 generation in different microsomal preparations as determined by AR/HRP in the end point mode assay.
| Rate (nmoles of H2O2/min)1 | |||
|---|---|---|---|
| per mg of protein | per 100 Units of NADPH cytochrome P450 reductase activity2 | per nmole cytochrome P450 | |
| Liver microsomes from control rats | 2.62 ± 0.19 | 0.99 ± 0.074 | 2.86 ± 0.21 |
| Recombinant human NADPH cytochrome P450 reductase | 0.91 ± 0.22 | 0.04 ± 0.01 | N/A3 |
| Supermix (recombinant NADPH-cytochrome P450 reductase plus mixture of recombinant cytochrome P450’s) | 3.78 ± 0.30 | 0.31 ± 0.13 | 12.67 ± 1.98 |
The rates of H2O2 generation by recombinant NADPH cytochrome P450 reductase are presented for 40 Units of the enzyme/sample. The rates for the Supermix microsomal enzymes are presented for 15 pmoles of the cytochrome P450’s and 10 Units of NADPH cytochrome P450 reductase activity/sample. The samples were incubated for 30 min at 37°C and AR/HRP mix was added every 5 min. The RFU values recorded after 45 sec were taken for calculations. Data (n = 3 ± SD) were calculated using the calibration curves shown in Fig 1.
One unit of NADPH cytochrome P450 reductase activity is defined as the reduction of 1.0 nmole of cytochrome c per minute at pH 7.7 at 30°C.
not applicable.
Quantification of H2O2 generation by recombinant human microsomal enzymes (Supersomes) using the AR/HRP assay
In our next series of experiments we assayed H2O2 using recombinant human NADPH-cytochrome P450 reductase coexpressed with cytochrome P450 enzymes typically found in native human liver microsomes. As observed with the liver microsomal enzymes, the reaction kinetics for H2O2 production using the recombinant enzymes was linear for at least 10 min in both the “continuous” and “fixed” mode assay conditions; the rates of H2O2 generation were also proportional to amount of protein of recombinant microsomal enzymes (Figs. 3A and B). The absolute rates (expressed as nmole/min/mg protein) of H2O2 generated by the recombinant enzymes containing cytochrome P450’s were generally similar to the activity of human and rat liver microsomes (Table 1).
Figure 3. Kinetics of H2O2 generation by recombinant human microsomal enzymes.

Assays mixes were prepared as described in the legend to Figure 2. Experiments were performed with a mixture of human drug metabolizing cytochrome P450’s coexpressed with human NADPH-cytochrome P450 reductase. Panel A. Continuous recording of H2O2 generation by human microsomal enzymes. Each well contained 10 μg/well (12 Units of NADPH-cytochrome P450 reductase, 1.5 pmol cytochrome P450) of protein. The data are the mean ± SD of three separate experiments in duplicate. Panel B. H2O2 generation by recombinant human microsomal enzymes at fixed time points. The data are the mean ± SD of two experiments with each microsomal protein concentration analyzed in triplicate. The dashed lines connecting the tops of the bars were generated by GraphPad Prism and were used for calculation of absolute rates of H2O2 production.
It is well established that NADPH-cytochrome P450 reductase by itself is a source of H2O2 formed via the production of superoxide anion [21–25]. This reaction is markedly increased in the presence of redox cycling chemicals [24]. We next assayed H2O2 formation via human recombinant NADPH-cytochrome P450 reductase in the absence of cytochrome P450’s. Unexpectedly, in the “continuous” detection mode, increases in resorufin fluorescence were found to be exponential, thus limiting our ability to calculate absolute rates of H2O2 production (Fig. 4A). In contrast, when H2O2 formation was assayed at “fixed” time points, amounts formed were linear with respect to time (Fig. 4B). In both assay modes, increases in fluorescence were abolished by the inclusion of catalase in the incubation mixes (not shown). Previous studies have shown that resorufin can redox cycle with NADPH-cytochrome P450 reductase [26, 27], a process that can generate H2O2, and this is likely to occur in our “continuous” mode assay with recombinant NADPH-cytochrome P450 reductase. To minimize resorufin redox cycling, we used paraquat (50 μM), which can compete with resorufin for redox cycling. Paraquat readily suppressed the exponential increases in H2O2 production detected using the “continuous” mode AR/HRP assay (Fig. 4C). In the absence of resorufin redox cycling, the reaction was linear and the absolute rates of H2O2 generation measured in “continuous” and “fixed” time points modes of the assay were nearly identical. As shown in Table 1, H2O2 production per Unit of recombinant NADPH-cytochrome P450 reductase was markedly lower when compared with preparations containing the same activity of NADPH-cytochrome P450 reductase coexpressed with human cytochrome P450’s.
Figure 4. Kinetics of H2O2 generation by recombinant recombinant human NADPH-cytochrome P450 reductase.
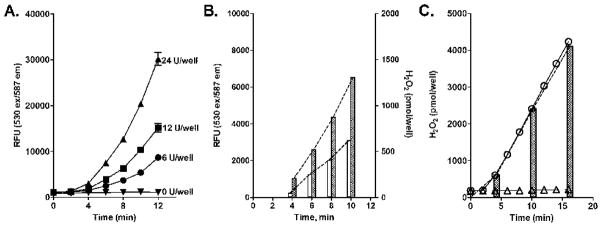
Assay mixes were prepared as described in the legend to Figure 3. Panel A. Continuous recording of H2O2 generation. The amounts of microsomal protein are indicated as Units of NADPH-cytochrome P450 reductase activity in cytochrome c reduction. The data are the mean ± SD of four experiments with each microsomal protein concentration analyzed in duplicate. Panel B. H2O2 generation by human NADPH-cytochrome P450 reductase assayed at fixed time points. The amounts of microsomal protein added were: open bars - 6 Units of NADPH-cytochrome P450 reductase/well; dotted bars - 12 Units of NADPH-cytochrome P450 reductase/well. The data are the mean ± SD of four separate experiments performed in duplicate. The dashed lines connected tops of the bars were generated by GraphPad Prism and used for calculation of absolute rates of H2O2 production. Panel C. Assay of H2O2 generation in the presence of 50 μM paraquat in continuous (circles) and fixed time points mode (black bars) assays. The amounts of microsomal protein added to the wells were 0.2 Units of NADPH-cytochrome P450 reductase/well. Bottom line (triangle) indicates the level of background fluorescence in wells with no proteins added. The data are the mean ± SD of two separate experiments performed in triplicate.
Discussion
In mammalian cells, the microsomal P450-related electron-transport system is not only a major site of xenobiotic transformation, but also an important source of reactive oxygen species, in particular, H2O2. The generation of ROS via microsomal P450 has been referred to as “uncoupling” or nonproductive activation of oxygen [4–6, 28]. Although this activity is well established, the relative contributions of cytochrome P450 enzymes and other components of the microsomal electron transport system on ROS production are unknown. Efforts at examining this have been hampered by a lack of sensitive methods available to detect H2O2. In the present studies, we describe the use of a highly sensitive assay using AR/HRP for analysis of H2O2 production by different components of the cytochrome P450 system. Previous studies have mainly used the ferrous-thiocyanate method to analyze H2O2 produced by microsomal enzymes [15,16,20]. However, this assay has low sensitivity for H2O2 due to low molar absorbtivity of the final product, a ferric-thiocyanate complex. The AR/HRP assay described in the present studies was at least 20 times more sensitive than the ferrous-thiocyanate method and could easily be modified for use in kinetic microplate readers. Using H2O2 standards, we found that the appearance of fluorescence was rapid and stable for at least 30 min under conditions that are optimal for many microsomal metabolic reactions. This allowed for the continuous monitoring of H2O2 generated during enzyme reactions, as well as detection of accumulated H2O2 at fixed time points. Another advantage of the AR/HRP assay is that resorufin, the product of the assay, is intensely fluorescent at physiological pH and thus, relatively easy to detect. This is in contrast to many other aromatic substrates for HRP which have fluorescence optimums at pH 10–11, and require strong alkaline buffers for detection of product [30,31]. Because the ferrous thiocyanate assay requires the addition of strong acids to solubilize iron salts, this assay can also only be used at fixed time points. Assays using “fixed time” points are required when there are interfering components in the reaction mix [32,33]. For example, NADPH, which is required for microsomal electron transport, can generate H2O2 non-enzymatically [18]. In this regard, we found that the addition of NADPH alone, or in combination with an NADPH regenerating system, in our AR/HRP assay resulted in increased fluorescence in the absence of added microsomal enzymes. However, this increase was relatively slow and represented less than 2–5% of the resorufin fluorescence detected in the presence microsomal enzymes. Using our modified AR/HRP assay, we confirmed that native microsomes generate H2O2. With relatively low amounts of microsomal proteins (2.0–15.0 μg/assay), we obtained absolute rates of H2O2 production by rat liver microsomes of 4 nmoles H2O2/min/mg protein, a value that is close to the rates previously reported for comparable microsomal preparations [15,19,20]. Similar rates of H2O2 production were obtained when the assay was performed in either a “continuous” or “fixed” time point mode, indicating that there was little or no interference from components of the assay mix.
It is generally thought that there are two components of the microsomal cytochrome P450-related electron transport system that generate reactive oxygen species: NADPH-cytochrome P450 reductase and cytochrome P450 enzymes. NADPH-dependent superoxide anion production by purified NADPH-cytochrome P450 reductase has been demonstrated using several methods including oxidation of epinephrine to adrenochrome [22], SOD inhibitable reduction of acetylated cytochrome c [28], and radical signals from spin-trapping probes [23]. This superoxide anion generation has been attributed to the slow autooxidation of semi-reduced flavins in NADPH-cytochrome P450 reductase, most likely FMN, and it has been hypothesized that in microsomes, NADPH-cytochrome P450 reductase catalyses the one electron reduction of oxygen if no other electron acceptors are present [34,35].
Although cytochrome P450 by itself cannot generate ROS, substantial amounts are produced when cytochrome P450 is combined with NADPH-cytochrome P450 reductase. Under these conditions, electrons flow from NADPH via NADPH-cytochrome P450 reductase to cytochrome P450 as the terminal electron acceptor, and unique oxygen activation reactions occur in the active centers of the cytochrome P450 enzymes. The mechanism mediating ROS production by cytochrome P450 are not completely understood, but it has been shown to occur at several distinct steps [4–6]. It was postulated that the process is initiated by substrate binding to cytochrome P450 [36]. This substrate-cytochrome P450 complex then receives electrons from NADPH-cytochrome P450 reductase. This is following by oxygen binding leading to the formation of an oxyferrous intermediate. The non-productive decay of this intermediate results in the generation of superoxide anion. Alternatively, reduction of the oxyferrous intermediate by a second electron followed by protonation forms a hydroperoxo intermediate; decay of this intermediate results in the generation of hydrogen peroxide [4,6].
The present studies demonstrate that H2O2 is also generated by recombinant NADPH-cytochrome P450 reductase in the presence of NADPH, albeit at relatively low rates. Significantly higher absolute rates of H2O2 production were noted when cytochrome P450 enzymes were coexpressed with NADPH-cytochrome P450 reductase. These data support the idea that cytochrome P450’s are a major source of ROS. Of particular interest was our observation that ROS production by the recombinant microsomal system did not require the addition of a cytochrome P450 substrate. This suggests that in recombinant microsomal enzymes, oxygen activation is not dependent on substrate binding, and that NADPH-cytochrome P450 reductase binding to cytochrome P450’s is sufficient to induce NADPH-mediated electron flow leading to H2O2 generation.
In summary, we have used a highly sensitive assay to quantify H2O2 production by microsomal enzymes. Using this assay, we showed that recombinant NADPH-cytochrome P450 reductase alone was capable of generating ROS in the presence of NADPH. This activity was markedly increased when NADPH-cytochrome P450 reductase was coexpressed with the cytochrome P450’s, a finding consistent with the fact that these proteins are terminal electron acceptors in the uncoupling reaction. We also found that substrate binding to cytochrome P450 is not required for initiation of oxygen binding and electron flow through the microsomal P450-related electron transport system. Further studies are needed to elucidate the precise mechanisms by which ROS are generated by each component of the microsomal system. At the present time it is not known if NADPH cytochrome P450 reductase itself, in fact, generates ROS following its interaction with cytochrome P450’s. It is possible that in the combined NADPH-cytochrome P450 reductase-cytochrome P450 system, all or only part of electron flow is diverted from NADPH-cytochrome P450 reductase to cytochrome P450’s to generate ROS. It is also not clear if all cytochrome P450’s are equally effective in generating ROS; the availability of recombinant preparations containing individual cytochrome P450’s should allow a better characterization of the source of ROS, and its control in the microsomal P450-related electron transport system.
Acknowledgments
Financial support: This work was supported in part by National Institutes of Health grants CA100994 (JDL), CA093798 (DEH), ES005022 (JDL, DLL), ES004738 (DLL, JDL), CA132624 (DLL, JDL), AR055073 (JDL, DLL, DEH) and GM034310 (DLL, JDL). This work was also funded in part by the National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (award #U54AR055073 to JDL). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government.
Abbreviations
- AR
Amplex Red
- DETAPAC
diethylenetriaminepentaacetic acid
- HRP
horseradish peroxidase
- LLD
lowest limit of detection
- LLQ
lowest limit of quantification, SOD, superoxide dismutase
- RFU
relative fluorescence units
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References List
- 1.Ortiz de Montellano PR. Cytochrome P450: structure, mechanism, and biochemistry. 3. Kluwer/Plenum; New York: 2005. [Google Scholar]
- 2.Guengerich FP. Cytochrome P450 and chemical toxicology. Chem Res Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 3.Hlavica P, Schulze J, Lewis DF. Functional interaction of cytochrome P450 with its redox partners: a critical assessment and update of the topology of predicted contact regions. J Inorg Biochem. 2003;96:279–297. doi: 10.1016/s0162-0134(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 4.Coon MJ. Cytochrome P450: nature’s most versatile biological catalyst. Annu Rev Pharmacol Toxicol. 2005;45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- 5.Poulos TL. Intermediates in P450 catalysis. Philos Transact A Math Phys Eng Sci. 2005;363:793–806. doi: 10.1098/rsta.2004.1537. [DOI] [PubMed] [Google Scholar]
- 6.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 7.Zangar RC, Davydov DR, Verma S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol. 2004;199:316–331. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 8.Kessova I, Cederbaum AI. CYP2E1: biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Curr Mol Med. 2003;3:509–518. doi: 10.2174/1566524033479609. [DOI] [PubMed] [Google Scholar]
- 9.Bast A. Is formation of reactive oxygen by cytochrome P-450 perilous and predictable? Trends in Pharmacological Sciences. 1986;7:266–270. [Google Scholar]
- 10.Crespi CL, Miller VP. The use of heterologously expressed drug metabolizing enzymes--state of the art and prospects for the future. Pharmacol Ther. 1999;84:121–131. doi: 10.1016/s0163-7258(99)00028-5. [DOI] [PubMed] [Google Scholar]
- 11.Tang W, Wang RW, Lu AY. Utility of recombinant cytochrome p450 enzymes: a drug metabolism perspective. Curr Drug Metab. 2005;6:503–17. doi: 10.2174/138920005774330602. [DOI] [PubMed] [Google Scholar]
- 12.Mohanty JG, Jaffe JS, Schulman ES, Raible DG. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J Immunol Methods. 1997;202:133–141. doi: 10.1016/s0022-1759(96)00244-x. [DOI] [PubMed] [Google Scholar]
- 13.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 14.Towne V, Will M, Oswald B, Zhao Q. Complexities in horseradish peroxidase catalyzed oxidation of dihydroxyphenoxazine derivatives: appropriate ranges for pH values and hydrogen peroxide concentrations in quantitative analysis. Anal Biochem. 2004;334:290–296. doi: 10.1016/j.ab.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 15.Thurman RG, Ley HG, Scholz R. Hepatic microsomal ethanol oxidation. Hydrogen peroxide formation and the role of catalase. Eur J Biochem. 1972;25:420–430. doi: 10.1111/j.1432-1033.1972.tb01711.x. [DOI] [PubMed] [Google Scholar]
- 16.Hildebrandt AG, Roots I, Tjoe M, Heinemeyer G. Hydrogen peroxide in hepatic microsomes. Methods Enzymol. 1978;52:342–350. doi: 10.1016/s0076-6879(78)52037-5. [DOI] [PubMed] [Google Scholar]
- 17.Gautschi K, Keller B, Keller H, Pei P, Vonderschmitt DJ. A new look at the limits of detection (LD), quantification (LQ) and power of definition (PD) Eur J Clin Chem Clin Biochem. 1993;31:433–440. doi: 10.1515/cclm.1993.31.7.433. [DOI] [PubMed] [Google Scholar]
- 18.Votyakova; TV, Reynolds; IJ. Detection of hydrogen peroxide with Amplex Red: interference by NADH and reduced glutathione auto-oxidation. Arch Biochem Biophys. 2004;431:138–144. doi: 10.1016/j.abb.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 19.Hildebraunt AG, Roots I. Reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent formation and breakdown of hydrogen peroxide during mixed function oxidation reactions in liver microsomes. Arch Biochem Biophys. 1975;171:385–397. doi: 10.1016/0003-9861(75)90047-8. [DOI] [PubMed] [Google Scholar]
- 20.Dostalek M, Hardy KD, Milne GL, Morrow JD, Chen C, Gonzalez FJ, Gu J, Ding X, Johnson DA, Johnson JA, Martin MV, Guengerich FP. Development of oxidative stress by cytochrome P450 induction in rodents is selective for barbiturates and related to loss of pyridine nucleotide-dependent protective systems. J Biol Chem. 2008;283:17147–17157. doi: 10.1074/jbc.M802447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aust SD, Roerig DL, Pederson TC. Evidence for superoxide generation by NADPH-cytochrome c reductase of rat liver microsomes. Biochem Biophys Res Commun. 1972;47:1133–1137. doi: 10.1016/0006-291x(72)90952-7. [DOI] [PubMed] [Google Scholar]
- 22.Mishin V, Pokrovsky A, Lyakhovich VV. Interactions of some acceptors with superoxide anion radicals formed by the NADPH-specific flavoprotein in rat liver microsomal fractions. Biochem J. 1976;154:307–310. doi: 10.1042/bj1540307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grover TA, Piette LH. Influence of flavin addition and removal on the formation of superoxide by NADPH-Cytochrome P-450 reductase: a spin-trap study. Arch Biochem Biophys. 1981;212:105–114. doi: 10.1016/0003-9861(81)90348-9. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki I, Piette LH, Grover TA. Kinetic studies on spin trapping of superoxide and hydroxyl radicals generated in NADPH-cytochrome P-450 reductase-paraquat systems. Effect of iron chelates. J Biol Chem. 1990;265:652–659. [PubMed] [Google Scholar]
- 25.Morehouse LA, Thomas CE, Aust SD. Superoxide generation by NADPH cytochrome P-450 reductase: the effect of iron chelators and the role of superoxide in microsomal lipid peroxidation. Arch Biochem Biophys. 1984;232:366–377. doi: 10.1016/0003-9861(84)90552-6. [DOI] [PubMed] [Google Scholar]
- 26.Dutton DR, Reed GA, Parkinson A. Redox cycling of resorufin catalyzed by rat liver microsomal NADPH-cytochrome P450 reductase. Arch Biochem Biophys. 1989;268:605–616. doi: 10.1016/0003-9861(89)90328-7. [DOI] [PubMed] [Google Scholar]
- 27.Balvers WG, Boersma MG, Vervoort J, Rietjens IM. Experimental and theoretical study on the redox cycling of resorufin by solubilized and membrane-bound NADPH-cytochrome reductase. Chem Res Toxicol. 1992;5:268–273. doi: 10.1021/tx00026a019. [DOI] [PubMed] [Google Scholar]
- 28.Estabrook RW, Kawano S, Werringloer J, Kuthan H, Tsuji H, Graf H, Ullrich V. Oxycytochrome P-450: its breakdown to superoxide for the formation of hydrogen peroxide. Acta Biol Med Ger. 1979;38:423–434. [PubMed] [Google Scholar]
- 29.Gillette JR, Brodie BB, La Du BN. The oxidation of drugs by liver microsomes: on the role of TPNH and oxygen. J Pharmacol Exp Ther. 1957;119:532–540. [PubMed] [Google Scholar]
- 30.Gomes A, Fernandes E, Lima JL. Fluorescence probes used for detection of reactive oxygen species. J Biochem Biophys Methods. 2005;65:45–80. doi: 10.1016/j.jbbm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Soh N. Recent advances in fluorescent probes for the detection of reactive oxygen species. Anal Bioanal Chem. 2006;386:532–543. doi: 10.1007/s00216-006-0366-9. [DOI] [PubMed] [Google Scholar]
- 32.Staniek K, Nohl H. H2O2 detection from intact mitochondria as a measure for one-electron reduction of dioxygen requires a non-invasive assay system. Biochim Biophys Acta. 1999;1413:70–80. doi: 10.1016/s0005-2728(99)00083-3. [DOI] [PubMed] [Google Scholar]
- 33.Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ Res. 2001;89:224–236. doi: 10.1161/hh1501.094365. [DOI] [PubMed] [Google Scholar]
- 34.Massey V. Activation of molecular oxygen by flavins and flavoproteins. J Biol Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 35.Murataliev MB, Feyereisen R, Walker FA. Electron transfer by diflavin reductases. Biochim Biophys Acta. 2004;1698:1–26. doi: 10.1016/j.bbapap.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Isin EM, Guengerich FP. Substrate binding to cytochromes P450. Anal Bioanal Chem. 2008;392:1019–10130. doi: 10.1007/s00216-008-2244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]