

Abstract
Animal models, particularly mouse models, play a central role in the study of the etiology, prevention and treatment of human prostate cancer (PCa). While tissue culture models are extremely useful in understanding the biology of PCa, they cannot recapitulate the complex cellular interactions within the tumor microenvironment that play a key role in cancer initiation and progression. The NCI Mouse Models of Human Cancers Consortium convened a group of human and veterinary pathologists to review the current animal models of PCa and make recommendations regarding the pathological analysis of these models. Over 40 different models with 439 samples were reviewed including genetically engineered mouse models, xenograft, rat and canine models. Numerous relevant models have been developed over the last 15 years and each approach has strengths and weaknesses. Analysis of multiple genetically engineered models has shown that reactive stroma formation is present in all the models developing invasive carcinomas. In addition, numerous models with multiple genetic alterations display aggressive phenotypes characterized by sarcomatoid carcinomas and metastases, which is presumably a histological manifestation of epithelial-mesenchymal transition. The significant progress in development of improved models of PCa has already accelerated our understanding the complex biology of PCa and promises to enhance development of new approaches to prevention, detection and treatment of this common malignancy.
Keywords: prostate cancer, animal models, transgenic, pathology, genetically engineered mice, xenograft
INTRODUCTION
Animal models, particularly mouse models, play a central role in the study of the etiology, prevention and treatment of human prostate cancer (PCa). While tissue culture models are extremely useful in understanding the biology of PCa, they cannot recapitulate the complex cellular interactions within the tumor microenvironment that play a key role in cancer initiation and progression. Immune cells, fibroblasts, myofibroblasts, blood vessels and nerves all interact with PCa cells and levels of circulating oxygen, nutrients and endocrine factors also play a dynamic role in regulating tumor growth. Furthermore, there is a paucity of available PCa cell lines compared to many other common cancers due to the difficulty in establishing such cell lines. Analysis of human PCa tissues has been a major focus of PCa research for the last two decades and the complex molecular alterations associated with various subtypes of human PCa are beginning to be well delineated (1). Within the next several years detailed molecular analysis of large numbers of PCas by genomic sequencing and other high throughput techniques will lay out the full complexity of human PCa at the molecular level. However, ultimately such studies are correlative and in vivo model systems to understand the underlying biology and to test novel prevention and treatment strategies are critically needed.
Mouse models of PCa can be divided into two broad categories; genetically engineered mouse (GEM) models and xenograft models. Over the last 20 years a significant number of GEM models of PCa have been created and analyzed. In most cases, these models have used prostate tissue restricted expression of oncogenes or Cre recombinase (to inactivate tumor suppressors) via probasin or other prostate specific promoters to create genetic lesions mimicking those identified in human PCa (See Fig 1). More recently, models using lox-stop-lox to engineer prostate specific expression of genes from androgen independent promoters has also been employed since cellular dedifferentiation during progression or therapies targeting androgen receptor can decrease probasin driven oncogene expression. Indeed, analysis of GEM models has been critical in validating the biological importance of the observed molecular alterations in human PCa. Over time, these models have been combined to mimic the multiple genetic alterations in human PCa and define the interactions between critical pathways in an in vivo context. GEM models have significant advantages in that they reflect tumor progression over time from the initiation of pre-invasive lesions to invasive and in some cases metastatic lesions within the prostatic microenvironment including a fully intact immune system. The chief disadvantage of these models is that in some cases there may be important biological differences in mouse versus human prostate and other tissues that may impact model phenotypes. For example, carcinogenesis is generally initiated at the time of sexual maturation in GEM models and so that aging associated changes in the tumor microenvironment are not present as they are in human PCa (2). Newer generation models employing chemically activated Cre-recombinase can allow for activation of carcinogenesis at later times, although the spectrum of aging associated changes in mouse stroma only partially overlaps with those seen in humans (3). At a practical level, GEM models also usually have higher costs and longer time frames to generate results compared to xenograft models, which acts as a barrier to their use.
Figure 1. Methods for generation of genetically engineered mouse models of prostate cancer.
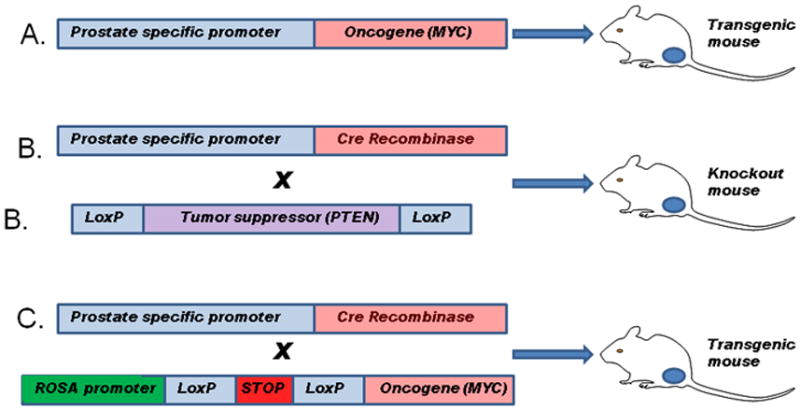
A. A prostate specific promoter (such as the enhanced ARR2Pb probasin promoter) is used to drive prostate specific expression of a gene of interest, typically an oncogene. The oncogene is expressed in the prostate epithelial cells at the onset of sexual maturity. B. Mice are generated with loxP sites flanking critical exons in a gene of interest, typically a tumor suppressor gene. These mice are then crossed with mice expressing the Cre recombinase under control of the probasin or other prostate specific promoter resulting in excision of key exons and inactivation of the targeted gene in prostatic epithelial cells bearing the targeted sequence in one or both alleles of the gene of interest. C. Mice are generated with the gene of interest downstream of a strong constitutive promoter such as ROSA26. Transcription/translation is inhibited by a lox-stop-lox cassette upstream of the gene of interest. These mice can be crossed to probasin Cre mice leading to excision of the stop sequence and expression of the gene of interest. This method has the advantage that expression of the gene of interest is no longer dependent on androgen receptor and other prostate specific factors whose activity may be altered by tumor progression or treatment.
In xenograft models, PCa cells are implanted in host mice. Most commonly, human PCa cell lines are implanted into immunocompromised (nude or SCID) mice either subcutaneously or injected orthotopically into the prostate. Orthotopic models have the advantage of growth within the prostate microenvironment and in a number of models metastasis occurs at high rates. A second class of xenograft models, known as tissue recombination models, uses epithelial cells (benign or malignant) combined with mesenchymal cells which are implanted either subcutaneously or under the renal capsule in immunocompromised mice. Such models represent a powerful method to study tumor-stromal interactions. In addition, in such models the epithelial and/or stromal compartment can be genetically manipulated to define the genetic lesions and paracrine signals capable of transforming stem cells from mouse or human prostate tissues (4–7). A third class of xenograft models are xenografts established from human PCa tissues and carried as xenografts. In most cases cell lines have not been established since, as described above, it is often quite difficult to establish PCa cell lines even from xenografts. Given the paucity of cell lines these xenografts provide valuable models to study the impact of therapies in a larger number of PCa models. Finally, mouse PCa xenografts can be established from mouse PCa cell lines in syngeneic, immunocompetent host mice. These models are mainstays of immunotherapy studies. In addition, these mouse xenografts in immunocompetent mice are useful for studying the role of immune cells in the tumor microenvironment in tumor progression and therapeutic resistance (8). Unfortunately, the number of mouse PCa cell lines is limited to date. The xenograft models described above have all proven extremely useful in evaluating the biology of human PCa and are used routinely to evaluate PCa therapies in a relatively quick and lower cost way of evaluating new therapies in PCa and mechanisms of therapeutic resistance. A major disadvantage of such models is that since they are almost all derived from advanced cancers so their applicability to prevention studies is questionable. Similarly, since most xenografts and PCa cell lines are derived from clinically aggressive lesions, they may not reflect the biology of precursor or less aggressive lesions. Finally, in almost all cases the tumor microenvironment is somewhat abnormal due to the location (subcutaneous or renal capsule) and/or the due to abnormalities of the host immune system.
Other animal models are used much less often in studies of PCa. Some rat strains have high rates of spontaneous carcinomas (9) and inducible carcinoma models using hormones and/or carcinogens in rats have been used by many groups (9). Transgenic rats can now be generated as well. An advantage of the rat over the mouse is the much larger size of the rat prostate. A disadvantage is that the number of analytical reagents for use in tumor analysis and transgenic and knockout lines for cross breeding is far smaller than for mice.
Finally, spontaneous PCa is relatively common in dogs and provides a potential useful natural model for evaluating novel PCa therapies. Such spontaneous models can be extremely useful but are less easily genetically manipulated than xenografts or mice, which limits their use.
Scope and Objectives
The National Cancer Institute Mouse Models of Human Cancers Consortium Prostate Cancer Steering Committee convened a panel of human and veterinary pathologists with expertise in PCa to review the current state of the art in animal models of PCa. A similar panel was convened more than 10 years ago resulting in the consensus report by Shappell et al (10). That consensus report defined the underlying principles and techniques for the pathological analysis of lesions in GEM models of PCa and provided detailed definitions for various lesions of the prostate in GEM models. In the subsequent 10 years numerous new models have been developed and the scientific community has gained extensive experience in analyzing these models. The goal of the current report is provide a consensus report updating the research community on current thinking in the pathological analysis of GEM models and to provide a detailed description of a number of important GEM models. In addition, we have extended our analysis to include xenograft models and a limited number of models in other species.
MATERIALS and METHODS
A team of veterinary and human pathologists with expertise in PCa was recruited by the senior investigators (RC and MI). Models to be reviewed were selected by the Mouse Models of Human Cancers Consortium Prostate Cancer Steering Committee and blocks and/or slides obtained from investigators. Slides were digitized using an Aperio ScanScope XT and uploaded to the Mouse Mutant Pathology Laboratory Spectrum public database (http://bit.ly/T9nHTN). The reader can view the accompanying digitized whole slide images (WSI) that are noted in the text by copying and pasting MC accession numbers (i.e MC12-0269) into the the search tool or the WSI can be viewed using the live links in Supplementary Tables 1. Detailed instructions for both approaches are appended to Supplementary Table 1. Pre-existing models submitted previously to this site were also reviewed. Review teams were assembled and digitized whole slide images were reviewed and annotated online. The review team met at Columbia University in New York City on April 17–18, 2012 and models were reviewed and discussed. Over 40 different models with 439 samples were reviewed including GEM, xenograft, rat and canine models. Following the review of the models, discussions were held regarding the optimal strategies for analysis of animal models of PCa, lessons learned since the last consensus meeting and emerging themes in the pathology of mouse models of prostate cancer.
There are several limitations to this study. First, it was not practical to review every GEM or xenograft model of PCa so this review is not encyclopedic in nature. Readers are referred to several excellent reviews for discussions of a number of models not represented in this report (11–14). Second, this report is focused on pathology rather than the biology of the cellular and molecular alterations that underlie the observed lesions. Third, the number of slides that could be reviewed for any model was limited so that conclusions regarding rates of progression, frequency of metastasis etc could not be assessed. The pathologists were able to determine if the interpretation of observed lesions in representative slides was concordant with the classification of the lesion by submitting investigator.
RESULTS
Genetically Engineered Mouse Models of Prostate Cancer
Methodological Considerations
The prior consensus report by Shappell et al (10) describes in detail pathology methods for analysis of GEM models of PCa. It is critical that a pathologist (MD or DVM) be involved in the pathology evaluation of the mouse model. Several key points need to be reiterated. First, the mouse prostate has a different anatomy and histology than the human prostate. It consists of the anterior (AP), ventral (VP), dorsal and lateral lobes (the latter two often combined as dorsolateral (DLP) (MC02-0695)). In contrast the human prostate is a single gland with different histological zones (peripheral, transition and central zones) (MC02-0713). The majority of human PCas arise in the peripheral zone. In the mouse, the glandular epithelial cells are surrounded by a very thin fibromuscular stroma, which is in contrast to the abundant fibromuscular stroma in the human prostate. The seminal vesicles of mice are large and occasionally the site of the development of proliferative lesions and tumors. The periurethral glands and bulbourethral glands can also be the site of tumor development in GEM models. GEM models have differential phenotypes in different prostate lobes that may depend on the underlying biology of the genetic alteration, the exact constructs used for expression, the site(s) of integration of transgenes and the mouse strain. It is the opinion of the panel that it is premature to conclude that lesions in any given lobe are more representative of human disease. Second, when initially analyzing models, it is preferable to submit the prostate and associated accessory sex glands and related organs en bloc for histological examination as described by Shappell et al (10). (MC02-0695). This allows examination of all prostate lobes as well as associated organs which may have neoplastic alterations which can extend into the prostate. Lobe specific harvesting can yield more tissue and is suitable for harvesting frozen tissue for molecular analysis and thus complements the en bloc method in models that have undergone initial characterization. Third, it is essential to carry out time course studies (serial sacrifice by age) to evaluate the full biological potential of the genetically engineered lesion. While the exact time course is model dependent, it should be recognized that a year or more may be required for the most advanced lesions to develop. Examination of the genitourinary organs should be complemented by necropsy and, if possible, imaging studies to evaluate local extension and metastasis. Examination of age matched controls of the same genetic background housed under the same conditions, preferably littermates, is essential.
Optimal pathological evaluation of mouse models (and human tissues) is dependent on well-fixed, thin, well-stained sections. In almost all cases neutral-buffered formalin is optimal for hematoxylin & eosin (H&E) sections and is compatible with most immunohistochemistry (with appropriate antigen retrieval). Obviously IHC studies to confirm the expression of a transgene or loss of expression of a targeted tumor suppressor in abnormal tissues are critical for confirming that the genetic alteration is actually present in the lesions seen. Additional studies can be useful to evaluate stromal alterations and invasion including trichrome stain to assess fibrosis and immunohistochemistry (IHC) for α-smooth muscle actin to assess for the integrity of the smooth muscle investment of the glands. Markers of neuroendocrine differentiation of epithelial cells such as chromogranin or synaptophysin are useful in confirming the diagnosis of neuroendocrine carcinoma (MC02-0699). Neuroendocrine cancers also display the typical perinuclear staining with anti-cytokeratin antibodies seen in human neuroendocrine carcinomas. The role of evaluation of basal cell specific markers is less clear. In human PCa, loss of basal cells is a hallmark of PCa and stains to evaluate basal cytokeratins and/or p63 are used routinely to evaluate problematic lesions. However, in mice there is evidence that basal cell markers can be expressed by cancer cells so the presence of staining for such markers does not exclude PCa. However, complete loss of basal cell markers in a focus that is histologically compatible with PCa supports this diagnosis. IHC is now a routine part of clinical practice in human pathology and inclusion of such studies enhances (but does not replace) histological evaluation of H&E sections. In this regard, studies to evaluate proliferation by IHC (such as Ki67) or apoptosis (by IHC or TUNEL) can be useful adjuncts to histological evaluation and can confirm the biological impact of a given genetic lesion. However, changes in proliferation or apoptosis are not the equivalent to neoplasia and increased proliferation and decreased apoptosis, while seen in neoplasia, are not adequate to define a lesion as neoplastic.
Analysis of progression in GEM models must be performed with care. Local progression requires careful dissection and at times en-bloc preparation of tumors and surrounding tissues (MC02-0695). Step and serial sections can often help define early lesions. The presence of metastatic lesions must be established by careful necropsy, which can be assisted by appropriate premortem imaging studies. Considerable caution must be taken to exclude spontaneous primary lesions at distant sites. Inbred mice, particularly older mice can be prone to such tumors, which are often strain and site specific. For example, primary lung neoplasms are common in inbred mice and can occur in up to 41% of 24 month old FVB mice (15) and can be multiple, mimicking metastasis (MC12-0377LA). Such lesions have characteristic morphology but IHC for lung specific markers such as surfactant protein can be used to confirm the lung origin of such tumor (Supplementary Fig 1). TTF-1 is also a useful marker for primary pulmonary tumors. It should be noted that transgene expression does not necessarily prove a distant lesion is metastatic since it is possible that a putatively prostate-specific promoter may have transcriptional activity in non-prostate cells that is sufficient to transform such cells, particularly if the genetic lesion has potent transforming activity. Use of mice with a global germline genetic lesion (i.e. Pten +/− knockout mice) can also confound interpretation of lesions at distant sites since the genetic change can lead to primary tumors at other sites. Similarly, use of non-prostate specific promoters (i.e. keratins), obviously has similar limitations.
Mouse models also may develop distant spread of tumor via intravascular spread without invasion (TG02-1510LA) at the distant sites, primarily in the lung. An example is shown in Supplementary Fig 2. Endothelial markers can be used to confirm the presence of the blood vessel around the tumor embolus if needed. Tumor emboli can be seen in other human cancers such as renal cell carcinoma but are rare in human PCa, although widespread intravascular spread in the lymphatics of the lungs is seen occasionally in advanced disease. The consensus panel felt that such lesions should be specifically identified as intravascular tumor emboli and not metastasis per se since the lesions do not invade the tissue at the distant site.
It should be noted that in a number of aggressive models extensive local invasion can occur. This is not considered metastasis since direct local invasion is pathologically and biologically distinct from spread to and colonization of distant sites by malignant cells. Direct local invasion of adjacent organs is analogous to pT4 lesions in human PCa as defined by the AJCC.
Classification of lesions of the prostate in genetically engineered mouse models
Shappell et al (10) have previously provided definitions and descriptions of the full spectrum of lesions seen the prostate. In this report we will more narrowly focus on the main lesions seen in GEM models and summarize new insights into these lesions that have accrued in the 10 years since the original consensus conference. Many of the newer models involve crosses of the various GEM lines and the pathology of such models may be the same or similar to one of the founder lines or may be unique. Lesions can be more progressive, more malignant and more metastatic in the new complex line.
Epithelial Hyperplasia
Epithelial hyperplasia is a non-neoplastic increase in epithelial cells within the prostate and can be manifested as simple hyperplasia, showing a single epithelial cell lining, or more complex, with architectural changes such as tufting, papillary and even cribriform changes (MC12-0306BY-recut-H&E-CL). It may be focal or diffuse. Epithelial hyperplasia may show some minimal nuclear atypia, which is often associated with inflammation, but it should not be marked. Diagnosis of epithelial hyperplasia, particularly if focal, should be based on quantitative comparison of number and extent of hyperplastic foci compared to age matched controls. Quantitative IHC analysis of proliferation markers and IHC/TUNEL for apoptosis can be useful for determination of biological cause of the observed hyperplasia.
Mouse prostatic intraepithelial neoplasia (mPIN)
Prostatic intraepithelial neoplasia (PIN) is characterized histologically by proliferation of atypical epithelial cells within pre-existing glandular spaces but without frank invasion. In humans, it is widely accepted that high-grade PIN (HGPIN) is the precursor to invasive carcinoma, although only a fraction of such lesions ultimately progress to invasive carcinoma. Similar lesions, characterized by proliferation of atypical cells within pre-existing glandular spaces, occur in GEM and are classified as mPIN. They are differentiated from hyperplastic lesions by their morphology and natural history in a specific GEM models. Examination of multiple models developed over the last 10 years since the prior consensus conference have shown that such lesions often progress to frankly invasive carcinoma. Thus, there is increased confidence that such lesions do indeed reflect pre-invasive neoplasia of the mouse prostate. mPIN has been seen in all prostate lobes in various models, although many publications do not clearly define which prostate lobes are actually involved with mPIN. A system developed by Cardiff and colleagues to recognize and classify mPIN has been published (16) and this system has proven useful as a framework for evaluating such lesions. Examples of mPIN are shown in Figure 2. In this system, mPIN is graded on a scale of 1–4 based on increasing degrees of architectural and cytological abnormalities. mPIN1 has 1–2 layers of cells and mild nuclear atypia (Fig 2A and 2B). Lesions of mPIN2 have increased nuclear atypia and contain two or more layers of cells often in papillary, tufting or cribriform arrangements (MC12-0385DF). mPIN3 lesions have obvious nuclear atypia and fill or almost fill the duct lumens in papillary or cribriform patterns. mPIN4 lesions are similar to mPIN3 but have even more severe atypia, fill the ductal lumens and may bulge into the surrounding stroma, but without clear invasion (MC12-0310BY). The consensus panel found that mPIN 1 and 2 are harder to reproducibly identify unless widespread while mPIN 3 and 4 are readily identified and are more consistently associated with the presence of/or progression to invasive lesions (MC12-0396DF; TG04-0319-HE). Thus, it is reasonable to equate mPIN1 and 2 to low grade PIN in humans while mPIN 3 and 4 are more like human HGPIN. It is generally agreed that the biological potential of low grade PIN in humans is not completely clear and that it can be difficult to reproducibly identify. As such, in human prostate pathology low grade PIN is generally not reported. In addition, mPIN1 and 2 can be seen focally in mice, particularly older mice (see Fig 2A). It is not advisable to report low grade PIN as an endpoint without extensive blinded pathological analysis of multiple animals and quantitative comparison with age matched controls and suitable statistical analysis. However, the presence of mPIN 1 and 2 lesions, evaluated in this manner, particularly if widespread in younger mice, may be a valuable first indication of the potential for the development of subsequent higher grade pathologies since a number of time course studies have shown that mPIN1 and 2 lesions precede the development of higher grade lesions (16–17). In contrast, high grade PIN (mPIN 3/4) is readily recognizable by trained pathologists and is often, but not always, associated with progression with age or biological complementation. In humans, HGPIN is more reproducibly recognizable and is often (but not always) associated with cancer. Thus the presence of mPIN3/4 is a useful pathological endpoint and should always be reported, although it is critical to always examine age matched control mice.
Figure 2. Prostatic intraepithelial neoplasia (PIN) in GEM mouse models.

A. Low grade mPIN in 73 week old wild type mouse. Arrow shows focal epithelial proliferation with mild atypia. B. Low grade mPIN in Pten null model (K5 promoter) 12 week old mouse C. High grade mPIN in Pten null model (K14 promoter). Note central necrosis and proliferation of atypical cells filling the duct but with well demarcated border (medium power). Chronic inflammation is noted (arrow). D. High grade mPIN in Pten null model. Arrow shows nucleus with striking pleomorphic atypia.
Adenocarcinoma
As described by Shappell et al (10) invasive carcinoma can be recognized by infiltrative and destructive growth pattern of atypical cells with at least focal glandular differentiation. In mice, this is usually associated with stromal desmoplasia, which is characterized by stroma with abundant fibrosis. The fibrotic stroma usually has increased numbers of spindle shaped cells (fibroblasts and/or myofibroblasts) as well as chronic inflammatory cells. Most invasive carcinomas in GEM models are adenocarcinomas. Examples are shown in Figures 3A and 3B.
Figure 3. Prostate malignancies in GEM of prostate cancer.

A. Invasive adenocarcinoma, Hi-myc model. B. Invasive adenocarcinoma, Pten null X Sox9 overexpression model. C. Microinvasive adenocarcinoma. Pten null Smad4 null model 10 weeks. Focal microinvasion (arrows). D. Intracystic carcinoma; APC model. Masses of poorly differentiated adenocarcinoma within a cystic space (arrow indicates wall of cystic space). E. Sarcomatoid carcinoma, Pten null p53 null model. Masses of atypical proliferating spindle cells entrapping residual glands with PIN. F. Neuroendocrine carcinoma, TRAMP model, metastatic to liver
Microinvasive adenocarcinoma in GEM mice can be difficult to reliably recognize due to tangential cutting, irregular out-foldings of epithelial structures and pre-existing stromal changes. This is also a problematic area in human pathology in many organ systems. Step or serial sections may be useful to identify frank invasion in mice where microinvasion is identified. Microinvasive carcinoma is not a commonly used diagnostic category in human prostate pathology and such lesions would usually be called “HGPIN with adjacent small atypical glands suspicious for adenocarcinoma”. The consensus panel agreed that microinvasive carcinoma an be diagnosed in unequivocal cases (Fig 3C) (MC12-0298ICR) but over-reliance on isolated foci of microinvasion as definitive pathological endpoint indicative of higher biological potential is discouraged without extensive well-controlled studies with appropriate statistical analysis. If only a microinvasive lesion is seen as the most advanced prostate lesion in a new line of mice, one should be cautious in diagnosing this as invasive adenocarcinoma unless unequivocal adenocarcinoma forming larger grossly visible tumors or metastases are found.
Intraductal adenocarcinoma is a well known entity in human prostate pathology although it is not common (18). Examples of lesions consistent with intraductal carcinoma were seen in some GEM models. These lesions are characterized by proliferating masses of atypical cells with marked expansion of pre-existing structures without obvious infiltrative growth (Fig 3D). The panel has advised using the term intracystic carcinoma for such lesions since its biological potential is not currently clear, unlike intraductal adenocarcinoma in humans, which is associated with aggressive disease. Additional studies will be needed to determine the true biological potential of intracystic carcinomas in mice.
A new development that was noted in several models was the presence of heterologous epithelial differentiation. This took the form of focal squamous or intestinal differentiation. Examples are shown in Supplemental Figure 3. Both squamous metaplasia and mucinous gland metaplasia can be seen benign human prostate. However, such heterologous differentiation is not commonly seen in human PCa; adenosquamous carcinoma of the prostate has been reported but is extremely rare. Presumably this heterologous differentiation represents divergent differentiation from pluripotent transformed precursor. Unless extensive, it was felt that such lesions should still be classified as adenocarcinoma in the same way that urothelial carcinomas are classified as such despite focal squamous or glandular differentiation. The biological significance of such changes is unclear, particularly since they were typically focal.
Sarcomatoid carcinoma
As will be described below, a number of GEM models, particularly models with aggressive behavior, show evidence of focal or extensive sarcomatoid differentiation within adenocarcinoma. This is characterized, as in human pathology, primarily by the presence of highly atypical spindle cells admixed with, or adjacent to, invasive adenocarcinoma. An example is shown in Fig 3E. In some models these lesions have been characterized by IHC and have been shown to co-express mesenchymal and epithelial markers to variable degrees (19) (EX09-0118-2). No instances of differentiation of the sarcomatoid elements into differentiated sarcomas such as osteosarcoma etc were observed in such lesions. While histologically identifiable sarcomatoid carcinoma is rare in human prostate cancer, there is evidence that epithelial-mesenchymal transition can promote PCa progression (20) although this concept remains controversial.
It should be noted that GEM models often have lesions of variable histological severity in the same lobe or gland, ranging from mPIN to adenocarcinoma and/or sarcomatoid carcinomas. Typically large numbers of prostate epithelial cells demonstrate activation of a transgene or loss of tumor suppressor genes at early times after activation of expression of the transgene or Cre recombinase. The individual cells presumably have variable rates of progression as they accumulate additional alterations and this presumably accounts for the variable histology seen. Human PCa also shows significant histological heterogeneity and neoplastic lesions can be geographically distinct within the prostate. There is evidence of molecular heterogeneity between these different tumor foci in human PCa. High grade PIN is also often multifocal. Thus there is evidence that GEM models and human PCa both may be multiclonal, although it seems likely that GEM models are more multiclonal than human PCa. At increasing times after tumor initiation, in mice and in humans, the fastest growing tumor may come to dominate the histology seen in the prostate.
Neuroendocrine carcinoma
Focal neuroendocrine differentiation is common in human prostatic adenocarcinoma (21). Frank neuroendocrine cancers are rare as primary cancers but are more common in heavily treated men with PCa dying of their disease (EX09-0054BI). Such neuroendocrine carcinomas are often admixed with more typical acinar adenocarcinoma and appear to arise from adenocarcinomas in most cases (21). Of note, they typically do not express androgen receptor (AR), unlike most castrate resistant PCa which continues to express AR that is active at castrate levels of circulating androgens. It is possible that more neuroendocrine cancers will be seen in the future as with the emergence of more effective therapies targeting AR and local AR ligand production. Neuroendocrine carcinomas are similar to human neuroendocrine carcinomas in appearance and are characterized by cells with high nuclear/cytoplasmic ratio, small amounts of cytoplasm and granular chromatin. IHC for neuroendocrine markers such as chromogranin or synaptophysin can be used to confirm neuroendocrine differentiation (Supplemental Fig 4), as is common practice in human pathology (MC02-0699). Neuroendocrine carcinomas in GEM mouse models are associated with rapid growth and metastasis and are highly lethal, as in human neuroendocrine PCa. They are often seen in TRAMP mice and crosses of them to other GEM. They are the most widely metastatic and aggressive mouse PCa (TG06-0293-6). An example is shown in Figure 3F (TG06-0394).
Pathology of Specific Genetically Engineered Mouse Models
PTEN/AKT Pathway
The PI3K/AKT pathway is a major signaling pathway that is activated in many human malignancies. Activation of PI3K/AKT pathway by a variety of mechanisms has been found in almost all of late stage PCas (1). PTEN is a major negative regulator of the PI3K/AKT pathway and early studies in germline Pten+/− heterozygous knockout mice showed mPIN lesions (22). To model the involvement of this pathway in human PCa, Hong Wu’s group at UCLA crossed Pten flox/flox mice to the ARR2-Probasin-Cre transgenic line, PB-Cre4, in which the Cre recombinase is under the control of an enhanced prostate specific probasin promoter, and generated mice with prostate-specific homozygous deletion of Pten (23). Such mice showed mPIN and ultimately invasive adenocarcinoma (23). An important feature of the model is that it recapitulates the disease progression seen in human PCa, with tumor initiation in the form of mPIN, followed by progression to invasive adenocarcinoma and metastasis, making this a widely used model for studying the PTEN/Akt pathway in PCa. As will be described below, sarcomatoid carcinoma develops in a number of PTEN deletion models with additional genetic alterations (MC12-0194NIH). Examples of pathology from models based on PI3K/PTEN/AKT pathway activation are shown in Figure 4. Additional images are shown in Supplementary Figures 5 and 6 with links and descriptions in Supplementary Table 2. The PTEN/AKT models have a very similar cytology characterized by nuclei that are relatively large and round to oval with a clear chromatin and abundant light pink cytoplasm. It should be noted that in PTEN deletion models that mPIN3/4, adenocarcinoma and sarcomatoid carcinoma can all be present in a single tumor. In addition, stromal fibrosis (desmoplasia) is almost always present in invasive disease.
Figure 4. Pathology in Pten deletion models.

A. HG mPIN in Pten/p53 null model. Elsewhere in this same tumor there were areas of invasive adenocarcinoma and sarcomatoid carcinoma. B. Invasive adenocarcinoma Pten/p53 null; C. Invasive adenocarcinoma Pten/Smad4 null model. The invasive adenocarcinomas both show obvious reactive stroma. D. Metastatic adenocarcinoma in lumbosacral lymph node. Pten/p53/Smad4 null model. E. Pten/p53 null telomerase reactivation model. Sarcomatoid carcinoma in the prostate. Area on left shows very poorly differentiated carcinoma with a pleomorphic spindle cell pattern on the right. F. Sarcomatoid carcinoma invading vertebral bone (Pten/p53 null telomerase model)
With the increased realization that PCa involves multiple signaling pathways, there have been many studies focused on engineering additional genetic lesions into the Pten null PCa model to study their potential cooperation with Pten loss in prostate carcinogenesis. A number of such models were examined by the panel members. In several of the models, introduction of another genetic lesion led to more aggressive tumors.
Yu Chen (Memorial Sloan Kettering Cancer Center) has studied a model that overexpressed ERG in Pten null PCa and found that in comparison to Pten loss alone, ERG significantly accelerated tumor development. Examination of tumors from this model revealed adenocarcinoma at 12–15 weeks of age and anaplastic invasive adenocarcinomas with sarcomatoid carcinomas by 25–30 weeks of age (MC12-0220S; Supplementary Fig 5). Studies by Carver et al (24) had previously shown that ERG expression in a germline Pten +/− mice resulted in development of adenocarcinoma, while mice with heterozygous loss of Pten alone only developed mPIN.
Amanda Swain (Institute of Cancer Research, UK) submitted a model with loss of Pten and activation of β-catenin. Two 12 week old mice were reviewed (MC12-0299ICR; MC12-0297ICR). The first was a mouse with loss of one Pten allele and overexpression of a stabilized form of β-catenin which showed high grade mPIN with focal squamous differentiation, while a mouse with loss of both Pten alleles and overexpression of a stabilized form of β-catenin showed multicentric invasive adenocarcinoma with focal squamous differentiation. The same investigator also submitted two slides from mice with loss of Pten and overexpression of SOX9 in the prostate. A 52-week-old mouse with loss of one Pten allele and overexpression of Sox9 showed microinvasive carcinoma in a background of high grade mPIN, while a 12 week-old mouse with loss of both Pten alleles and overexpression of Sox9 showed multicentric, highly invasive adenocarcinoma (Fig 3B). See also Supplementary Figs 5 and 6. These findings are concordant with prior studies from the Swain laboratory implicating Sox 9 in enhanced PCa progression (25)
Charles Sawyer’s group generated mice in which the prostate was Pten null and overexpressed human MYC (26) (MC12-0189S; MC12-0188S). The prostates reviewed from these bigenic mice showed adenocarcinoma with focal intestinal metaplasia in a background of high grade mPIN, involving all lobes of the prostate. Concerning distribution and histomorphology of neoplastic lesions, bigenic mice are very similar to Pten null mice (i.e. involvement of all the prostate lobes and neoplastic epithelial cells with abundant eosinophilic cytoplasm) but are remarkably different from MYC transgenic mice, which showed multiple invasive adenocarcinomas predominantly involving the lateral lobe of the prostate and composed of epithelial cells with abundant clear vacuolated cytoplasm, large vesicular nuclei with prominent nucleoli and marked apoptotic activity.
Hong Wu’s group has developed mice with expression of mutant K-ras (G12D/WT) expression and Pten homozygous deletion in the prostate and showed that, while RAS activation alone was not sufficient to cause malignant transformation in the prostate, combination of Pten deletion and RAS activation led to accelerated progression of PCa in comparison to Pten deletion alone (27). The slides reviewed by the panel demonstrated adenocarcinoma with focal squamous differentiation at 10 weeks (MC12-0376LA-recut-H&E-CL). Between 35 and 50 weeks, the tumors showed features of sarcomatoid carcinoma (MC12-0106LA-HE-KB). Metastasis was observed with 100% penetrance in lungs (see Supplementary Fig 1) (MC12-0106LA-2-HE-KB). Of note, these investigators provide detailed molecular analysis of the sarcomatoid carcinomas which are consistent with mesenchymal differentiation due to epithelial to mesenchymal transition (EMT). Thus activation of the MAP kinase pathway in the Pten homozygous knockout model accelerates tumor progression and is associated with EMT
Cory Abate-Shen’s group has reported similar observations regarding the role of the MAPK pathway in tumor progression using a different strategy (28). Slides from mice with Nkx3.1CreERT2 knock-in allele simultaneously inactivating one allele of Nkx3.1 while driving tamoxifen-dependent Cre that induce conditional deletion of Pten were examined. The anterior prostate (AP) from a 24-months-old Nkx3.1 CreERT2/+; Ptenflox/flox intact mouse showed diffuse high grade PIN with adenocarcinoma, while the AP from a 17 months-old Nkx3.1 CreERT2/+ ;Ptenflox/flox castrated mouse showed adenocarcinoma with focal squamous differentiation. The addition of a mutant K-ras allele activation (Nkx3.1CreERT2/+;Pten flox/flox;KrasLSL/+ mouse) with tamoxifen treatment initiated at 2 months of age showed adenocarcinoma with focal intestinal metaplasia in a background of high grade mPIN at 18 weeks (MC12-0263CA-recut-H&E-CL). Examination of slides of tumors from mice with a mutant BRAF (V600E) with the same knockout background (Nkx3.1CreERT2/+; Ptenflox/flox; BrafLSL/+), with tamoxifen treatment initiated at 4 months, showed sarcomatoid carcinoma at 35 weeks (MC12-0265CA-recut-H&E-CL). Metastasis was reported in 30% of these mice. Thus MAPK activation was again associated with aggressive sarcomatoid PCa in a PTEN knockout background.
While p53 is the most commonly mutated tumor suppressor gene in human tumors, deletion of p53 alone does not produce a tumor phenotype in the prostate (29). The Pandolfi group has shown combined homozygous loss of Pten and p53 resulted in significantly more penetrant and rapidly developing prostate cancer than Pten deletion alone (29). Examination of a 7 month old mouse with homozygous loss of both Ptenand p53 revealed sarcomatoid carcinoma (Supplementary Fig 5). The Kelly group has also generated mice with simultaneous homozygous deletion of Pten and p53 (30). There was diffuse development of high grade mPIN in all lobes by 8–10 weeks of age, with invasive adenocarcinoma as early as 12 weeks (Fig 3B). Adenocarcinoma predominated before approximately 16 weeks with subsequent development of sarcomatoid carcinoma (Fig 3E). Sarcomatoid carcinoma is the predominant pattern in end stage mice (18–22 wks) with small foci of residual adenocarcinoma and entrapped high grade PIN glands. There is also occasional squamous differentiation. Rare metastasis to sub-lumbar lymph nodes and occasional tumor emboli in pulmonary capillaries were observed. Clonal cell lines were derived from these mice to investigate the differentiation and metastatic potential of different tumor initiating cell populations when injected orthotopically into nude mice. A bi-potential clonal line capable of luminal and basal differentiation gave rise to adenosquamous carcinoma with a high rate of lung metastasis. Another clonal line with a luminal phenotype in vitro (CK8+/Vimentin−) gave rise to orthotopic sarcomatoid carcinoma (CK8+/Vimentin+) with no distant metastasis. Slides submitted by Akash Patnaik (Beth Israel Deaconess Medical Center) demonstrated phenotypes similar to other Pten/p53 null models described above (See Supplementary Fig 6).
The DePinho group has generated a mouse model with homozygous deletion of Smad4 and Pten in the mouse prostate (31). This led to the emergence of invasive, metastatic and lethal prostate cancers with 100% penetrance with metastases to regional lymph nodes and lungs (31). Examination of slides from this model revealed highly glandular adenocarcinomas at 13 weeks (Fig 4C) and metastases to lumbosacral lymph nodes (Fig 4D) (MC12-0387DF) and lung (Supplementary Fig 6) at 13 and 23 weeks respectively. The same group has made an inducible telomerase reverse transcriptase (mTert) allele that was activated in a mice with homozygous loss of Pten and p53 tumor suppressors in a telomerase null background carried for 3–4 generations prior to mTert induction (32). Telomerase reactivation in these mice resulted in very aggressive cancers. We reviewed five digital slides showing primary tumor masses and lesions involving vertebral bone and bone marrow identified as a metastasis. The prostate showed adenocarcinoma and highly pleomorphic sarcomatoid carcinomas (Fig 4E). A large mass of sarcomatoid carcinoma adhered to the spine, invaded spinal muscle and spinal cord via spinal nerves and bone marrow (Fig 4F) (MC12-0381-1DF). We did not see metastatic lesions in bone marrow compatible with hematogenous or lymphatic spread but rather the tumor cells in spinal bone and bone marrow appeared to have originated via direct invasion and spread from the primary prostate mass.
More recently, the Pandolfi group (Harvard) has generated a mouse with homozygous deletion of the Lrf (Pokemon) transcription factor along with homozygous deletion of the Pten gene. Histological examination of a tumor from a 7 month old mouse revealed sarcomatoid carcinoma similar the mouse with Pten and p53 homozygous deletion described above, consistent with enhanced progression due to Lrf deletion (MC12-0289BIL-recut-HE-EH).
Using the Pten null mouse model, Hong Wu’s lab has recently demonstrated reciprocal feedback regulation of PI3K and androgen receptor (AR) signaling (33). AR deletion in prostate epithelial cells of Pten null mice promoted robust adenocarcinoma development in the dorsolateral lobes. The slide submitted to the panel showed invasive adenocarcinoma of the prostate with focal intestinal metaplasia. Dr. Chawnshang Chang submitted one slide from germine Pten heterozygous knockout mice with deletion of stromal AR (34). The phenotype appeared similar to the control Pten +/− mouse with multifocal high grade mPIN at 28 weeks of age.
Pten deletion has been used to study the cellular origin of prostate cancer in mice. Li Xin and colleagues studied the cell of origin of PCa by deleting Pten in basal and luminal cells, respectively (17). A 41 to 42-week-old K14-CreER;Pten flox/flox bigenic mouse (Pten deletion in basal cells) showed multifocal intermediate to high grade PIN which was more severe in the DLP and VP. A 30 to 36-week-old K8-CreER;Ptenflox/flox bigenic mouse (PTEN deletion in luminal cells) showed adenocarcinoma in a background of diffuse high grade mPIN which was also more severe in the DLP and VP. Michael Shen (Columbia University) asked the same question using different promoters to drive Cre expression in basal and luminal cells, respectively. CK5CreERT2/+;Pten flox/flox mice (Pten deletion in basal cells) showed microinvasive carcinoma in a background of high grade PIN detected at 6 months, while Nkx3.1CreERT2/+;Ptenflox/flox mice (Pten deletion in luminal cells) showed microinvasive carcinoma in a background of high grade PIN detected at 3 months. Their results suggest that both basal and luminal cells can be an origin for prostate cancer in mice.
Early studies of this pathway used a transgenic approach with prostate specific expression of constitutively active myristoylated-Akt (myr-AKT) showed mPIN lesions (35). The panel reviewed a Tet-inducible myr-AKT under the control of a probasin promoter. The prostate showed diffuse hyperplasia and low grade mPIN after induction but no invasive carcinoma (Supplementary Fig 5). To date activated AKT is less potent at inducing neoplasia in the mouse prostate than homozygous Pten deletion.
MYC pathway
Increased copy number of the Myc oncogene and overexpression of MYC protein are common in human PCa (1). The Sawyers group has shown that overexpression of MYC in the prostate resulted in mPIN that progressed to invasive adenocarcinoma thereby delineating MYC’s functional role in prostate cancer initiation and progression (Hi-Myc model) (36). The reproducible kinetics and high penetrance of mPIN followed adenocarcinoma combined with shared molecular signatures with human PCa has made this a powerful mouse model to study MYC’s oncogenic program. An example of adenocarcinoma in a MYC transgenic is shown in Fig 3A. It is of interest to note that the De Marzo group has shown by IHC that the characteristic cytological features seen in mPIN are seen as soon as the MYC transgene is expressed, implying that they are a direct result of MYC activity and do not require additional genetic lesions. As in Pten null adenocarcinoma, there is a prominent stromal response with fibrosis and infiltration of chronic inflammatory cells (MC12-0192S-H&E-recut-KB).
Sarki Abdulkadir’s group has developed a model which included prostate-specific, Cre-inducible, focal overexpression of MYC and floxed Pten allele and p53 alleles allowing Cre-mediated heterozygous or hemizygous loss to these two tumor suppressor genes (COMPP model) (37) The slides submitted showed extensive PCa in all lobes(MC12-0026V-H&E-CL). This is in contrast, to the Hi-Myc model which generally developed tumors in the dorsolateral and ventral lobes. The COMPP model retained histomorphological features associated with MYC overexpression such as prominent nucleoli and large vesicular nuclei.
ERG Pathway
The TMPRSS2/ERG fusion gene is present in half of all human PCas and results in expression of a slightly truncated ERG oncogene under the control of the androgen responsive TMPRSS2 promoter (38–39). Yu Chen’s laboratory (Memorial Sloan-Kettering Cancer Center) has developed ERG-overexpressing C57BL/6 transgenic mice by knocking ERG-IRES-nlsGFP into the 1st intron of the endogenous TMPRSS2 locus. The slides submitted for panel review showed earlier and more numerous foci of low grade mPIN in the transgenic ERG mice when compared to controls. At around 20 weeks of age, low grade mPIN lesions were noted in some of the transgenic mice, but not controls, and at around 70 weeks of age nearly all transgenic mice showed low grade mPIN lesions while controls showed only rare low grade mPIN lesions. Although neither high grade mPIN nor adenocarcinomas were noted in the transgenic mice overexpressing ERG only, ERG overexpression in Pten null mice induced earlier and more significant lesions, including sarcomatoid carcinomas, as noted above (see “PTEN/Akt pathway”, above).
The Kelly laboratory has generated a transgenic ERG mouse using a recombinant bacterial artificial chromosome that incorporated a 25Kb human TMPRSS2 promoter plus exons 1 and 2 adjacent to the human ERG genomic region downstream of intron 7/exon 8 (40). These mice were mated to Pb-Cre4; Pten flox/flox mice to produce TMPRSS2-ERG; Pten+/− mice. There were no significant lesions in the prostates of TMPRSS2-ERG mice at 1 year of age. At 28 weeks there was an increased number and severity of mPIN lesions in the prostates of TMPRSS2-ERG; Pb-cre; Pten+/− mice as compared to Pb-cre; Pten+/− alone mice. However, this difference disappeared by 1 year of age as there was no significant difference in number or severity of mPIN lesions at 52 weeks.
The Vasioukhin laboratory has previously reported on their transgenic ERG mouse model, which overexpresses an N-terminally truncated ERG protein using a modified probasin promoter (41). In these mice, focal mPIN lesions (generally low grade) are detected in mice beginning around 5–6 months. The number and severity of lesions tends to increase with age, and at around 18 months adenocarcinomas are noted. At two years of age, high grade pleomorphic and spindle cell tumors were present, consistent with sarcomatoid carcinomas (MC12-0114FH-2-HE-KB). The reason for the more aggressive phenotype noted by the Vashioukhin group is unclear but may be related to the very high levels of ERG transgene expression in this model (Vasioukhin, unpublished observations).
Retinoblastoma (Rb) Pathway
The Van Dyke laboratory has developed a model of Rb inactivation via ARR2-Pb driven SV40 T121 expression and Pten/p53 deletion in prostate under the control of Pb-Cre4 promoter (Pten and p53 floxed). Slides submitted show that by 17 weeks there is diffuse high grade mPIN in all lobes with early adenocarcinoma. By 20 weeks adenocarcinoma was common (MC12-0323NCI) and there are poorly differentiated foci with evidence of early sarcomatoid carcinoma. In older mice (>21wks) sarcomatoid carcinoma was seen. In the anterior prostate there was often intracystic carcinoma that filled the entire lumen of the gland with no invasion into the underlying stroma. Lung metastases were occasionally observed.
Robert Matusik’s group submitted multiple slides from different crosses on their LADY (LPb-Tag-12t7) backbone with mice with activating lesions of oncogenic pathways including. MYC, β-catenin (42) and IKB. The LADY model expresses SV40 large-T Antigen (without small t) which inactivates both Rb and p53 and these mice reproducibly develop mPIN. All models showed progression from mPIN to invasive adenocarcinoma, with extensive involvement of all murine prostate lobes. Some models exhibited a biphasic adenocarcinoma-neuroendocrine carcinoma pattern. Another interesting cross with LADY has been reported by Klezovitch et al (43). In this model, LADY mice were crossed with transgenic mice overexpressing hepsin, a serine protease that is expressed at increased levels in human PCa, using a probasin promoter. This model displayed multifocal metastasis to multiple organs and notably to bone. The metastatic lesions all express neuroendocrine markers. The panel reviewed a metastatic lesion in bone which showed infiltration by tumor cells (Fig 5A). The tumor cells had more abundant cytoplasm than is typically seen in neuroendocrine carcinomas but expressed markers characteristic of neuroendocrine differentiation.
Figure 5. Pathology of genetically engineered mouse models.

A. Metastasis to femur in LADY X Hepsin transgenic mouse. Sheets of epithelial cells within bone are shown. B. Sheets of poorly differentiated adenocarcimoma with focal squamous areas. AR Osr1 model. C. Adenocarcinoma in ubi-CAT mouse D, HG mPIN in mouse with loss of stromal TGF-β signaling.
Fibroblast growth factor (FGF) Pathway
There is abundant correlative evidence from the analysis of human PCa that fibroblast growth factors (FGFs) and FGF receptors play an important role in PCa (44). A number of mouse models expressing either FGF ligands and activated receptors have been developed (45–49). The most aggressive model is ligand-inducible FGFR1 transgenic mouse model, JOCK1 (juxtaposition of CID and kinase1). In this model, a transgene consisting of an FGFR1 kinase domain that contains a drug binding domain is localized to prostate epithelium via the ARR2PB promoter. In the presence of lipid-permeable dimerizer, AP20187 (a chemical inducer of dimerization, CID), the intracellular signaling domains of FGFR1 oligomerize and activate unrestrained FGFR1 signaling by juxtaposing FGFR1 kinase domains that contain a drug binding domain. When mice are treated with dimerizing drug the develop hyperplasia, mPIN, adenocarcinoma and sarcomatoid carcinomas (19). Marked desmosplastic stroma response was noted (MC12-0080BY-H&E-CL).
Androgen receptor pathway (MC12-0261ST-H&E-KB)
The androgen receptor (AR) plays a central role in the biology of PCa in humans. The Sun laboratory submitted mice with an inserted human AR transgene with a LoxP-stop-loxP (LSL) cassette into the mouse ROSA26 locus (50). Conditional transgene activation is driven by Cre mediated recombination of the LSL cassette. The Osr1 promoter was used to drive expression of Cre recombinase at day 11.5 in the urogenital sinus epithelium resulting in Ar expression in prostate epithelium. There is slow development of focal low grade mPIN lesions with rare high grade PIN was observed with increased frequency in the dorsal and anterior prostate. Large intracystic adenocarcinomas were observed in the anterior prostate (MC12-0258ST-H&E-KB). Despite their large size, there was only minimal invasion into the underlying stroma, however focal lymphovascular invasion was observed in one mouse. Within these large adenocarcinomas there were occasional foci of squamous differentiation (Fig 5B).
WNT/β-catenin/APC pathway
One published model of adenomatosis polyposis coli (APC) gene deletion mediated prostate neoplasia was submitted to the panel for review. In this model, Probasin-Cre mediated deletion of APC was engineered in an undefined mixed strain of mice that contained proportions of C57BL/6J and 129Sv/J strains (51). Extensive mPIN was identified by the panel in submitted samples, and adenocarcinoma was observed in supplementary samples from aged animals. Adenocarcinomas were predominantly intracystic with areas of microinvasion (MC02-0241). Distant metastases were neither observed nor reported by the investigators. The investigators reported in their paper that one feature of the model is that androgen is required for tumor formation but not maintenance.
Slides submitted by the Spencer group (Baylor College of Medicine) of mice with ubiquitous expression (MHC1 promoter) of an inducible LRP5 construct, which induces constitutive β-catenin signaling when treated with dimerizer drug (see JOCK1 above), were examined (52). Slides from mice showed adenocarcinoma (Fig 5C) and sarcomatoid carcinoma associated with reactive stroma in some mice.
RAS/RAF/MAP kinase pathway
Mutations of RAS have been observed in PCa but appear to be uncommon, at least in men in the US. Similarly, RAF mutations are uncommon. However, activation of the MAP kinase pathway is common in PCa and introduction of mutant RAS or RAF oncogenes can activate this pathway. As described above, such lesions can enhance progression and the development of sarcomatoid carcinomas in mice with PTEN loss. Patrick Humbert’s group submitted slides from their model of Cre-driven Scribble deficiency in combination with G12D K-ras (53). The Scribble complex is involved in establishing and maintaining epithelial polarity. Biallelic loss of Scribble was noted to result in mPIN in the slides reviewed. The group reviewed slides from the cross with K-rasG12D mutation. This combination resulted in mPIN and extensive desmoplasia and foci highly suspicious for microinvasion.
TGF-β pathway
Transforming growth factor-β (TGF-β) has a well established role in promoting carcinogenesis, including prostate carcinogenesis. Neil Bhowmick’s group has generated mice with partial Tgfbr2 knock out in stromal fibroblasts by crossing Tgfbr2floxE2/floxE26 and Col 1α2-Cre-ER mice in a C57BL/6 background. Systemic administration of tamoxifen activates Cre mediated recombination, yielding mice deficient in stromal Tgfbr2 mediated cell signaling. Slides from these mice showed high grade mPIN associated with stromal chronic inflammation (Fig 5D) (MC12-0047CSM-H&E-CL). Thus loss of TGF-β signaling in stroma induces paracrine signaling that is sufficient drive the initial stages of prostate carcinogenesis. Deletion of Smad4, a central mediator of TGF-β signaling, alone does not cause morphologic changes in the prostate (31); however in conjunction with Pten deletion as reported above causes an epithelial carcinoma with high metastatic potential.
SV40 T-antigen
TRAMP mice have been widely used in a variety of studies of the biology of PCa with over 400 publications to date. This model expresses a complete SV40-T antigen (small and large-T). TRAMP mice have a high incidence of neuroendocrine tumors arising in the prostate that are highly metastatic to lung, liver and other tissues (TG06-0394-3). TRAMP mice crossed to other GEM also have such lesions. TRAMP mice also have tumors of the seminal vesicles also develop polypoid tumors of epithelial cells admixed with stroma admixed (phylloides tumors). They are often multiple and quite large. Invasion is not common but some lesions have small foci of possible invasion. Addition of additional genetic lesions can inhibit development of the neuroendocrine carcinoma and metastasis. An example is the model reported by the Ronai group which was submitted to the group. In this model knockout of the ubiquitin ligase Siah2 suppresses formation of neuroendocrine tumors (54). Similar suppression has been seen with other crosses of knockout mice with TRAMP (55).
Xenograft models
Pure xenograft models
Wang and colleagues have developed xenograft models of PCa by engrafting fresh, histologically intact human primary prostate cancer tissues in severe combined immunodeficient (SCID) mice (56). Specifically, the investigators evaluated the graft take rate and the histopathologic features of cancer tissues implanted in the subcutaneous, sub-renal capsule, and orthotopic sites. While the efficiency of viable graft recovery was high at the sub-renal capsule and prostatic orthotopic sites (~95% and ~ 70%, respectively), the take rate of subcutaneously grafted tissues was only around 50%. Of note, the histopathologic features of the PCa grafts overlapped with those of the pre-implantation cancer tissues. Moreover, the differentiation marker PSA was expressed at similar levels in the tumor specimens before grafting and in the tumor grafts grown in testosterone-supplemented SCID hosts (MC12-0118BC-AR_PSA-EH). Overall, these data suggest that patient-derived PCa xenograft models could be valuable not only for investigating PCa biology but also for preclinical evaluation of experiments therapeutics in vivo. More recently, Wang’s group was able to successfully propagate PCa sub-renal capsule grafts and generate several well-characterized transplantable xenograft lines. Most of these xenografts lines, when engrafted under the renal capsules of SCID mice, consist of tumor cells that express AR and PSA, show local invasion into adjacent host kidney parenchyma, and display androgen dependent in vivo growth (Fig 6A). A subset of the grafts has the capability of spreading to distal organs, mostly as intravascular tumor emboli (Fig 6A). The investigators also successfully generated xenograft lines that are initially sensitive to castration in vivo, but subsequently become castration resistant. The castration-resistant tumors consist of small round cells that express neuroendocrine markers, are negative for AR and PSA, and display androgen-independent growth in vivo. Finally, a xenograft line derived from a metastatic lesion of a small cell carcinoma of the prostate (i.e. LTL-352) is also available. The sub-renal capsule tumors show histopathologic features of poorly differentiated neuroendocrine carcinoma (Fig. 6B), and are characterized by invasive growth into the host kidney parenchyma and metastases to distant organs. Additional details can be obtained at livingtumorlab.com
Figure 6. Representative microscopic images of patient-derived prostate carcinoma xenografts.

A. Tissue sections of a poorly differentiated PCa xenograft stained with hematoxylin and eosin (H&E) or immunostained for AR or PSA. Upper panels show invasive growth into the host kidney parenchyma. Lower panels show intravascular tumor emboli in the host lungs. B. Sub-renal capsule xenograft derived from a poorly differentiated neuroendocrine carcinoma of the prostate. Tissue sections stained with H&E or immunostained for AR, PSA, or synaptophysin (SYN) are shown.
The Vessella laboratory has generated 28 novel prostate cancer xenograft lines designated as the LuCaP series which were generated by implanting small fragments of human PCa tissues subcutaneously in male BALB/c nu/nu mice. Their overall yield in achieving established xenografts is approximately 18%. About 15% of the xenografts have been derived from primary PCa and the remainder from metastatic lesions obtained either at surgery or through the rapid autopsy program. Most of the LuCaP xenografts produce PSA that is detectable in the mouse sera, often up to several hundred nanograms per milliliter. Characterization of many of these xenografts including phenotypic, molecular, and response-to-therapy data has been reported in numerous publications. For example, Kumar et al (57) have described whole exome sequencing of a number of these xenografts revealing both recurrent and novel mutations and the origin of a number of these xenografts is described in that report. Histological examination revealed moderately to poorly differentiated adenocarcinomas, undifferentiated carcinoma and, in some tumors, histology compatible with neuroendocrine carcinoma (see Supplementary Fig 7) (MC12-0085UW-HE-KB).
Tissue recombination models
Prostate tissue recombinants engrafted under the kidney capsule of immunodeficient murine hosts are valuable tools to study the role of mesenchymal-epithelial interactions in both normal prostate development and prostate tumorigenesis (58) Several years ago, Hayward and colleagues established a non-tumorigenic immortalized human prostate epithelial cell line (BPH-1) by expressing the SV40 large T antigen in primary epithelial cell cultures. Tissue recombinants generated using BPH-1 cells and normal human prostatic fibroblasts show the presence of small epithelial nests and cords exhibiting squamous differentiation. Tissue recombinants generated using BPH-1 cells and fetal rat urogenital sinus mesenchyme (rUGM) display the presence of large nests of keratinizing squamous epithelium that, however, do not invade the host kidney parenchyma (Figure 7A). Of note, BPH-1 cells are able to form invasive tumors when recombined with human prostatic carcinoma-associated fibroblasts (CAFs). These tumors do not histologically resemble prostatic adenocarcinoma but display morphological characteristics of squamous cell carcinoma (Figure 7B) (59) (MC12-0169V-H&E-recut-KB). To develop models that more closely mimic key futures of PCa in patients, Hayward’s group recently established a spontaneously immortalized normal human prostate epithelial cell line (NHPrE1). Recombinants of NHPrE1 with rUGM show the presence of benign glandular structures that express prostate differentiation markers including AR, PSA and NKX3.1 (Figure 7C)(60). The investigators are now planning to expose NHPrE1 cells to multiple genetic and environmental insults to develop a model of multistep prostate tumorigenesis.
Figure 7. Tissue recombination models of prostate cancer.

A–C. Representative microscopic images of H&E stained sections of tissue recombinants generated using BPH-1 cells with rUGM (A), or CAFs (B). C. Representative microscopic image of H&E stained sections of tissue recombinants generated using NHPrE1 cells and rUGM. D. DRS model. Nests of tumors cells surrounded by blood vessels and stroma with eosinophilic material derived from Matrigel.
The Rowley laboratory has developed a tissue recombination model known as the differential reactive stoma (DRS) model using prostate stromal cells lines derived from normal human prostate (61) (MC12-0313BY-recut-HE-EH). The LNCaP cell line is normally poorly tumorigenic when injected subcutaneously in nude mice but when mixed with stromal cell lines tumorigenesis is significantly enhanced. Of note, only certain lines robustly enhance tumorigenesis. Matrigel is also added in the “3-way” DRS model. This model has been used to dissect stromal factors supporting PCa tumorigenesis (62–63). Stromal lines have also been developed from mouse prostates and can be derived from GEM mice with knockout of specific genes. Both the epithelial and stromal compartments can be manipulated to decrease or increase expression of specific genes allowing dissection of tumor stroma interactions. Histologically, tumors consist of nests of malignant epithelial cells admixed with stromal cells and blood vessels (Fig 7D). Eosinophilic masses of Matrigel may also be present.
Non-murine models
Rat prostate cancer models
Transgenic rat models of PCa have been developed. We examined a transgenic rat model with the SV40 T antigen under probasin promoter control, allowing prostate-specific gene expression (64). Male rats are said to develop PCa at 100% incidence before they are 15 weeks old. The slides examined show focal and diffuse hyperplasia and PIN and adenocarcinoma (Fig 8A) (MC12-0302DV) An area of neuroendocrine carcinoma was also seen (Fig 8B).
Figure 8. Non-murine models of prostate cancer.

A. Transgenic rat expressing SV40 large-T antigen. Adenocarcinoma with associated chronic inflammation. B. Transgenic rat expressing SV40 large-T antigen. Neuroendocrine carcinoma infiltrating around focal area of hyperplasia (arrow). C. Poorly differentiated adenocarcinoma of canine prostate with chronic inflammatory infiltrate.
Canine prostate cancer (MC12-0313BY-recut-HE-EH)
Pet dogs have been identified as an animal model that can contribute significantly to our understanding of cancer and to the treatment of spontaneous neoplasms. Dogs develop spontaneous HGPIN and carcinomas of the prostate gland that are often both aggressive and metastatic, although generally androgen insensitive (65). Lai et al have reviewed the histology of prostate cancer in the dog and identified multiple variants of adenocarcinoma, undifferentiated carcinoma and sarcomatoid carcinoma (66). Examples submitted showed poorly differentiated adenocarcinoma (Fig 8C) or undifferentiated carcinoma. Reactive stroma and inflammation were prominent. Clearly, there are important differences between the anatomy and histology of the canine and human prostate glands and in the neoplasms arising in them. However, human and canine prostate glands share a common embryologic origin and have many homologous anatomic and micro-anatomic structures, and even though prostate tumors in dogs tend to be hormone insensitive and many of the tumors arise in castrated dogs, they may perhaps provide a model of human advanced castrate resistant tumors. Regardless of differences between tumors of dogs and humans, the dog may be a useful complementary model to studies of human prostate cancer. The domestic dog shares a common environment with humans and is subject to sporadic neoplasia of presumptive multifactorial etiology that develops slowly over time influenced by an intact immune system, as is the case in humans. Additionally dog breeds present a useful genetic resource in the study of diseases such as cancer. The NCI has identified pet animals as a potential population to incorporate into studies of new therapeutics (67). The utility of the dog in studies of cancer therapeutics is further enhanced by the existence of resources for molecular studies in dogs; for example, the canine genome sequence, completed in 2005, represents an invaluable resource for studies of neoplasia in dogs. The dog’s important contribution to translational research directed at helping humans can potentially benefit pet dogs themselves.
DISCUSSION
Over the last 15 years numerous animal models of PCa, particularly GEM models, have been developed. The comprehensive analysis of the GEM models has identified two issues related to the pathology of these models that bears further discussion. The first is the observation that invasive cancers in the GEM models are associated with reactive stroma characterized by the presence of spindle cells (fibroblasts and/or myofibroblasts) and chronic inflammatory cells. Of note, some stromal changes are also noted in association with high grade mPIN in the GEM models. Most human PCa is not associated with histologically evident reactive stroma and tends to infiltrate the preexisting fibromuscular stroma with little histologically obvious reaction, although changes in gene and protein expression occur in this normal appearing stroma (68). Ayala and colleagues have identified a subset of human PCa with histologically evident reactive stroma (69–70). The degree of reactive stroma formation can be graded and cancers with the most extensive reactive stroma (reactive stroma Grade 3) have significantly worse outcome (69–70). Of note, reactive stroma formation was seen in stroma adjacent to some foci of HGPIN, similar to the observation in GEM models. Thus, the stroma of GEM models of PCa most closely resembles the stroma of this aggressive subgroup of human PCas. Clearly further work is needed to evaluate the biological similarities and differences between the tumor microenvironment in human PCa and GEM models. Of note, the tissue recombination xenograft models described above will be extremely useful in dissecting the relevant biology, at least for carcinoma associated fibroblasts and myofibroblasts.
Another common pathological phenotype noted was the high incidence of sarcomatoid carcinomas in the most aggressive GEM models. Of note, this phenotype was identified by the authors the published reports of some models while in other models it was not specifically commented on. Sarcomatoid carcinomas were associated with aggressive local growth and invasion and increased metastasis in many GEM models. We noted this phenotype on multiple Pten null models with additional genetic lesions. It was also identified in older mice in GEM models with unrestrained FGFR1 signaling (JOCK1) and high level ERG overexpression. However, it should be noted that the presence of sarcomatoid carcinoma was not always associated with increased metastasis. In a Pten null p53 null GEM model with sarcomatoid carcinoma in 100% of mice with end stage disease, lymphovascular invasion was ubiquitous, however local lymph node metastasis was rare and distant metastases were not observed (21). In addition, an orthotopic transplant model that was developed using these PCa cells demonstrated that a clonal cell line which gave rise to sarcomatoid carcinoma did not develop distant metastasis. It is unclear whether all GEM models will necessarily display sarcomatoid differentiation when they display aggressive behavior and this will need to be carefully evaluated moving forward. The histological evidence of sarcomatous differentiation has been shown to be due to EMT in several models. Histologically evident sarcomatous differentiation is not uncommon in a number of cancers such as lung and renal cell carcinoma and is associated with aggressive clinical behavior, but is uncommon in human PCa. Careful studies are needed to determine the extent to which this EMT reflects molecular events in aggressive human PCa.
In general, specific genetic lesions were associated with characteristic histological patterns which are readily recognizable. Models associated with the PTEN/AKT pathway develop PIN and high grade, invasive lesions with characteristic cytological features that differ from similar lesions in the other models. The PTEN/AKT signature phenotype can be identified by the large nuclei with a delicate chromatin pattern and relatively small nucleoli (MC12-0699BIL). The light pinkish cytoplasm tends to be relatively abundant with well-defined cell borders (MC02-0699). In contrast, the TRAMP neuroendocrine tumors have small compact nuclei with dense hyperchromatic nuclei and very scanty cytoplasm without defined cytoplasmic membranes. The signature MYC pathway cells have round to oval nuclei with a course chromatin rim with prominent nucleoli (TG04-0319-HE) as seen commonly in human PCa. The cytoplasm is relatively sparse and frequently clear (TG04-0319LA). The lesions associated with the Ras oncogene display a wide variety of phenotypes rarely seen with the other neoplastic phenotypes. For example, many Ras-associated tumors have goblet cells indicative of intestinalization (MC02-0696). Squamous metaplasia was found in Ras-associated neoplasms (MC12-0254CA; MC12-0248CA). In addition, Ras expression was associated with sarcomatoid carcinomas. Other phenotypes were observed but the limited number of slides available with other models precluded definitive conclusions.
The use of the animal models has significantly enhanced our understanding of the pathobiology of PCa. Accurate pathological analysis of these models is critical for their optimal use. Moving forward it will be important to make use of these models to develop improved methods of preventing, detecting and treating human PCa. The optimal model or models for such studies is not yet clear and it is anticipated that correlation of results in animal models and human patients will be the ultimate test of the utility of these models.
Supplementary Material
Acknowledgments
Grant Support: NCI: U01 Mouse Models of Human Cancers Consortium (U01CA141497; U01 CA141582); P30CA125123 (M.I.); UCLA, Pacific Northwest and DF/HCC SPOREs in Prostate Cancer, P01 CA89021 (M.L.) and RO1 CA131945 (M.L); UWCCC Core Grant P30 CA014520 (RS) P30 CA069533 13S5 through the OHSU-Knight Cancer Institute (G.V.T.); Department of Defense Prostate Cancer Research Program (W81XWH-11-1-0227 J.H.); Prostate Cancer Foundation
The technical assistance of Katie Bell, Judy Walls (Histology) and Ed Hubbard and Arishneel Ram (WSI) at University of California, Davis is gratefully acknowledged.
References
- 1.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell 1proliferation through paracrine mechanisms. Cancer Res. 2006;66:794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi-Frias D, Vakar-Lopez F, Coleman IM, Plymate SR, Reed MJ, Nelson PS. The effects of aging on the molecular and cellular composition of the prostate microenvironment. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A. 2009;106:12465–12470. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell. 2007;12:572–585. doi: 10.1016/j.ccr.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci U S A. 2006;103:7789–7794. doi: 10.1073/pnas.0602567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–305. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shirai T, Takahashi S, Cui L, Futakuchi M, Kato K, Tamano S, et al. Experimental prostate carcinogenesis - rodent models. Mutat Res. 2000;462:219–226. doi: 10.1016/s1383-5742(00)00039-9. [DOI] [PubMed] [Google Scholar]
- 10.Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 11.Wang F. Modeling human prostate cancer in genetically engineered mice. Prog Mol Biol Transl Sci. 2011;100:1–49. doi: 10.1016/B978-0-12-384878-9.00001-7. [DOI] [PubMed] [Google Scholar]
- 12.Jeet V, Russell PJ, Khatri A. Modeling prostate cancer: a perspective on transgenic mouse models. Cancer Metastasis Rev. 2010;29:123–142. doi: 10.1007/s10555-010-9212-9. [DOI] [PubMed] [Google Scholar]
- 13.van Weerden WM, Bangma C, de Wit R. Human xenograft models as useful tools to assess the potential of novel therapeutics in prostate cancer. Br J Cancer. 2009;100:13–18. doi: 10.1038/sj.bjc.6604822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irshad S, Abate-Shen C. Modeling prostate cancer in mice: something old, something new, something premalignant, something metastatic. Cancer Metastasis Rev. 2012 doi: 10.1007/s10555-012-9409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahler JF, Stokes W, Mann PC, Takaoka M, Maronpot RR. Spontaneous lesions in aging FVB/N mice. Toxicol Pathol. 1996;24:710–716. doi: 10.1177/019262339602400606. [DOI] [PubMed] [Google Scholar]
- 16.Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, Shen MM, et al. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161:727–735. doi: 10.1016/S0002-9440(10)64228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell. 2012;21:253–265. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson B, Magi-Galluzzi C, Zhou M. Intraductal carcinoma of the prostate. Arch Pathol Lab Med. 2012;136:418–425. doi: 10.5858/arpa.2011-0519-RA. [DOI] [PubMed] [Google Scholar]
- 19.Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12:559–571. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol. 2011;8:428–439. doi: 10.1038/nrurol.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y, Niu J, Huang J. Neuroendocrine differentiation in prostate cancer. Am J Transl Res. 2009;1:148–162. [PMC free article] [PubMed] [Google Scholar]
- 22.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 24.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomsen MK, Ambroisine L, Wynn S, Cheah KS, Foster CS, Fisher G, et al. SOX9 elevation in the prostate promotes proliferation and cooperates with PTEN loss to drive tumor formation. Cancer Res. 2010;70:979–987. doi: 10.1158/0008-5472.CAN-09-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clegg NJ, Couto SS, Wongvipat J, Hieronymus H, Carver BS, Taylor BS, et al. MYC cooperates with AKT in prostate tumorigenesis and alters sensitivity to mTOR inhibitors. PLoS One. 2011;6:e17449. doi: 10.1371/journal.pone.0017449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Kobayashi T, Floc’h N, Kinkade CW, Aytes A, Dankort D, et al. B-Raf Activation Cooperates with PTEN Loss to Drive c-Myc Expression in Advanced Prostate Cancer. Cancer Res. 2012;72:4765–4776. doi: 10.1158/0008-5472.CAN-12-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin P, Liu YN, Pierce R, Abou-Kheir W, Casey O, Seng V, et al. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am J Pathol. 2011;179:422–435. doi: 10.1016/j.ajpath.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–273. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, et al. Telomerase Reactivation following Telomere Dysfunction Yields Murine Prostate Tumors with Bone Metastases. Cell. 2012;148:896–907. doi: 10.1016/j.cell.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai KP, Yamashita S, Huang CK, Yeh S, Chang C. Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol Med. 2012;4:791–807. doi: 10.1002/emmm.201101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci U S A. 2003;100:7841–7846. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 37.Kim J, Roh M, Doubinskaia I, Algarroba GN, Eltoum IE, Abdulkadir SA. A mouse model of heterogeneous, c-MYC-initiated prostate cancer with loss of Pten and p53. Oncogene. 2012;31:322–332. doi: 10.1038/onc.2011.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Cai Y, Ren C, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 40.Casey OM, Fang L, Hynes PG, Abou-Kheir WG, Martin PL, Tillman HS, et al. TMPRSS2- driven ERG expression in vivo increases self-renewal and maintains expression in a castration resistant subpopulation. PLoS One. 2012;7:e41668. doi: 10.1371/journal.pone.0041668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:2105–2110. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu X, Wang Y, DeGraff DJ, Wills ML, Matusik RJ. Wnt/beta-catenin activation promotes prostate tumor progression in a mouse model. Oncogene. 2011;30:1868–1879. doi: 10.1038/onc.2010.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klezovitch O, Chevillet J, Mirosevich J, Roberts RL, Matusik RJ, Vasioukhin V. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell. 2004;6:185–195. doi: 10.1016/j.ccr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 44.Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer. 2004;11:709–724. doi: 10.1677/erc.1.00535. [DOI] [PubMed] [Google Scholar]
- 45.Wang F, McKeehan K, Yu C, Ittmann M, McKeehan WL. Chronic activity of ectopic type 1 fibroblast growth factor receptor tyrosine kinase in prostate epithelium results in hyperplasia accompanied by intraepithelial neoplasia. Prostate. 2004;58:1–12. doi: 10.1002/pros.10311. [DOI] [PubMed] [Google Scholar]
- 46.Jin C, McKeehan K, Guo W, Jauma S, Ittmann MM, Foster B, et al. Cooperation between ectopic FGFR1 and depression of FGFR2 in induction of prostatic intraepithelial neoplasia in the mouse prostate. Cancer Res. 2003;63:8784–8790. [PubMed] [Google Scholar]
- 47.Freeman KW, Welm BE, Gangula RD, Rosen JM, Ittmann M, Greenberg NM, et al. Inducible prostate intraepithelial neoplasia with reversible hyperplasia in conditional FGFR1-expressing mice. Cancer Res. 2003;63:8256–8263. [PubMed] [Google Scholar]
- 48.Freeman KW, Gangula RD, Welm BE, Ozen M, Foster BA, Rosen JM, et al. Conditional activation of fibroblast growth factor receptor (FGFR) 1, but not FGFR2, in prostate cancer cells leads to increased osteopontin induction, extracellular signal-regulated kinase activation, and in vivo proliferation. Cancer Res. 2003;63:6237–6243. [PubMed] [Google Scholar]
- 49.Elo TD, Valve EM, Seppanen JA, Vuorikoski HJ, Makela SI, Poutanen M, et al. Stromal activation associated with development of prostate cancer in prostate-targeted fibroblast growth factor 8b transgenic mice. Neoplasia. 2010;12:915–927. doi: 10.1593/neo.10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu C, Luong R, Zhuo M, Johnson DT, McKenney JK, Cunha GR, et al. Conditional expression of the androgen receptor induces oncogenic transformation of the mouse prostate. J Biol Chem. 2011;286:33478–33488. doi: 10.1074/jbc.M111.269894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruxvoort KJ, Charbonneau HM, Giambernardi TA, Goolsby JC, Qian CN, Zylstra CR, et al. Inactivation of Apc in the mouse prostate causes prostate carcinoma. Cancer Res. 2007;67:2490–2496. doi: 10.1158/0008-5472.CAN-06-3028. [DOI] [PubMed] [Google Scholar]
- 52.Shahi P, Park D, Pond AC, Seethammagari M, Chiou SH, Cho K, et al. Activation of Wnt signaling by chemically induced dimerization of LRP5 disrupts cellular homeostasis. PLoS One. 2012;7:e30814. doi: 10.1371/journal.pone.0030814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pearson HB, Perez-Mancera PA, Dow LE, Ryan A, Tennstedt P, Bogani D, et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin Invest. 2011;121:4257–4267. doi: 10.1172/JCI58509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell. 2010;18:23–38. doi: 10.1016/j.ccr.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polnaszek N, Kwabi-Addo B, Peterson LE, Ozen M, Greenberg NM, Ortega S, et al. Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer Res. 2003;63:5754–5760. [PubMed] [Google Scholar]
- 56.Wang Y, Revelo MP, Sudilovsky D, Cao M, Chen WG, Goetz L, et al. Development and characterization of efficient xenograft models for benign and malignant human prostate tissue. Prostate. 2005;64:149–159. doi: 10.1002/pros.20225. [DOI] [PubMed] [Google Scholar]
- 57.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108:17087–17092. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayward SW, Haughney PC, Rosen MA, Greulich KM, Weier HU, Dahiya R, et al. Interactions between adult human prostatic epithelium and rat urogenital sinus mesenchyme in a tissue recombination model. Differentiation. 1998;63:131–140. doi: 10.1046/j.1432-0436.1998.6330131.x. [DOI] [PubMed] [Google Scholar]
- 59.Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135–8142. [PubMed] [Google Scholar]
- 60.Jiang M, Strand DW, Fernandez S, He Y, Yi Y, Birbach A, et al. Functional remodeling of benign human prostatic tissues in vivo by spontaneously immortalized progenitor and intermediate cells. Stem Cells. 2010;28:344–356. doi: 10.1002/stem.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tuxhorn JA, McAlhany SJ, Dang TD, Ayala GE, Rowley DR. Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res. 2002;62:3298–3307. [PubMed] [Google Scholar]
- 62.Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005;65:8887–8895. doi: 10.1158/0008-5472.CAN-05-1702. [DOI] [PubMed] [Google Scholar]
- 63.Yang F, Strand DW, Rowley DR. Fibroblast growth factor-2 mediates transforming growth factor-beta action in prostate cancer reactive stroma. Oncogene. 2008;27:450–459. doi: 10.1038/sj.onc.1210663. [DOI] [PubMed] [Google Scholar]
- 64.Cho YM, Takahashi S, Asamoto M, Suzuki S, Inaguma S, Hokaiwado N, et al. Age-dependent histopathological findings in the prostate of probasin/SV40 T antigen transgenic rats: lack of influence of carcinogen or testosterone treatment. Cancer Sci. 2003;94:153–157. doi: 10.1111/j.1349-7006.2003.tb01412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leroy BE, Northrup N. Prostate cancer in dogs: comparative and clinical aspects. Vet J. 2009;180:149–162. doi: 10.1016/j.tvjl.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 66.Lai CL, van den Ham R, van Leenders G, van der Lugt J, Mol JA, Teske E. Histopathological and immunohistochemical characterization of canine prostate cancer. Prostate. 2008;68:477–488. doi: 10.1002/pros.20720. [DOI] [PubMed] [Google Scholar]
- 67.Khanna C, London C, Vail D, Mazcko C, Hirschfeld S. Guiding the optimal translation of new cancer treatments from canine to human cancer patients. Clin Cancer Res. 2009;15:5671–5677. doi: 10.1158/1078-0432.CCR-09-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res. 1999;5:1063–1071. [PubMed] [Google Scholar]
- 69.Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, et al. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res. 2003;9:4792–4801. [PubMed] [Google Scholar]
- 70.Ayala GE, Muezzinoglu B, Hammerich KH, Frolov A, Liu H, Scardino PT, et al. Determining prostate cancer-specific death through quantification of stromogenic carcinoma area in prostatectomy specimens. Am J Pathol. 2011;178:79–87. doi: 10.1016/j.ajpath.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.