

Abstract
Hepatocellular carcinoma (HCC) is one of the most common and deadly human cancers and it remains poorly managed. Human HCC development is often associated both with elevated expression of inducible nitric oxide synthase (iNOS) and with genetic deletion of the major denitrosylase S-nitrosoglutathione reductase (GSNOR/ADH5). However, their causal involvement in human HCC is not established. In mice, GSNOR deficiency causes S-nitrosylation and depletion of the DNA repair protein O6-alkylguanine-DNA-alkyltransferase (AGT) and increases rates of both spontaneous and DEN carcinogen-induced HCC. Here we report that administration of 1400W, a potent and highly selective inhibitor of iNOS, blocked AGT depletion and rescued the repair of mutagenic O6-ethyldeoxyguanosines following DEN challenge in livers of GSNOR-deficient (GSNOR−/−) mice. Notably, short-term iNOS inhibition following DEN treatment had little effect on carcinogenesis in wild-type mice, but was sufficient to reduce HCC multiplicity, maximal size, and burden in GSNOR−/− mice to levels comparable with wild-type controls. Furthermore, increased HCC susceptibility in GSNOR−/− mice was not associated with an increase in interleukin 6, tumor necrosis factor-α, oxidative stress, or hepatocellular proliferation. These results suggested that GSNOR deficiency linked to defective DNA damage repair likely acts at the tumor initiation stage to promote HCC carcinogenesis. Together, our findings provide the first proof of principle that HCC development in the context of uncontrolled nitrosative stress can be blocked by pharmacological inhibition of iNOS, possibly providing an effective therapy for HCC patients.
Keywords: S-nitrosylation, Nitric oxide, S-nitrosoglutathione reductase, O6-alkylguanine-DNA-alkyltransferase, Tumor initiation
Introduction
Hepatocellular carcinoma (HCC) is one of the most lethal human cancers and is the third leading cause of cancer deaths worldwide (1). The major risk factors for HCC are chronic hepatitis and cirrhosis caused by hepatitis B and C virus infection and heavy alcohol consumption, which affect hundreds of millions of people (2). Because of the spread of hepatitis C virus infection, HCC incidence and mortality in many parts of the world including the United States are rapidly increasing (2). The molecular mechanisms through which risk factors contribute to hepatocarcinogenesis remain poorly understood (3), resulting in a paucity of effective therapeutic approaches and extremely poor prognosis for HCC patients.
The development of human HCC is often associated with elevated production of nitric oxide (NO) and an increase in nitrosative stress. Inducible nitric oxide synthase (iNOS), the enzyme responsible for the high production of NO in the innate immune response and inflammation, is often substantially increased at both mRNA and protein levels in hepatocytes of patients with chronic hepatitis B and C virus infection (4–6), hemochromatosis (7), and alcoholic cirrhosis (8), all of which are established risk factors of HCC (2). Furthermore, iNOS is expressed at high levels in hepatocytes within HCC (6, 9), and HCC patients exhibit elevated concentrations of plasma nitrite/nitrate, the stable end-products of NOS activity (10, 11). Expression of iNOS has also been described in the development of fibrosis-associated (12) or DEN-induced (13, 14) HCC in animal models. NO affects functions of a wide range of proteins through S-nitrosylation, the covalent modification of cysteine thiols (15). Protein S-nitrosylation is increased by NOS activity but down-regulated by S-nitrosoglutathione reductase (GSNOR), a ubiquitous and highly conserved denitrosylase (16–18). By preventing excessive protein S-nitrosylation, GSNOR plays an evolutionarily conserved, critical role in protecting against nitrosative stress (16, 17). The human GSNOR gene (ADH5) is located at approximately 4q23, a region in which chromosomal deletion occurs frequently in cirrhotic and dysplastic hepatocytes and in HCC (19–23). Furthermore, the abundance and activity of GSNOR are significantly decreased in cancer samples from ~50% of patients with HCC (24). Interestingly, gene-expression profiling showed that both GSNOR deficiency and iNOS overexpression in the liver are closely associated with de novo hepatocarcinogenesis after tumor resection and a poor prognosis in HCC patients (25). Despite the accumulating evidence suggesting that excessive S-nitrosylation and nitrosative stress from concurrent GSNOR deficiency and iNOS overexpression in the liver may contribute critically to human HCC, their precise involvement in the etiology of HCC has not been well studied and remains poorly defined.
Our previous studies using a mouse line with targeted deletion of the GSNOR gene suggested that S-nitrosylation from GSNOR deficiency promotes both spontaneous and DEN-induced HCC, thereby establishing a model for studying the important roles of dysregulated protein S-nitrosylation in hepatocarcinogenesis (24). In this model, we showed that during inflammatory responses following intraperitoneal injection of DEN or lipopolysaccharide (LPS), GSNOR deficiency led to S-nitrosylation, ubiquitination, and proteasomal degradation of O6-alkylguanine-DNA-alkyltransferase (AGT). AGT is the major DNA repair enzyme responsible for repair of highly mutagenic and cytotoxic O6-alkylguanines (26) and is important for protecting against dialkylnitrosamine-induced HCC (27, 28). AGT, not susceptible to inactivation by tyrosine nitration, is highly susceptible to inactivation by S-nitrosylation (29). We showed that in the livers of DEN-challenged GSNOR−/− mice, the repair of carcinogenic O6-ethyldeoxyguanosine was impaired, leading to their persistent elevation. Predisposition to HCC, S-nitrosylation and depletion of AGT, and accumulation of O6-ethyldeoxyguanosine due to GSNOR deficiency were all largely abrogated by concurrent deletion of the iNOS gene in GSNOR−/−iNOS−/− mice, suggesting an important role for iNOS-derived S-nitrosylation in AGT inactivation and liver carcinogenesis in GSNOR−/− mice (24). Moreover, hepatocyte-specific deletion of GSNOR caused nitrosative inactivation of liver AGT and increased DNA damage and mortality after DEN challenge (30), suggesting the importance of GSNOR regulation of S-nitrosylation in liver parenchymal cells. However, GSNOR, previously known as alcohol dehydrogenase class III, has been shown in vitro to have enzymatic activity towards long-chain primary alcohols as well as glutathione-dependent formaldehyde dehydrogenase activity (16, 31). In addition, findings derived by comparison of GSNOR−/− and GSNOR−/−iNOS−/− mice could be confounded by potential differences in genetic background or animal development between the two mouse lines. Thus, it is possible that dysregulated S-nitrosylation may not be the only mechanism through which GSNOR deficiency contributes to hepatocarcinogenesis. Furthermore, it is currently unclear during which stage of tumorigenesis, initiation or promotion, GSNOR deficiency has the critical effect on HCC development.
In the present study, we sought to determine whether increased hepatocarcinogenesis from GSNOR deficiency could be reduced by pharmacological inhibition of NO production, which may represent a novel therapeutic approach to human HCC. As such, we used the highly selective pharmacologic inhibitor of iNOS, 1400W (32), to temporarily block iNOS activity during tumor initiation in the DEN-induced HCC model. We found that short-term iNOS inhibition after DEN-challenge reduces the acute genotoxic effects of DEN and significantly decreases the subsequent development of HCC.
Materials and Methods
Animals
GSNOR−/− mice (17) are in a C57BL/6 background after ten consecutive backcrosses with the wild-type mice. Both GSNOR−/− and C57BL/6 mice were kept in ventilated filter-top cages with centralized water supplies and maintained on normal mouse chow (5058 PicoLab Mouse Diet 20) in a specific pathogen-free facility at the University of California at San Francisco (UCSF). The experimental protocol was approved by the Institutional Animal Care and Use Committee of UCSF.
Acute DEN treatment and iNOS inhibition
Male GSNOR−/− and wild-type C57BL/6 mice were given at postnatal day 15 a single intraperitoneal injection of DEN (50 μg/g body weight). For 4 days after DEN treatment, the DEN-challenged mice were given intraperitoneal injections of phosphate-buffered saline (PBS) or 1400W (1 μg/g) twice daily to inhibit iNOS activity as we did previously (17). The inhibitor was not given orally because of its poor oral bioavailability (33). On day 5, the mice were euthanized and liver samples were collected. Levels of alanine aminotransferase (ALT) were analyzed by University of California Davis Comparative Pathology Laboratory (Davis, CA).
Chemical hepatocarcinogenesis
Male GSNOR−/− and wild-type C57BL/6 mice, 3 to 5 per group, were given at postnatal day 15 a single intraperitoneal injection of DEN (5 μg/g). Starting at 6 hours after DEN challenge, half of the mice in a cage were intraperitoneally injected with 1400W (1 μg/g) once daily for four days, whereas the other half of the mice were injected with PBS. Mice are weaned at postnatal day 21–22 and kept under standard barrier conditions. Mice were sacrificed nine months after DEN challenge, and livers were harvested and examined for tumor growth. Visible superficial tumors were counted and measured. Tumor burden was estimated by the sum of the calculated areas (πr2) of counted tumors.
Immunoblot
Liver lysates were prepared by homogenization of samples on ice in a lysis buffer containing 50 mM Tris-HCl, pH 8.0, 0.1% NP-40, 1 mM EDTA, 0.1 mM DTPA, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Roche, Basel, Switzerland). Proteins (25 – 50 μg) were separated by SDS-PAGE under reducing conditions, transferred to nitrocellulose membranes, and probed with a goat antiserum against AGT (R&D Systems, Minneapolis, MN), or a monoclonal antibody against β-actin (A5441; Sigma, St. Louis, MO) followed by detection with corresponding secondary antibodies coupled to Alexa Fluor 680 (Invitrogen, Carlsbad, CA) or IRDye 800 (Rockland Immunochemicals, Gilbertsville, PA), and imaged and quantified using an infrared fluorescence imaging system (Odyssey; LICOR Biosciences, Lincoln, NE) (30). The fluorescent signal corresponding to AGT, normalized to that of β-actin, was compared to the mean of the control in each experiment. A rabbit polyclonal antibody (kindly provided by Dr. Harry Ischiropoulos) was used for the detection of nitrotyrosine.
Immuno-slot blot
O6-ethyldeoxyguanosine (O6-etdG) and O2-ethyldeoxythymidine (O2-etdT) lesions were detected as previously described (24). Briefly, genomic DNA was isolated from the livers of mice injected with DEN (50 μg/g intraperitoneally, at postnatal day 15) and 0 or 1 μg/g of 1400W (for 4 days). Ethylnitrosourea-treated calf thymus DNA was used as a positive control. The DNA was sonicated, denatured, and transferred to nitrocellulose membrane using a Bio-Dot slot blotting apparatus (Bio-Rad). Lesions were detected using a rat monoclonal antibody against O6-etdG (ER6, Axxora) or a mouse monoclonal antibody against O2-etdT (EM 4-1, Axxora) and imaged using Femto Chemiluminescent Substrate (Thermo Scientific) on Hyperfilm ECL film (GE Healthcare, Buckinghamshire, UK). The optical density of the scanned image was quantified using Odyssey Application Software. The signal corresponding to O6-etdG in a 1400W-treated sample was compared to the mean of the PBS-treated control in each experiment.
Glutathione, glutathione disulfide, and glutathione reductase activity
Glutathione (GSH) levels are determined by a glutathione disulfide (GSSG) reductase recycling assay modified from Griffith (34). Liver samples are homogenized by sonication in 5 volumes per gram of wet tissue weight of 5% sulfosalicylic acid. The liver homogenates are centrifuged, and the supernatants are measured immediately for total glutathione (GSH + GSSG). To determine glutathione disulfide, GSH in the sample is first masked by 2-vinglagridine. Glutathione levels are normalized with protein contents in the liver homogenates. Glutathione reductase activity was measured essentially as described by Smith et al. (35).
8-hydroxy-2′-deoxyguanosine (8-OHdG) measurement
Genomic DNA from the livers of DEN-treated mice was purified (GenElute Mammalian Genomic DNA kit, Sigma) in the presence of 0.1 mM desferal, an iron chelator to prevent autooxidation of DNA in vitro. Single-strand DNA was generated by boiling followed by rapid cooling on ice. After addition of sodium acetate (pH 5.3, 100 mM final), DNA was digested with 1U nuclease P1 at 50°C for 2h. The pH was adjusted with 1M Tris-HCl (pH 8.0) and the DNA was treated with alkaline phosphatase at 37°C for 1h. The digest was centrifuged for 5 min at 10,000 × g. The supernatant containing nucleotides was assayed for 8-OHdG using an EIA kit (Catalog number 589320, Cayman Chemical) according to manufacturer protocol. Absorbance at 410 nm in the absence (B0) or presence (B) of DNA, through the logit transformation, ln [(B/B0)/(1 − B/B0)], was used to generate the standard curve and calculate the concentration of 8-OHdG in the samples.
Real-time quantitative PCR
RNA from the livers of DEN-treated mice was isolated by Trizol/Chloroform extraction followed by silica-cartridge purification (PureLink RNA Mini Kit, Ambion). RNA quantity and quality were determined by UV absorbance at 260nm and A260/280 ratio, respectively using a NanoDrop spectrophotometer. Quantitative RT-PCR was performed using the Power SYBR Green RNA-to-Ct 1-Step Kit (Applied Biosystems) according to manufacturer’s protocol. Primers used are as follows: interleukin 6 (IL-6): 5′-TAGTCCTTCCTACCCCAATTTCC-3′ and 5′-TTGGTCCTTAGCCACTCCTTC-3′ (PrimerBank ID 13624311A1); tumor necrosis factor-α (TNF-α): 5′-CCCTCACACTCAGATCATCTTCT-3′ and 5′-GCTACGACGTGGGCTACAG-3′ (PrimerBank ID 7305585A1); glyceraldehyde 3-phosphate dehydrogenase (GAPDH): 5′-AATGGTGAAGGTCGGTGTG-3′ and 5 ′-GTGGAGTCATACTGGAACATGTAG-3′ (PrimeTime Mm.PT.39a.1, IDT). The specificity of PCR product was determined by melt curve and agarose gel analyses.
Cytokines
Liver IL-6 and TNF-α expression was quantified using the Luminex Cytokine assay (Invitrogen) per manufacturer protocol.
Statistical analysis
Tumor number, maximal size, and burden of each group were compared using a two-tailed, unpaired Student’s t-test. Comparison of the results using the Mann-Whitney U-test yielded similar results. Other data were analyzed with a two-tailed, unpaired Student’s t-test.
Results
Pharmacologic inhibitor of iNOS prevents AGT depletion in DEN-challenged GSNOR−/−mice
We employed the DEN model to determine whether acute nitrosative inactivation of AGT in GSNOR−/− mice can be prevented by pharmacological inhibition of iNOS. While AGT decrease after nitrosamine treatment largely recovers by day 4 in wild-type mice (36), AGT level remains low in GSNOR−/− mice 4 to 6 days after DEN challenge (24). Therefore, we treated the DEN-challenged GSNOR−/− mice with 1400W twice a day for 4 days (Fig. 1). Livers were harvested on day 5 and analyzed for AGT. As expected, DEN challenge in GSNOR−/− mice resulted in a significant reduction in liver AGT levels (24, 30) (Fig. 1). AGT levels were rescued in the DEN-challenged GSNOR−/− mice by 1400W treatment (Fig. 1). Thus, our scheme of iNOS inhibition was effective in protecting AGT in GSNOR−/− mice. These data provide pharmacological evidence that AGT is highly susceptible to nitrosative inactivation.
Figure 1.

Inhibition of iNOS by 1400W reverses AGT depletion after DEN treatment. A, Postnatal day 15 GSNOR−/− mice were injected intraperitoneally with DEN. Starting at 6 hours after DEN injection, 1400W (+) or PBS (−) was administered intraperitoneally twice daily for 4 days. Livers were collected on day 5, and lysates were analyzed by immunoblot using antibodies against AGT and β-actin. Each lane represents an individual mouse. B, Quantification of AGT protein detected by immunoblot. AGT levels (mean ± SE) in 1400W-treated mice (n = 4) were significantly higher than that in control mice (n = 4; P = 0.004, two-tailed Student’s t-test).
Inhibition of iNOS after DEN challenge reduces O6-ethyldeoxyguanosine lesions in the liver of GSNOR−/− mice
To assess whether the protection of AGT by iNOS inhibition rescues the repair of O6-alkylguanines in the DEN-challenged GSNOR−/− mice, liver genomic DNA was analyzed for the presence of O6-alkylguanines by immuno-slot blot using a monoclonal antibody against O6-etdG (Fig. 2). We detected substantial amounts of O6-etdG after DEN treatment, and the levels of O6-etdG were decreased approximately five-fold by 1400W treatment (Fig. 2). Levels of O2-etdT, a DEN-induced lesion not repaired by AGT, were not significantly affected by iNOS inhibition (Fig. 2). Thus, pharmacologic inhibition of iNOS activity by 1400W after DEN challenge in GSNOR−/− mice specifically rescued the capacity to repair O6-etdG lesions likely through the protection of AGT.
Figure 2.
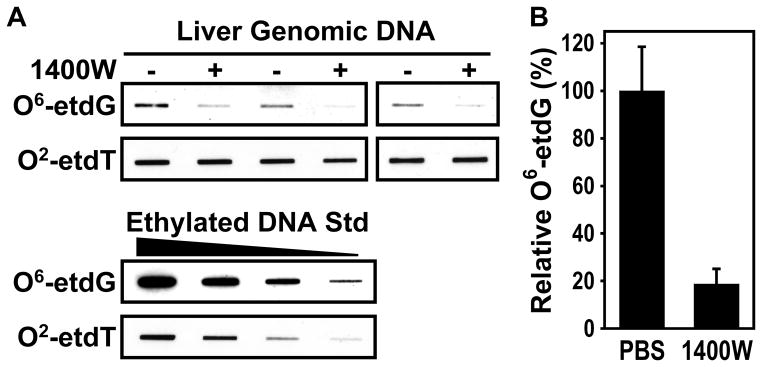
Inhibition of iNOS by 1400W reduces DEN-induced O6-ethyldeoxyguanosine (O6-etdG) lesions. A, Genomic DNA was isolated from the liver samples described in Fig. 1. The DNA (1 μg per lane) was analyzed by immuno-slot blot using monoclonal antibodies against O6-etdG and O2-ethyldeoxythymidine (O2-etdT) (upper panel). Ethylnitrosourea-alkylated calf thymus DNA (40, 20, 10, and 5 ng) was used as a positive control (lower panel). B, Quantification of relative O6-etdG levels. O6-etdG levels (mean ± SE) in 1400W-treated mice (n = 3) were significantly lower than that in control mice (n = 3; P = 0.013, two-tailed Student’s t-test).
Short-term 1400W treatment during tumor initiation reduces DEN-induced tumorigenesis in GSNOR−/− mice
Since we showed that short-term inhibition of iNOS by 1400W was capable of protecting AGT and preventing accumulation of O6-alkylguanines, we used the DEN-induced HCC model according to the scheme in Fig. 3A to determine whether short-term inhibition of iNOS for 4 days will reduce long-term tumorigenesis. As shown in Fig. 3B, we found that DEN-challenged GSNOR−/− mice typically developed numerous large liver tumors after 9 months. However, fewer and smaller tumors developed in GSNOR−/− mice treated with 1400W (Fig. 3B). Histopathological analysis showed in 1400W-treated and control mice the development of both hepatocellular adenomas and carcinomas (Fig. 3C).
Figure 3.

DEN-induced tumorigenesis following short-term treatment with 1400W. A, Schematic of the HCC study. Postnatal day 15 GSNOR−/− mice were injected intraperitoneally with DEN followed by intraperitoneal injection of 1400W or vehicle (PBS) once daily for 4 days. The mice were sacrificed 9 months after DEN treatment. Livers were harvested and examined for evidence of tumor growth. B, Livers from representative mice treated with DEN followed by PBS or 1400W (Bar = 10 mm). C, Hematoxylin and eosin staining of liver tumor sections (Bar = 100 μm). Data are representative of three 1400W-treated and three control GSNOR−/− mice.
We quantitatively evaluated the effect of iNOS inhibition on DEN-induced liver tumorigenesis in both wild-type and GSNOR−/− mice (Fig. 4). Consistent with previous reports (24), DEN challenge in absence of iNOS inhibition resulted in significantly higher tumor numbers in GSNOR−/− mice (n = 15) than in wild-type mice (n = 15) (P = 4.4 × 10−6, Fig. 4A). Importantly, while almost all DEN-treated GSNOR−/− mice developed liver tumors, short-term 1400W treatment following DEN-challenge (n = 17) resulted in a 70% reduction of tumor numbers in GSNOR−/− mice (P = 8.1 × 10−6, Fig. 4A). Similar 1400W treatment in DEN challenged wild-type mice (n = 13) had no effect on tumor multiplicity. Interestingly, 1400W treatment of GSNOR−/− mice brings the tumor number down to the level of wild-type mice. Thus, these data strongly suggest that tumor multiplicity in DEN-challenged GSNOR−/− mice critically depends on nitrosative stress in the initiation phase of tumorigenesis.
Figure 4.
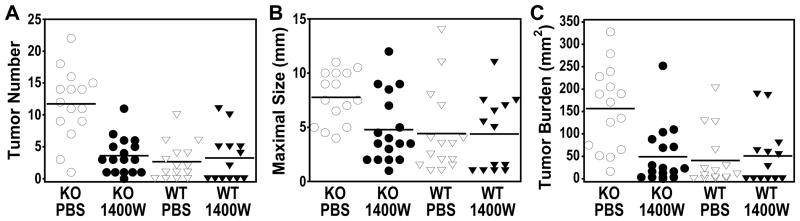
Short-term 1400W treatment during tumor initiation reduces DEN-induced tumorigenesis in GSNOR−/− mice. DEN-induced tumorigenesis as described in Fig. 3A was analyzed in the following four groups: wild-type (WT) and GSNOR−/− (KO) mice treated with 1400W or PBS. Each data point represents an individual animal, and the mean of each group is represented by a line. A, Number of tumors (≥ 2mm) on the liver surface in each animal. The tumor number was significantly higher in the PBS-treated KO mice compared to each of the other three groups (P < 8 × 10−5), which was not significantly different from each other. B, The maximal tumor diameter was significantly higher in the PBS-treated KO mice compared to each of the other three groups (P < 0.009). C, The tumor burden, calculated as the sum of the areas of surface tumors, was significantly higher in the PBS-treated KO mice compared to each of the other three groups (P < 0.003).
We next analyzed tumor size in the four experimental groups (Fig. 4B). Consistent with previous reports (24), DEN challenge resulted in significant increase in maximal tumor size in GSNOR−/− versus wild-type mice (Fig. 4B, P = 0.0085). Short-term 1400W treatment following DEN-challenge had no effect on maximal tumor size in wild-type mice (Fig. 4B), and 1400W did not affect HCC cell growth in culture (Supplemental Fig. 1). However, short-term 1400W treatment resulted in a significant reduction of maximal tumor size in GSNOR−/− mice (Fig. 4B, P = 0.0058). Indeed, 1400W treatment of GSNOR−/− mice brings the maximal tumor size down to the level of wild-type mice. Thus inhibition of nitrosative stress during tumor initiation in DEN-challenged GSNOR−/− mice reduced maximal tumor size.
For the four experimental groups, we also analyzed tumor burden, which was calculated as the sum of the tumor area for each animal (Fig. 4C). DEN challenge led to an over 3-fold increase in tumor burden in GSNOR−/− mice when compared to wild-type control (P = 0.00042). Again, short-term 1400W treatment following DEN challenge had no effect on wild-type mice but significantly reduced tumor burden in GSNOR−/− mice, decreasing it to the level of wild-type mice. Thus inhibition of nitrosative stress during tumor initiation in GSNOR−/− mice also reduced tumor burden.
Liver injury, inflammation, and proliferation following DEN treatment are comparable in wild-type and GSNOR−/− mice
DEN can cause liver damage and inflammation, resulting in compensatory proliferation, which may contribute to DEN-induced HCC (37). We found that serum ALT levels, a measure of liver injury, are comparable between DEN-challenged GSNOR−/− mice and wild-type controls (Fig. 5A). Since inflammatory IL-6 and TNF-α were previously shown to promote HCC (37, 38), we analyzed livers from DEN-treated mice by quantitative PCR for the expression levels of IL-6 and TNF-α mRNA (Fig. 5B). The expression levels of IL-6 and TNF-α in DEN-challenged GSNOR−/− and wild-type mice did not differ significantly (Fig. 5B). Similarly, IL-6 and TNF-α protein levels in the liver were not significantly different between DEN-challenged wild-type and GSNOR−/− mice either (Fig. 5C). Thus, GSNOR deficiency does not appear to directly affect IL-6 and TNF-α expression after DEN challenge. To assess whether proliferation differed between DEN-treated wild-type and GSNOR−/− mice, we performed immunohistochemical analysis of Ki-67, a marker of proliferating cells. Again, we found no significant difference in proliferation in DEN-challenged wild-type and GSNOR−/− mice (Fig. 5D). Thus, these parameters do not appear to contribute significantly to the increase in HCC promoted by GSNOR deficiency.
Figure 5.
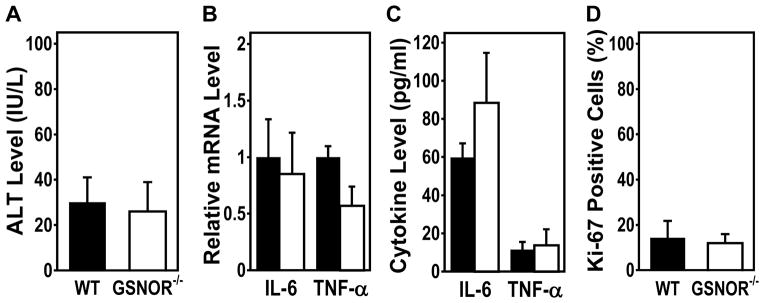
Serum ALT, liver IL-6 and TNF-α expression, and proliferation in DEN-treated wild-type and GSNOR−/− mice. A, Serum ALT levels assessed two days after DEN challenge (25 μg/g). The data (mean ± SD) are from five wild-type and five GSNOR−/− mice. B, Relative IL-6 and TNF-α mRNA levels after DEN treatment in four paired wild-type (filled) and GSNOR−/−(open) mice. IL-6 and TNF-α mRNA levels, analyzed by quantitative RT-PCR and normalized to GAPDH mRNA levels, are not statistically different between wild-type and GSNOR−/− mice (PIL-6 = 0.54, PTNF-α = 0.1). C, IL-6 and TNF-α protein levels after DEN treatment in four paired wild-type (filled) and GSNOR−/− (open) mice. The cytokine levels are not statistically different between wild-type and GSNOR−/− mice (PIL-6 = 0.08, PTNF-α = 0.64). D, Quantification of immunohistochemical staining for Ki-67 in livers of DEN-treated mice. Liver samples were collected 48 hours after DEN injection (25 μg/g) into 15-day-old wild-type (n = 5) and GSNOR−/− (n = 6) mice. In all panels, the data are mean ± SD.
DEN treatment may cause acute oxidative stress (39); persistent oxidative damage from deficiency in the antioxidant enzyme CuZn-superoxide dismutase can lead to increased incidence of HCC (40). We found that the levels of glutathione, glutathione disulfide, and glutathione reductase activity in the livers of GSNOR−/− mice did not differ significantly from that in wild-type control (Supplemental Fig. 2). Neither DEN alone nor DEN with 1400W affected glutathione reductase activity (Supplemental Fig. 2B). Moreover, the levels of 8-OHdG, a major DNA product from oxidative stress, are comparable in the livers of DEN-treated GSNOR−/− and wild-type mice (Supplemental Fig. 3). We also analyzed protein tyrosine nitration in the livers of DEN-treated mice by immunoblot and found no increase in overall protein tyrosine nitration by GSNOR deficiency (Supplemental Fig. 4). Thus, the livers of GSNOR−/− mice, compared to wild-type controls, do not appear to suffer significantly increased oxidative stress.
Discussion
We previously showed that GSNOR deficiency resulted in an iNOS-dependent increase in susceptibility to spontaneous and DEN-induced HCC (24). In the present study, we investigated whether during the stage of tumor initiation, S-nitrosylation and nitrosative stress from GSNOR deficiency has the critical effect on HCC development. We demonstrated that pharmacologic inhibition of iNOS activity during DEN challenge in GSNOR−/− mice prevented both the depletion of the important DNA repair protein AGT and persistent elevation of carcinogenic O6-alkylguanines. Importantly, we showed that short-term iNOS inhibition during the stage of tumor initiation by DEN was sufficient to reduce HCC multiplicity, maximal size, and burden in GSNOR−/− mice to levels observed in wild-type mice. Furthermore, we found that increased HCC susceptibility in GSNOR−/− mice was not associated with an increase in IL-6, TNF-α, oxidative stress, or hepatocellular proliferation. Thus, the results suggest that S-nitrosylation from GSNOR deficiency, likely through the inhibition of DNA damage repair, promotes hepatocarcinogenesis at the stage of tumor initiation. Our findings provide the first proof of principle that HCC development in the context of uncontrolled nitrosative stress can be inhibited by pharmacological inhibitor of iNOS.
Employing a pharmacological inhibitor specific to iNOS in DEN-challenged GSNOR−/− mice, we provided evidence for a critical role of nitrosative stress in HCC development at the stage of tumor initiation. Previously we showed that the increase in susceptibility to both spontaneous and DEN-induced HCC from GSNOR deficiency was abolished in GSNOR−/−iNOS−/− mice, providing genetic evidence for an important role of iNOS-derived S-nitrosylation in HCC development (24). Nevertheless, the GSNOR−/− and GSNOR−/−iNOS−/− mice are not expected to have exactly the same genetic background, and genetic deletion of iNOS might result in compensatory changes in the development of GSNOR−/−iNOS−/− mice. These genetic and possible developmental differences may compromise interpretation of the results obtained from the two mouse lines. Furthermore, the genetic approach is not amenable to dissecting the role of dysregulated S-nitrosylation in the various stages of tumor development. The issues inherently associated with comparison of two mouse lines were avoided in the present study, which was performed in a single GSNOR−/− line. Our results with iNOS inhibition also addressed the concern raised by in vitro studies that GSNOR has dehydrogenase activities towards chemicals unrelated to S-nitrosylation. More importantly, the present work established a carcinogenic role for nitrosative stress specifically at tumor initiation. This is supported by the findings that the important DNA repair protein AGT is highly susceptible to S-nitrosylation-dependent inactivation (24, 29). We showed recently that whereas the frequency of 8-OHdG-inducible G:C to T:A mutations was not elevated in DEN-challenged GSNOR−/− mice compared to wild-type control, the frequency of the transition from G:C to A:T, a mutation deriving from DEN-induced O6-ethylguanines that are normally repaired by AGT, was significantly increased by GSNOR deficiency (41). Overall, our findings suggest that in the mouse model, S-nitrosylation-dependent AGT inactivation as a result of GSNOR-deficiency is a major contributor to increased mutagenesis and hepatocarcinogenesis. As S-nitrosylation can affect functions of a wide range of proteins, including many important to tumorigenesis (42), it remains to be explored that dysregulated S-nitrosylation from GSNOR deficiency may promote HCC by altering activities or functions of additional proteins during tumor initiation or other stages of hepatocarcinogenesis.
We found that inhibition of iNOS activity by 1400W did not affect HCC multiplicity, maximal size, or burden in DEN-challenged wild-type mice. Although cytotoxicity from NO or related activity, particularly at high levels provided from exogenous sources, has been shown for numerous cells including HCC cell lines (43), it is well established that NO produced endogenously by NOSs inhibits death of hepatocytes (44). Using iNOS−/− mice in both spontaneous and fibrosis-associated models of HCC, Denda and colleagues showed that iNOS activity has neither promoting nor inhibitory effect on hepatocarcinogenesis (12). Consistent with these findings, our current study further showed that in the presence of wild-type GSNOR, iNOS activity has little effect on hepatocellular tumor initiation in the DEN model. Thus, the pro-hepatocarcinogenic activity of iNOS, demonstrated both genetically (24) and pharmacologically, is normally prevented by GSNOR. These results suggest that potential therapeutic approaches utilizing iNOS inhibition in HCC patients should take GSNOR status into consideration.
Our findings of a critical role for GSNOR deficiency during tumor initiation may have important implications for understanding the pathogenesis of human HCC. Substantially elevated expression of iNOS has been extensively described in hepatocytes in both HCC and many pathological conditions predisposing to HCC (4–9). Somatic deletion of the human GSNOR gene (ADH5) occurs frequently in cirrhotic and dysplastic hepatocytes and in HCC (19–23), and significant reductions in the levels of both GSNOR protein and activity are apparently common in human HCC (24). Hoshida and colleagues showed through mRNA-expression profiling that in nontumoral liver tissues adjacent to resected primary tumors in HCC patients, an increase in iNOS expression and a decrease in GSNOR expression are both closely associated with de novo hepatocarcinogenesis after tumor resection and a poor prognosis (25). The underling mechanism of the association of iNOS and GSNOR with human HCC remains unclear, but our previous results and present findings suggest that excessive S-nitrosylation from concurrent GSNOR deficiency and iNOS overexpression may cause nitrosative inactivation of DNA repair systems, providing a driving force for mutagenesis and hepatocarcinogenesis.
Preventing increased hepatocarcinogenesis from GSNOR deficiency by pharmacological inhibitor of iNOS, demonstrated here as a proof of principle in a mouse model, may provide the basis for a potential therapeutic approach to HCC patients. In patients having concurrent GSNOR deficiency and iNOS overexpression in liver tissues adjacent to primary HCC, iNOS inhibition after tumor resection may be particularly attractive as an adjuvant therapeutic approach, which may provide a significant survival benefit. In addition, our findings suggest that loss of GSNOR in pre-neoplastic liver tissues could increase overall risk of HCC and therefore may have prognostic value. Inhibition of excessive S-nitrosylation in these patients may help to prevent HCC development.
Supplementary Material
Acknowledgments
Grant Support
This work was supported by the National Institutes of Health (R01CA55578 and R01CA122359 to L.L.).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: C. Tang, W. Wei, L. Liu
Development of methodology: C. Tang, W. Wei, L. Liu
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C. Tang, W. Wei, M.A. Hanes, L. Liu
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C. Tang, W. Wei, M.A. Hanes, L. Liu
Writing, review, and/or revision of the manuscript: C. Tang, W. Wei, M.A. Hanes, L. Liu
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C. Tang, W. Wei, M.A. Hanes, L. Liu
Study supervision: L. Liu
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–76. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 3.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 4.Majano PL, Garcia-Monzon C, Lopez-Cabrera M, Lara-Pezzi E, Fernandez-Ruiz E, Garcia-Iglesias C, et al. Inducible nitric oxide synthase expression in chronic viral hepatitis. Evidence for a virus-induced gene upregulation. J Clin Invest. 1998;101:1343–52. doi: 10.1172/JCI774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kane JM, 3rd, Shears LL, 2nd, Hierholzer C, Ambs S, Billiar TR, Posner MC. Chronic hepatitis C virus infection in humans: induction of hepatic nitric oxide synthase and proposed mechanisms for carcinogenesis. J Surg Res. 1997;69:321–4. doi: 10.1006/jsre.1997.5057. [DOI] [PubMed] [Google Scholar]
- 6.Rahman MA, Dhar DK, Yamaguchi E, Maruyama S, Sato T, Hayashi H, et al. Coexpression of inducible nitric oxide synthase and COX-2 in hepatocellular carcinoma and surrounding liver: possible involvement of COX-2 in the angiogenesis of hepatitis C virus-positive cases. Clin Cancer Res. 2001;7:1325–32. [PubMed] [Google Scholar]
- 7.Hussain SP, Raja K, Amstad PA, Sawyer M, Trudel LJ, Wogan GN, et al. Increased p53 mutation load in nontumorous human liver of wilson disease and hemochromatosis: oxyradical overload diseases. Proc Natl Acad Sci U S A. 2000;97:12770–5. doi: 10.1073/pnas.220416097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNaughton L, Puttagunta L, Martinez-Cuesta MA, Kneteman N, Mayers I, Moqbel R, et al. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc Natl Acad Sci U S A. 2002;99:17161–6. doi: 10.1073/pnas.0134112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeguchi M, Ueta T, Yamane Y, Hirooka Y, Kaibara N. Inducible nitric oxide synthase and survivin messenger RNA expression in hepatocellular carcinoma. Clin Cancer Res. 2002;8:3131–6. [PubMed] [Google Scholar]
- 10.Moussa YI, Plevris JN, Hayes PC. Plasma nitrites/nitrates in HCV infection and hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2000;12:159–63. doi: 10.1097/00042737-200012020-00005. [DOI] [PubMed] [Google Scholar]
- 11.Moriyama A, Masumoto A, Nanri H, Tabaru A, Unoki H, Imoto I, et al. High plasma concentrations of nitrite/nitrate in patients with hepatocellular carcinoma. Am J Gastroenterol. 1997;92:1520–3. [PubMed] [Google Scholar]
- 12.Denda A, Kitayama W, Kishida H, Murata N, Tamura K, Kusuoka O, et al. Expression of inducible nitric oxide (NO) synthase but not prevention by its gene ablation of hepatocarcinogenesis with fibrosis caused by a choline-deficient, L-amino acid-defined diet in rats and mice. Nitric Oxide. 2007;16:164–76. doi: 10.1016/j.niox.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Ahn B, Han BS, Kim DJ, Ohshima H. Immunohistochemical localization of inducible nitric oxide synthase and 3-nitrotyrosine in rat liver tumors induced by N-nitrosodiethylamine. Carcinogenesis. 1999;20:1337–44. doi: 10.1093/carcin/20.7.1337. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Zhang JJ, Wang X, Bu XY, Lou YQ, Zhang GL. Effect of berberine on hepatocyte proliferation, inducible nitric oxide synthase expression, cytochrome P450 2E1 and 1A2 activities in diethylnitrosamine- and phenobarbital-treated rats. Biomed Pharmacother. 2008;62:567–72. doi: 10.1016/j.biopha.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–4. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–28. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Wang ZE, Doulias PT, Wei W, Ischiropoulos H, Locksley RM, et al. Lymphocyte Development Requires S-nitrosoglutathione Reductase. J Immunol. 2010;185:6664–9. doi: 10.4049/jimmunol.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagai H, Pineau P, Tiollais P, Buendia MA, Dejean A. Comprehensive allelotyping of human hepatocellular carcinoma. Oncogene. 1997;14:2927–33. doi: 10.1038/sj.onc.1201136. [DOI] [PubMed] [Google Scholar]
- 20.Patil MA, Gutgemann I, Zhang J, Ho C, Cheung ST, Ginzinger D, et al. Array-based comparative genomic hybridization reveals recurrent chromosomal aberrations and JAB1 as a potential target for 8q gain in hepatocellular carcinoma. Carcinogenesis. 2005 doi: 10.1093/carcin/bgi178. [DOI] [PubMed] [Google Scholar]
- 21.Wong N, Lai P, Lee SW, Fan S, Pang E, Liew CT, et al. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: relationship to disease stage, tumor size, and cirrhosis. Am J Pathol. 1999;154:37–43. doi: 10.1016/S0002-9440(10)65248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeh SH, Chen PJ, Shau WY, Chen YW, Lee PH, Chen JT, et al. Chromosomal allelic imbalance evolving from liver cirrhosis to hepatocellular carcinoma. Gastroenterology. 2001;121:699–709. doi: 10.1053/gast.2001.27211. [DOI] [PubMed] [Google Scholar]
- 23.Raidl M, Pirker C, Schulte-Hermann R, Aubele M, Kandioler-Eckersberger D, Wrba F, et al. Multiple chromosomal abnormalities in human liver (pre)neoplasia. J Hepatol. 2004;40:660–8. doi: 10.1016/j.jhep.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 24.Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci Transl Med. 2010;2:19ra13. doi: 10.1126/scitranslmed.3000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pegg AE. Repair of O(6)-alkylguanine by alkyltransferases. Mutat Res. 2000;462:83–100. doi: 10.1016/s1383-5742(00)00017-x. [DOI] [PubMed] [Google Scholar]
- 27.Iwakuma T, Sakumi K, Nakatsuru Y, Kawate H, Igarashi H, Shiraishi A, et al. High incidence of nitrosamine-induced tumorigenesis in mice lacking DNA repair methyltransferase. Carcinogenesis. 1997;18:1631–5. doi: 10.1093/carcin/18.8.1631. [DOI] [PubMed] [Google Scholar]
- 28.Nakatsuru Y, Matsukuma S, Nemoto N, Sugano H, Sekiguchi M, Ishikawa T. O6-methylguanine-DNA methyltransferase protects against nitrosamine-induced hepatocarcinogenesis. Proc Natl Acad Sci U S A. 1993;90:6468–72. doi: 10.1073/pnas.90.14.6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Xu-Welliver M, Kanugula S, Pegg AE. Inactivation and degradation of O(6)-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002;62:3037–43. [PubMed] [Google Scholar]
- 30.Wei W, Yang Z, Tang CH, Liu L. Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis. 2011;32:973–7. doi: 10.1093/carcin/bgr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen DE, Belka GK, Du Bois GC. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem J. 1998;331:659–68. doi: 10.1042/bj3310659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, et al. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–63. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 33.Rebello S, Zhu B, McMonagle-Strucko K, Pulicicchio C, Merrill J, Luo Y, et al. Pharmacokinetic and pharmacodynamic evaluation of inhibitors of inducible nitric oxide synthase (iNOS) in mice. AAPS PharmSci. 2002;4(Suppl 1) ( http://www.aapsj.org/abstracts/AM_2002/AAPS2002-002237.pdf) [Google Scholar]
- 34.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–12. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 35.Smith IK, Vierheller TL, Thorne CA. Assay of glutathione reductase in crude tissue homogenates using 5,5′-dithiobis(2-nitrobenzoic acid) Anal Biochem. 1988;175:408–13. doi: 10.1016/0003-2697(88)90564-7. [DOI] [PubMed] [Google Scholar]
- 36.Souliotis VL, van Delft JH, Steenwinkel MJ, Baan RA, Kyrtopoulos SA. DNA adducts, mutant frequencies and mutation spectra in lambda lacZ transgenic mice treated with N-nitrosodimethylamine. Carcinogenesis. 1998;19:731–9. doi: 10.1093/carcin/19.5.731. [DOI] [PubMed] [Google Scholar]
- 37.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 38.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 39.Nakae D, Kobayashi Y, Akai H, Andoh N, Satoh H, Ohashi K, et al. Involvement of 8-hydroxyguanine formation in the initiation of rat liver carcinogenesis by low dose levels of N-nitrosodiethylamine. Cancer Res. 1997;57:1281–7. [PubMed] [Google Scholar]
- 40.Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–80. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 41.Leung J, Wei W, Liu L. S-nitrosoglutathione reductase deficiency increases mutagenesis from alkylation in mouse liver. Carcinogenesis. 2013 doi: 10.1093/carcin/bgt031. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang CH, Wei W, Liu L. Regulation of DNA repair by S-nitrosylation. Biochim Biophys Acta. 2012;1820:730–5. doi: 10.1016/j.bbagen.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen L, Zhang Y, Kong X, Lan E, Huang Z, Peng S, et al. Design, synthesis, and antihepatocellular carcinoma activity of nitric oxide releasing derivatives of oleanolic acid. J Med Chem. 2008;51:4834–8. doi: 10.1021/jm800167u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, Stamler JS. NO: an inhibitor of cell death. Cell Death Differ. 1999;6:937–42. doi: 10.1038/sj.cdd.4400578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.