

Abstract
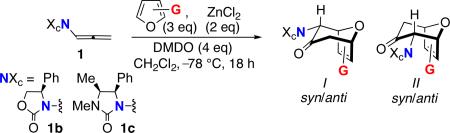
The regioselectivities and stereoselectivities of ZnCl2-catalyzed (4+3) cycloadditions between chiral oxazolidinone-substituted oxyallyls and unsymmetrical disubstituted furans have been determined. The substitution pattern on the furan is found to provide a valuable tool for controlling the stereochemistry (endo-I or endo-II) of the 7-membered cycloadduct. While cycloadditions with monosubstituted furans usually favor endo-I products, from addition of the furan to the more crowded face of the oxyallyl, cycloadditions with 2,3- and 2,5-disubstituted furans instead favor the endo-II stereochemistry. Density functional theory calculations are performed to account for the selectivities. For monosubstituted furans, the crowded transition state leading to the endo-I cycloaduct is stabilized by an edge-to-face interaction between the furan and the oxazolidinone 4-Ph group, but this stabilization is overcome by steric clashing if the furan bears a 2-CO2R group or is 2,3-disubstituted.
INTRODUCTION
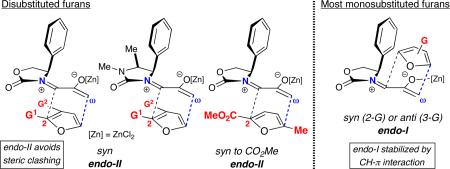
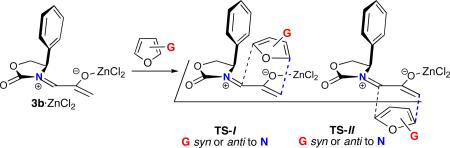
Oxazolidinone-stabilized oxyallyls (3) are a fascinating class of reactive intermediates, generated by oxidation of allenamides (1).1,2 Several valuable synthetic routes to 7-membered carbocycles rely on (4+3) cycloadditions between oxyallyl intermediates and dienes,3, 4 and we have found that the cycloadditions of oxyallyls 3 with monosubstituted furans (Schemes 1 and 2) display distinctive regioselectivities and stereoselectivities.5,6 These reactions may be performed either under thermal conditions or with catalysis by ZnCl2. The regioselectivities are summarized in Scheme 1, which shows the achiral oxyallyl 3a; cycloadditions of 3a with 2-substituted furans (G = Me or CO2Me) favor the “syn” regiochemistry, while reactions with 3-substituted furans favor “anti” regiochemistry. The same regioselectivities are obtained with the chiral oxyallyl 3b (Scheme 2), but in this case the reactions also feature a novel mode of stereoinduction. Surprisingly, reactions of 3b with most monosubstituted furans were found to favor the endo-I stereochemistry, which entails addition of the furan to the more crowded face of 3b. Density functional theory calculations6 showed that the crowded transition state is favored because it contains a stabilizing edge-to-face aromatic interaction (CH–π) between furan and Ph. Of the monosubstituted furans studied, only those containing a 2-CO2R group did not follow this behavior.
Scheme 1.

Regioselectivities of (4+3) Cycloadditions of Achiral Oxyallyl 3a with Monosubstituted Furans.6b
Scheme 2.

Stereoselectivities of (4+3) Cycloadditions of Chiral Oxyallyl 3b with Monosubstituted Furans.6a,c
In order to explore the generality of these stereochemical and regiochemical control elements, we have examined the (4+3) cycloadditions of 3b with more complex, unsymmetrical disubstituted furans. We report that these reactions often proceed with high levels of regio- and stereocontrol, even when the selectivities cannot be easily predicted from individual substituent effects. Complete control over stereoselectivity can be obtained through strategic choice of substituents.
RESULTS AND DISCUSSION
We investigated the (4+3) cycloadditions of oxyallyls 3 with a range of unsymmetrical disubstituted furans containing Me and CO2Me substituents at different positions. The cycloadditions were performed under ZnCl2-catalyzed conditions. Results are given in Table 1.
Table 1.
(4+3) Cycloadditions of Oxyallyls with Disubstituted Furans.a
Isolated yields are quoted. Isomer ratios are given in parentheses and were determined by 1H and/or 13C NMR.
From reaction with allenamide 1b.
From reaction with allenamide 1c.
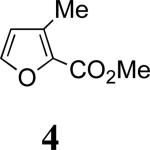
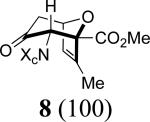
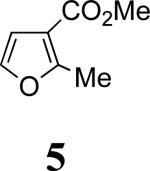
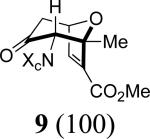
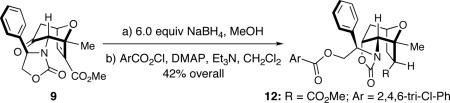
Our work commenced with the 2-CO2Me,3-Me-substituted furan, 4. Based on the selectivities previously observed with monosubstituted furans (Schemes 1 and 2), it is not possible to predict the major product for 4; a 2-CO2Me substituent favors syn-II but a 3-Me substituent favors anti-I. The cycloaddition of 3b with 4 was found to give exclusively the syn-II cycloadduct, 8, in 67% yield. The structure of 8 was assigned unambiguously by X-ray (Figure 1). The cycloaddition of 3b with the 2-Me,3-CO2Me-substituted furan 5 also gave exclusively a syn-II adduct (9). Confirmation of the structure of 9 was obtained following reduction/esterification, which afforded a rearranged derivative (12) whose X-ray structure was determined (Figure 1).7 The regioselectivities observed for both 4 and 5 match those previously obtained6b with the achiral oxyallyl 3a, which displayed syn selectivities of ≥95:5.
Figure 1.

X-Ray structures of cycloadduct 8, ester 12 (derived from 9), and cycloadducts 10a and 10b.
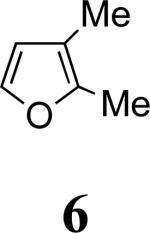
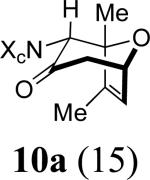
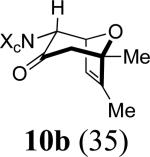
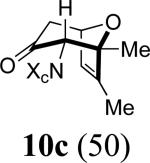
We next examined the 2-Me,3-Me-substituted furan, 6. In order to deal with competing oxidation of this more electron-rich furan, we had to switch to the more reactive allenamide, 1c,8 which contains Close's chiral imidazolidinone auxiliary.9 Previous experiments5 indicated that Close's imidazolidinone controls stereoselectivity in the same way as 4-Ph-oxazolidinone. Cycloaddition of 3c with 6 afforded mainly the syn-II product, 10c, but also gave two other cycloadducts: syn-I (10a) and anti-I (10b). X-ray structures were determined for the two minor products (Figure 1), while the major product was assigned unambiguously by NOESY and COSY (see Supporting Information).
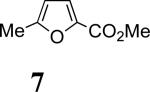
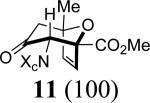
In 2,3-disubstituted furans, the substituents have opposite regio-directing influences (Scheme 1) and the selectivities obtained with 4–6 indicate that it is the 2-substituent that dictates the outcome. In order to discern the relative directing power of Me and CO2Me groups at the 2-position, we examined the reaction of 3b with 7. The only cycloadduct detected was 11, where CO2Me is syn to nitrogen. Its structure was confirmed by NOESY and COSY.
The regioselectivities and stereoselectivities observed here with disubstituted furans are quite distinct from those reported previously for monosubstituted furans.6 We performed density functional theory calculations to explore the selectivities. Transition states for ZnCl2-catalyzed cycloadditions of the oxyallyls 3b and 3c with furans 4–7 were computed at the B3LYP/6-31G(d) level (with LANL2DZ on Zn), as we have previously done in our studies of related oxyallyl (4+3) cycloadditions. The transition states for the reaction of 3b with 4 are representative and are shown in Figure 2. Regardless of the regiochemistry, the transition states each show the same sense of asynchronicity, with more advanced bonding at the unsubstituted terminus of the oxyallyl. The HOMO coefficient of the oxyallyl is larger at this terminus.
Figure 2.

Transition structures for ZnCl2-catalyzed cycloadditions of oxyallyl 3b with furan 4, optimized with B3LYP. Below each structure are given the activation energies obtained from M06-2X single-point calculations. Distances in Å, ΔH‡ and ΔG‡ in kcal/mol.
We previously found that B3LYP activation energies provide good predictions of regioselectivity and stereoselectivity for oxyallyl (4+3) cycloadditions. In order to provide a better treatment of dispersion energies in the cycloadditions of 4–7, which are likely to vary significantly between stereoisomeric transition states I and II, we calculated the activation barriers from single-point energies at the M06-2X/6-311+G(d,p) level. Solvent effects were modeled by means of CPCM calculations. The M06-2X activation energies in the gas phase and in dichloromethane are given in Table 2.
Table 2.
Calculated Activation Energies for ZnCl2-Catalyzed Cycloadditions of 3b with Disubstituted Furans.a
| ||||
|---|---|---|---|---|
| Reactants | ΔH‡ (ΔG‡) [ΔG‡soln] |
|||
| syn-I | anti-I | syn-II | anti-II | |
| 4 + 3b·ZnCl2 | –13.2 (5.2) [11.5] | –12.7 (4.9) [10.1] | –16.1 (2.2) [7.2] | –9.9 (6.8) [11.0] |
| 5 + 3b·ZnCl2 | –13.2 (4.3) [12.5] | –15.5 (3.6) [11.9] | –14.6 (2.6) [7.3] | –13.0 (5.6) [14.1] |
| 6 + 3b·ZnCl2 | –12.8 (3.9) [8.4] | –15.0 (2.3) [7.4] | –12.1 (3.6) [8.0] | –12.4 (3.9) [8.0] |
| 6 + 3c·ZnCl2 | –7.0 (9.7) [14.3] | –8.3 (9.0) [14.4] | –6.1 (9.6) [14.2] | –6.0 (10.4) [14.9] |
| 7 + 3b·ZnCl2 | –9.6 (8.4) [16.1] | –11.5 (4.8) [9.9] | –14.4 (3.8) [11.0] | –8.3 (7.0) [11.4] |
M06-2X/6-311+G(d,p)//B3LYP/6-31G(d)–LANL2DZ. Solution-phase data incorporate CPCM solvation energies in CH2Cl2, computed at the M06-2X/6-31G(d)–LANL2DZ level. ΔH† and ΔG‡ in kcal/mol at 298.15 K.
The calculated values of ΔG‡ in the gas phase and in solution predict a strong preference for the observed product (syn-II) for both furans 4 and 5. The syn-II TS from 4 is favored by ≥2.9 kcal/mol in dichloromethane, and the syn-II TS from 5 is favored by ≥4.6 kcal/mol. Lower selectivity is predicted for furan 6. Indeed, the cycloaddition of 6 with 3b is calculated to favor the anti-I TS by 0.6 kcal/mol. However, the cycloaddition of the same furan with 3c, which was studied experimentally, is correctly predicted to favor syn-II. The syn-II TS is preferred by 0.1–0.2 kcal/mol over syn-I and anti-I. Experimentally, a mixture of products was obtained containing the latter two isomers as minor products. The agreement between experiment and theory is not as good for 7, where the calculations predict a 1.1 kcal/mol preference for anti-I in dichloromethane. The selectivity in the gas phase does correctly predict syn-II, however. Experimentally, the syn-II cycloadduct was obtained in 40% yield.
Activation energies were also computed with a second dispersion-inclusive functional, B3LYP-D3 (Supporting Information). The predicted selectivities mirror those from M06-2X. B3LYP alone also correctly predicts the major products for furans 4–6 (and from 7 in the gas phase), but fails to predict the presence of minor products in the cycloaddition involving 6.
Our previous calculations showed that a 2-substituent on the furan favors syn regiochemistry because the shorter forming bond in the transition state is located at the less-hindered carbon (C-5) of the furan.6 When the 2-substituent is electron-withdrawing, this steric effect is reinforced by a strong oxyallyl–furan interaction energy, related to the large furan LUMO coefficient at C-5. For 3-substituted furans, by contrast, the anti regiochemistry is favored because its transition state avoids steric clashing between the 3-substituent and the oxazolidinone.
The syn selectivities obtained experimentally from furans 4–7 (Table 1) indicate that the regio-directing influence of a 2-substituent outweighs that of a 3-substituent, regardless of whether the substituent is Me or CO2Me. A 2-CO2Me group exerts a stronger regio-directing effect than a 2-Me group (cf. 7), because the oxyallyl–furan interaction in the TS is stronger when CO2Me is syn to nitrogen.
The stereoselectivities observed with furans 4–7 cannot be predicted from those of the corresponding monosubstituted furans. Most monosubstituted furans favor diastereomer I, due to the stabilizing edge-to-face aryl–aryl interaction in the crowded (I) transition state (Scheme 2).6a,c Only for 2-CO2Me-furan was II favored; in that case, the (syn-)I TS is destabilized by electrostatic repulsion between the CO2Me group and the Ph π-cloud.
A similar type of repulsive electrostatic effect influences the stereoselectivities for 4 and 7, favoring II. More generally, however, the stereoselectivities for all three 2,3-disubstituted furans (4–6) are dictated by the furan 3-substituent. This group eliminates the possibility of an edge-to-face CH–π interaction in the crowded syn-I TS, and replaces it with a repulsive electrostatic interaction (3-CO2Me) or a weaker CH–π interaction (3-Me). The result is a preference for II. Only for 6, where no CO2Me group is present, are isomers of I found as minor products.
CONCLUSION
The investigations detailed herein provide a valuable new approach to controlling the stereoselectivities of (4+3) cycloadditions. The different classes of substituent effects operating for monosubstituted and disubstituted furans are outlined in Scheme 3, and can be summarized as follows: cycloaddition of oxyallyl 3b or 3c with a 2-substituted furan (including a disubstituted furan) favors the syn-I cycloadduct, unless (a) the 2-substituent is CO2R or (b) there is also a substituent present at furan C-3. In the latter two cases, syn-II is preferred instead. These principles enable cycloadditions of oxyallyls 3 with complex furans to be achieved with high levels of regioselectivity and stereoselectivity.
Scheme 3.

Summary of Substituent-Controlled Regioselectivity and Stereoselectivity in (4+3) Cycloadditions of Oxyallyl 3b with Furans
EXPERIMENTAL SECTION
General Procedure for Allenamide (4 + 3) Cycloadditions
To a solution of a respective allenamide in CH2Cl2 (0.1 M) was added 3-9 equiv of the appropriate furan and 4Å powdered molecular sieves [0.5 g]. The reaction solution was cooled to –78 °C, and 2.0 equiv of ZnCl2 (1.0 M in Et2O) was added. 4.0 - 6.0 equiv of dimethyldioxirane [DMDO] in acetone was then added as a chilled solution [at –78 °C] via a syringe pump over 3-4 h. The syringe pump was cooled using dry ice during the entire addition time. After the addition, the reaction mixture was stirred for an additional 14 h before it was quenched with sat aq NaHCO3, filtered through Celite™, concentrated in vacuo, and extracted with CH2Cl2 [4 × 20 mL]. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude residue was purified via silica gel column chromatography [gradient eluent: 10% to 75% ethyl acetate in hexane].
8: Reaction Scale: 0.50 mmol of allenamide 1b; Yield: 67%; mass: 119.6 mg. Rf = 0.13 (50% EtOAc in hexane); [α]D23 –169.6 ° [c 4.6, CH2Cl2]; white solid; mp = 119 – 120 °C; 1H NMR (400 MHz, CDCl3) δ 2.04 (s, 3H), 2.54 (d, 1H, J = 16.0 Hz), 2.83 (dd, 1H, J = 5.4, 16.0 Hz), 3.29 (s, 3H), 3.91 (s, 1H), 4.20 (t, 1H, J = 9.0 Hz), 4.62 (t, 1H, J = 9.0 Hz), 4.79 (t, 1H, J = 9.0 Hz), 4.90 (dt, 1H, J = 5.2, 1.6 Hz), 7.00 (d, 1H, J = 1.6 Hz), 7.31 – 7.45 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 15.3, 44.3, 52.6, 65.9, 68.5, 70.5, 76.8, 88.3, 128.7, 129.3, 129.7, 130.2, 136.6, 142.9, 158.5, 166.9, 200.9; IR (thin film) cm-1 3280w, 2911w, 1766s, 1437w; mass spectrum (APCI): m/e (% relative intensity) 358.1 (100) (M + H)+; HRMS (MALDI-TOF): m/e calcd for C19H19NO6Na+ (M+Na+) 380.1105, found 380.1097.
9: Reaction Scale: 1.00 mmol of allenamide 1b; Yield: 60%; mass: 214.2 mg. Rf = 0.25 (50% EtOAc in hexane); [α]D23 –74.5 ° [c 4.0, CH2Cl2]; 1H NMR (500 MHz, CDCl3) δ 1.30 (s, 3H), 2.60 (d, 1H, J = 17.5 Hz), 2.88 (dd, 1H, J = 6.0, 17.5 Hz), 3.45 (s, 1H), 3.80 (s, 3H), 4.36 (t, 1H, J = 9.0 Hz), 4.71 (t, 1H, J = 9.0 Hz), 4.83 (t, 1H, J = 9.0 Hz), 4.99 (dd, 1H, J = 6.0, 2.0 Hz), 7.08 (d, 1H, J = 0.25 Hz), 7.48 – 7.52 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 15.4, 44.3, 52.6, 66.0, 68.6, 70.6, 76.9, 88.4, 126.4, 128.8, 129.4, 129.8, 130.3, 143.0, 158.6, 167.0, 201.0; IR (thin film) cm-1 2954s, 2930s, 1764s, 1726s; mass spectrum (APCI): m/e (% relative intensity) 358.2 (100) (M + H)+; HRMS (MALDI-TOF): m/e calcd for C19H19NO6Na+ (M+Na+) 380.1105, found 380.1095.
10a: Reaction Scale: 0.44 mmol of allenamide 1c; Yield: 9%; mass: 13.5 mg. Rf = 0.22 (50% EtOAc in hexane); [α]D23 +14.9 ° [c 0.75, CH2Cl2]; mp = 162 – 163 °C; 1H NMR (400 MHz, CDCl3) δ 0.79 (d, 1H, J = 6.8 Hz), 1.46 (s, 3H), 1. 81 (s, 3H), 2.49 (d, 1H, J = 17.8 Hz), 2.60 (dd, 1H, J = 5.8, 17.8 Hz), 2.69 (s, 3H), 3.29 (s, 1H), 3.82 (dq, 1H, J = 6.8, 9.2 Hz), 4.70 (d, 1H, J = 9.2 Hz), 4.75 (d, 1H, J = 5.8 Hz), 5.91 (s, 1H), 7.13 (br, 1H), 7.32 (br, 4H); 13C NMR (100 MHz, CDCl3) δ 14.0, 15.5, 21.4, 29.3, 43.8, 56.6, 65.2, 69.3, 75.9, 86.8, 128.5, 128.8, 128.9, 129.5, 135.3, 144.8, 159.3, 200.9; IR (thin film) cm-1 2929m, 1725s, 1687s, 1432m, 1263m, 1161m, 736m, 694s; mass spectrum (APCI): m/e (% relative intensity) 341.2 (60) (M + H)+; HRMS (MALDI-TOF): m/e calcd for C20H24N2O3Na+ (M+Na+) 363.1679, found 363.1680.
10b: Reaction Scale: 0.44 mmol of allenamide 1c; Yield: 21%; mass: 31.4 mg. Rf = 0.17 (50% EtOAc in hexane); [α]D23 –184.3 ° [c 0.79, CH2Cl2]; white solid; mp = 194 – 195 °C; 1H NMR (400 MHz, CDCl3) δ 0.71 (d, 1H, J = 6.8 Hz), 1.34 (s, 3H), 1.59 (s, 3H), 2.44 (d, 1H, J = 15.6 Hz), 2.57 (d, 1H, J = 15.6 Hz), 2.73 (s, 3H), 3.96 (dq, 1H, J = 6.8, 8.8 Hz), 4.46 (d, 1H, J = 8.8 Hz), 4.49 (d, 1H, 4.2 Hz), 4.66 (s, 1H), 5.10 (d, 1H, J = 4.2 Hz), 7.03 (br, 1H), 7.31-7.33 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 12.1, 15.3, 21.6, 29.3, 50.8, 57.2, 59.7, 64.5, 79.2, 85.9, 125.6, 127.4, 128.4, 129.2, 138.8, 146.3, 162.6, 203.8; IR (thin film) cm-1 2941m, 1728s, 1686s, 1431m, 1255m, 1196s, 1025m, 855s, 735s, 700s; mass spectrum (APCI): m/e (% relative intensity) 339.2 (45) (M - H)-; HRMS (MALDI-TOF): m/e calcd for C20H24N2O3Na+ (M+Na+) 363.1679, found 363.1678.
10c: Reaction Scale: 0.44 mmol of allenamide 1c; Yield: 31%; mass: 46.4 mg. Rf = 0.23 (50% EtOAc in hexane); [α]D23 -105.7 ° [c 0.83, CH2Cl2]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.85 (d, 1H, J = 6.8 Hz), 1.13 (s, 3H), 1. 89 (s, 3H), 2.47 (d, 1H, J = 17.2 Hz), 2.71 (dd, 1H, J = 5.8, 17.2 Hz), 2.79 (s, 3H), 3.43 (s, 1H), 3.92 (dq, 1H, J = 6.8, 9.2 Hz), 4.61 (d, 1H, J = 9.2 Hz), 4.74 (d, 1H, J = 5.8 Hz), 5.86 (s, 1H), 7.27-7.34 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 14.4, 15.1, 21.4, 29.1, 44.7, 56.3, 68.1, 74.9, 75.9, 87.7, 121.1, 128.5, 128.8, 129.5, 137.0, 146.9, 161.7, 204.2; IR (thin film) cm-1 2933m, 1703s, 1431, 1400s, 1262m, 1159m, 762m; mass spectrum (APCI): m/e (% relative intensity) 341.2 (60) (M + H)+; HRMS (MALDI-TOF): m/e calcd for C20H24N2O3Na+ (M+Na+) 363.1679, found 363.1674.
11: Reaction Scale: 0.35 mmol of allenamide 1b; Yield: 40%; mass: 50.0 mg. Rf = 0.22 (50% EtOAc in hexane); [α]D23 –31.4 ° [c 6.4, CH2Cl2]; 1H NMR (500 MHz, CDCl3) δ 1.43 (s, 3H), 2.59 (d, 1H, J = 16.0 Hz), 2.68 (dd, 1H, J = 5.2, 16.0 Hz), 3.65 (s, 3H), 3.93 (s, 1H), 4.18 (t, 1H, J = 9.0 Hz), 4.65 (t, 1H, J = 9.0 Hz), 4.82 (t, 1H, J = 9.0 Hz), 6.07 (d, 1H, J = 6.0 Hz), 6.66 (d, 1H, J = 6.0 Hz), 7.28 – 7.41 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 23.2, 51.3, 64.4, 67.2, 70.5, 72.8, 86.1, 89.4, 126.4, 128.4, 129.9, 133.4, 135.6, 136.5, 158.1, 167.5, 199.8; IR (thin film) cm-1 3520w, 2957w, 2927w, 1759s, 1726s; mass spectrum (APCI): m/e (% relative intensity) 358.2 (35) (M + H)+; HRMS (MALDI-TOF): m/e calcd for C19H19NO6Na+ (M+Na+) 380.1105, found 380.1090.
Synthesis of Ester 12 from Cycloadduct 9
To a solution of cycloadduct 9 (53.6 mg, 0.150 mmol) in anhyd MeOH (5 mL) was added NaBH4 (34.2 mg, 0.904 mol) portion-wise within 1 h at 0 °C. The reaction mixture was further stirred at room temperature for 6 h until the consumption of the starting material. Sat aq NaCl (10 mL) was added to quench the reaction. After removal of the volatile solvent under reduced pressure, the aqueous residue was extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was dried over Na2SO4 and concentrated in vacuo, and the crude residue was directly used for the next step.
The above crude residue was taken up in CH2Cl2 (5 mL), and to this solution were added DMAP (1.1 mg, 0.009 mmol) and Et3N (0.06 mL, 0.431 mmol) with vigorous stirring under ice-water cooling bath. After which, 2,4,6-trichlorobenzoyl chloride (0.07 mL, 0.450 mmol) was then added in one portion. The reaction mixture was stirred at rt for 30 h, and was then quenched with cold H2O (10 mL), and extracted with CH2Cl2 [4 × 20 mL]. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude residue was purified via silica gel column chromatography [gradient eluent: 10% to 30% EtOAc in hexane] to give the pure ester 12 (35.3 mg, 0.062 mmol, 42% over two steps) as a white solid.
12: Rf = 0.21 (30% EtOAc in hexane); [α]D23 + 29.9 ° [c 1.05, CH2Cl2]; white solid; mp = 176 – 177 °C; 1H NMR (400 MHz, CDCl3) δ 1.66 (s, 3H), 2.01 – 2.14 (m, 2H), 2.29 (dt, 1H, J = 8.4, 12.0 Hz), 2.56 (ddd, 1H, J = 2.4, 8.8, 12.0 Hz), 2.79 (dd, 1H, J = 8.8, 11.6 Hz), 3.35 (s, 3H), 3.37 (d, 1H, J = 7.2 Hz), 4.35 (br, 1H); 4.39 (t, 1H, J = 6.4 Hz); 4.84 – 4.89 (m, 2H); 4.95 (q, 1H, J = 7.2 Hz), 7.54 (d, 1H, J = 1.4 Hz), 7.52 (d, 1H, J = 1.4 Hz), 7.35 – 7.41 (m, 3H), 7.31 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 26.8, 31.2, 33.7, 51.8, 52.3, 60.9, 63.7, 64.4, 72.0, 74.1, 82.2, 128.3, 129.1, 129.2, 131.7, 132.8, 135.2, 136.7, 159.3, 163.6, 169.8 (one carbon peak was missing due to overlapping); IR (thin film) cm-1 2955m, 1755s, 1579m, 1373m, 1265s, 1209m, 1117m, 734m; mass spectrum (APCI): m/e (% relative intensity) 574.0 (7) (M + 6 + H)+; 572.1 (33) (M + 4 + H)+; 569.2 (30) (M + 2)+, 568.1 (96) (M + H)+; 567.1 (90) (M)+, 419.4 (9), 344.2 (22), 329.0 (85), 328.1 (21), 327.0 (100); HRMS (MALDI-TOF): m/e calcd for C26H24Cl3NO7Na+ (M+Na+) 590.0516, found 590.0511.
THEORETICAL CALCULATIONS
Density functional theory calculations were performed in Gaussian 09.10 Geometries were optimized in the gas phase using the B3LYP11 functional, with a mixed basis set consisting of LANL2DZ on Zn and 6-31G(d) on all other atoms. The lowest-energy conformer of each species was identified through conformational searching. The B3LYP vibrational frequencies were used to characterize species as minima or transition states, and to obtain scaled12 zero-point energies and thermal contributions to enthalpy and entropy. Single-point energies were then computed at the M06-2X/6-311+G(d,p) level,13 and used in conjunction with the B3LYP thermochemical corrections to obtain gas-phase activation enthalpies and free energies. Free energies of activation in dichloromethane were calculated by incorporating CPCM14 solvation energies computed at the M06-2X/6-31G(d)–LANL2DZ level (UAKS radii). A standard state of 1 mol/L was used. Activation energies were also computed from single-point energy calculations at the B3LYP-D315 level, with zero-damping. The activation energies predicted by B3LYP and B3LYP-D3 for cycloadditions involving 3b are provided in the Supporting Information, along with M06-2X activation energies for cycloaditions of 3c with 4–7.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH and Australian Research Council for generous financial support (GM-36700 to K.N.H., GM-66055 to R.P.H., and DP0985623 to E.H.K.), and the National Computational Infrastructure National Facility (Australia) and University of Melbourne for computer resources. E.H.K. also thanks the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology for financial support. We thank Dr. Vic Young and Mr. Gregory T. Rohde of The University of Minnesota for X-ray crystallography.
Footnotes
Supporting Information
NMR spectra and characterizations for all new compounds, X-ray Crystallographic Information Files and thermal ellipsoid plots, optimized geometries and energies, computed barriers at the B3LYP and B3LYP D3 levels for cycloadditions of 3b and M06-2X barriers for 3c. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.For a key review on allenamide chemistry, see: Wei L-L, Xiong H, Hsung RP. Acc. Chem. Res. 2003;36:773–782. doi: 10.1021/ar030029i.
- 2.For recent reviews on heteroatom-substituted oxyallyl intermediates, see: Lohse AG, Hsung RP. Chem. Eur. J. 2011;17:3812–3822. doi: 10.1002/chem.201100260.Harmata M. Chem. Commun. 2010;46:8904–8922. doi: 10.1039/c0cc03621h.Harmata M. Recent Res. Devel. Org. Chem. 1997;1:523–535.
- 3.For general reviews on (4+3) cycloadditions, see: Harmata M. Chem. Commun. 2010;46:8886–8903. doi: 10.1039/c0cc03620j.Harmata M. Adv. Synth. Catal. 2006;348:2297–2306.Battiste MA, Pelphrey PM, Wright DL. Chem. Eur. J. 2006;12:3438–3447. doi: 10.1002/chem.200501083.Hartung IV, Hoffmann HMR. Angew. Chem. Int. Ed. 2004;43:1934–1949. doi: 10.1002/anie.200300622.Rigby JH, Pigge FC. Org. React. 2004:351–478.Harmata M, Rashatasakhon P. Tetrahedron. 2003;59:2371–2395.Harmata M. Acc. Chem. Res. 2001;34:595–605. doi: 10.1021/ar000064e.Davies HML. In: Advances in Cycloaddition. Harmata M, editor. Vol. 5. JAI; Stamford, CT: 1999. pp. 119–164.West FG. In: Advances in Cycloaddition. Lautens M, editor. Vol. 4. JAI; Greenwich, CT: 1997. pp. 1–40.Harmata M. Tetrahedron. 1997;53:6235–6280.Katritzky AR, Dennis N. Chem. Rev. 1989;89:827–861.
- 4.For (4+3) cycloadditions of donor-substituted oxyallyls, including regioselective examples, see: Föhlisch B, Krimmer D, Gehrlach E, Kaeshammer D. Chem. Ber. 1988;121:1585–1594.Murray DH, Albizati KF. Tetrahedron Lett. 1990;31:4109–4112.Walters MA, Arcand HR, Lawrie DJ. Tetrahedron Lett. 1995;36:23–26.Lee JC, Jin S, Cha JK. J. Org. Chem. 1998;63:2804–2805.Harmata M, Rashatasakhon P. Synlett. 2000:1419–1422.Beck H, Stark CBW, Hoffmann HMR. Org. Lett. 2000;2:883–886. doi: 10.1021/ol991386p.Myers AG, Barbay JK. Org. Lett. 2001;3:425–428. doi: 10.1021/ol006931x.Harmata M, Ghosh SK, Hong X, Wacharasindhu S, Kirchhoefer P. J. Am. Chem. Soc. 2003;125:2058–2059. doi: 10.1021/ja029058z.MaGee DI, Godineau E, Thornton PD, Walters MA, Sponholtz DJ. Eur. J. Org. Chem. 2006:3667–3680.Chung WK, Lam SK, Lo B, Liu LL, Wong W-T, Chiu P. J. Am. Chem. Soc. 2009;131:4556–4557. doi: 10.1021/ja807566t.Lo B, Chiu P. Org. Lett. 2011;13:864–867. doi: 10.1021/ol102897d.Liu LL, Chiu P. Chem. Commun. 2011;47:3416–3417. doi: 10.1039/c1cc00087j.
- 5.For our contributions to the field of (4+3) cycloadditions, see: Xiong H, Hsung RP, Berry CR, Rameshkumar C. J. Am. Chem. Soc. 2001;123:7174–7175. doi: 10.1021/ja0108638.Xiong H, Hsung RP, Shen L, Hahn JM. Tetrahedron Lett. 2002;43:4449–4453.Rameshkumar C, Xiong H, Tracey MR, Berry CR, Yao LJ, Hsung RP. J. Org. Chem. 2002;67:1339–1345. doi: 10.1021/jo011048d.Xiong H, Huang J, Ghosh SK, Hsung RP. J. Am. Chem. Soc. 2003;125:12694–12695. doi: 10.1021/ja030416n.Rameshkumar C, Hsung RP. Angew. Chem. Int. Ed. 2004;43:615–618. doi: 10.1002/anie.200352632.Huang J, Hsung RP. J. Am. Chem. Soc. 2005;127:50–51. doi: 10.1021/ja044760b.Antoline JE, Hsung RP, Huang J, Song Z, Li G. Org. Lett. 2007;9:1275–1278. doi: 10.1021/ol070103n.Antoline JE, Hsung RP. Synlett. 2008:739–744.Lohse AG, Hsung RP, Leider MD, Ghosh SK. J. Org. Chem. 2011;76:3246–3257. doi: 10.1021/jo200147h.
- 6.a Krenske EH, Houk KN, Lohse AG, Antoline JE, Hsung RP. Chem. Sci. 2010;1:387–392. doi: 10.1039/C0SC00280A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lohse AG, Krenske EH, Antoline JE, Houk KN, Hsung RP. Org. Lett. 2010;12:5506–5509. doi: 10.1021/ol1023745. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Antoline JE, Krenske EH, Lohse AG, Houk KN, Hsung RP. J. Am. Chem. Soc. 2011;133:14443–14451. doi: 10.1021/ja205700p. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
7.During the synthesis of ester 12 from cycloadduct 9, an acyl transfer occurred during the esterification step.
- 8.a Wei L-L, Hsung RP, Xiong H, Mulder JA, Nkansah NT. Org. Lett. 1999;1:2145–2148. [Google Scholar]; b Berry CR, Hsung RP, Antoline JE, Petersen ME, Challeppan R, Nielson JA. J. Org. Chem. 2005;70:4038–4042. doi: 10.1021/jo0503411. [DOI] [PubMed] [Google Scholar]
- 9.a Close WJ. J. Org. Chem. 1950;15:1131–1134. [Google Scholar]; b Roder H, Helmchen G, Peters E-M, Peters K, von Schnering H-G. Angew. Chem. 1984;96:895–896. [Google Scholar]
- 10.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 11.a Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; b Becke AD. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]; c Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 12.Merrick JP, Moran D, Radom L. J. Phys. Chem. A. 2007;111:11683–11700. doi: 10.1021/jp073974n. [DOI] [PubMed] [Google Scholar]
- 13.a Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]; b Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 14.a Barone V, Cossi M. J. Phys. Chem. A. 1998;102:1995–2001. [Google Scholar]; b Cossi M, Rega N, Scalmani G, Barone V. J. Comput. Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 15.Grimme S, Antony J, Ehrlich S, Krieg H. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.