

Abstract
The major advantages of using the baculovirus–insect cell system for recombinant protein production are its ability to produce large amounts of recombinant proteins and its ability to provide eucaryotic modifications, such as glycosylation. However, the glycans linked to recombinant glycoproteins produced by this system typically differ from those found on native mammalian products. This is an important problem because glycans on mammalian glycoproteins can influence their functions in many different ways. The inability of baculovirus-infected insect cells to produce glycans identical to those found on native mammalian glycoproteins is due, in part, to the absence of functional levels of certain glycosyltransferases in insect cells. Thus, the purpose of this study was to engineer these activities into Tn-5B1-4, an established insect cell line that is widely used as a host for baculovirus-mediated protein production. Expression plasmids were constructed in which cDNAs encoding mammalian β1,4-galactosyltransferase and α2,6-sialyltransferase were placed under the transcriptional control of a baculovirus immediate early promoter. These plasmids were then used to isolate two different transgenic Tn-5B1-4 derivatives and the biological and biochemical properties of these cell lines were examined. The results show that both of the engineered insect cell lines have improved glycoprotein-processing capabilities, relative to the parental cell line.
Keywords: glycosylation, insect cells, baculovirus, metabolic engineering, glycosyltransferases
INTRODUCTION
The baculovirus-insect cell expression system is used routinely to produce recombinant glycoproteins for many different applications in basic and applied research (Jarvis, 1997; O’Reilly et al., 1992; Summers and Smith, 1987). The major advantages of this binary expression system are that baculovirus expression vectors can induce production of large amounts of recombinant proteins and insect cells have eucaryotic protein-processing capabilities. Protein glycosylation is among the most important of eucaryotic protein-processing pathways, as the addition of covalently linked glycans can dramatically influence protein functions in many different ways (Varki et al., 1999). However, insect cells generally fail to glycosylate proteins in precisely the same manner as mammalian cells (Altmann et al., 1999; Jarvis, 1997; Jarvis et al., 1998; Marchal et al., 2000; Marz et al., 1995) and this is widely recognized as a major limitation of the baculovirus-insect cell expression system.
In mammalian cells, protein N-glycosylation is carried out by an elaborate, but well-characterized metabolic pathway (Kornfeld and Kornfeld, 1985; Montreuil et al., 1995; Varki et al., 1999). Preassembled N-glycan precursors are transferred from a lipid carrier to newly synthesized proteins in the rough endoplasmic reticulum (RER) and the glycan precursor is then converted to one of several possible endproducts by a series of sequential processing reactions that occur along the secretory pathway. Minor trimming of the precursor by glucosidases in the RER produces the simplest endproducts, known as “high-mannose” N-glycans. However, high-mannose N-glycans also can be intermediates, which are further trimmed and elongated by mannosidases and glycosyltransferases in the RER and Golgi apparatus. These enzymes convert high-mannose N-glycans to a variety of different endproducts, which are generally known as “hybrid” or “complex” N-glycans. In the context of the present study, it is important to note that elongation of high-mannose to hybrid and complex N-glycans involves the addition of galactose and, often, sialic acid residues by glycosyltransferases found in mammalian cells. However, these enzymes are found to only a limited extent, or not at all, in insect cell systems (Altmann et al., 1993; Butters et al., 1981; Stollar et al., 1976; van Die et al., 1996). Thus, the absence of functional levels of these enzymes accounts, at least in part, for the inability of the baculovirus–insect cell system to produce recombinant mammalian glycoproteins with structurally authentic N-glycans.
The overall purpose of this study was to use metabolic engineering to improve protein N-glycosylation in Tn-5B1-4 cells. This insect cell line has gained wide recognition in recent years as an excellent host for baculovirus-mediated recombinant protein production because it can provide higher production levels than other host cell lines (Davis et al., 1992; Wickham et al., 1992). Thus, Tn-5B1-4 cell lines engineered for improved protein N-glycosylation would be of considerable interest to the scientific community. Toward this end, we constructed immediate early expression plasmids encoding mammalian glycosyltransferases and used these plasmids to isolate two new cell lines derived from Tn-5B1-4. Analysis of these engineered cell lines demonstrated that both have improved glycoprotein-processing capabilities relative to the parental cell line.
MATERIALS AND METHODS
Cells and Viruses
The Trichoplusia ni BTI-Tn-5B1-4 cell line (Tn-5B1-4; Wickham et al., 1992), also known as High Five (Invitrogen, Carlsbad, CA), was routinely maintained as either adherent or suspension cultures in antibiotic-free TNM-FH medium (Hink, 1970) supplemented with 10% (v/v) fetal bovine serum (Hyclone, Logan, UT) and 0.1% (w/v) Pluronic F-68 (BASF Wynaandotte Corp., Parsippany, NJ; see Murhammer and Goochee, 1988). This growth medium was designated complete TNM-FH. Tn-5B1-4 cells were seeded at densities of 0.5 × 106 cells per flask in 25-cm2 T-flasks (Corning Inc., New York, NY) or 0.3 × 106 cells/mL in 50-mL spinner flasks (Bellco Glass, Inc., Vineland, NJ) and cultured at 28°C to densities of about 1.5 to 2.0 × 106 cells/mL. Tn5β4GalT and Tn5β4GalT/ST6 cells were maintained under selective pressure as adherent cultures in complete TNM-FH containing 0.5 mg/mL of hygromycin (Sigma, St. Louis, MO). These cells were seeded into 25-cm2 T-flasks at a density of 0.5 × 106 cells and cultured at 28°C to a density of about 2.0 to 2.5 × 106 cells per flask. The baculovirus used for these studies was Autographa californica multicapsid nuclear polyhedrosis virus (AcMNPV) strain E2 (Summers and Smith, 1978). Baculovirus stocks were produced by infecting the Sf9 subclone of IPLB-SF21-AE cells (Vaughn et al., 1977) at a multiplicity of about 0.01 plaque-forming units (pfu) per cell. Budded virus was harvested at 3 to 5 days postinfection, and titered by plaque assays on Sf9 cells, as described previously (Summers and Smith, 1987).
Plasmid Constructions
pAcIE1, pIE1HRGalT, pIE1HRST6, and pIE1Hygro are immediate early expression plasmids (Jarvis et al., 1996) that encode AcMNPV IE1 (Guarino and Summers, 1987), bovine β1,4-galactosyltransferase (β1,4GalT; Shaper et al., 1986), rat α2,6-sialyltransferase (α2,6SiaT; Weinstein et al., 1987), or hygromycin phosphotransferase (Yates et al., 1985), respectively, under the transcriptional control of the AcMNPV ie1 promoter (Guarino and Summers, 1987) and hr5 enhancer (Guarino et al., 1986). The construction of each of these plasmids has been described previously (Hollister and Jarvis, 2000; Hollister et al., 1998). pDIEHR1 is a new immediate early expression plasmid, which contains two back-to-back copies of the AcMNPV ie1 promoter separated by the hr5 enhancer element. This plasmid was constructed by deleting the restriction sites from SacII to NotI, inclusive, in the multiple cloning site of pIE1HR4 (Jarvis et al., 1996), then inserting a blunt-ended ie1 promoter fragment from pAcP(−)IE1TV6 (Jarvis et al., 1996) into the unique SmaI site of the intermediate plasmid, pIE1HR4ΔS/N (Fig. 1). The bovine β1,4GalT cDNA (Shaper et al., 1986) was subsequently inserted as a BamHI fragment into the unique BglII site downstream of one of the ie1 promoters in pDIEHR1 to produce pDIE1HRGalT. In parallel, the SV40 poly A signal was added to the rat α2,6SiaT cDNA (Weinstein et al., 1987) in pBSKS (Strata-gene, La Jolla, CA) to produce pBSKS/ST6polyA. Finally, the entire α2,6SiaT–poly A sequence was subcloned as a SacII fragment from pBSKS/ST6polyA into the unique SacII site downstream of the other ie1 promoter in pDIE1HRGalT, to produce pDIE1GalT/ST6. The genetic structure of each plasmid was confirmed by restriction mapping and each was prepared from large-scale bacterial cultures for all subsequent experiments by alkaline lysis (Birnboim and Doly, 1979) followed by equilibrium centrifugation on continuous ethidium bromide–CsCl gradients (Sambrook et al., 1989).
Figure 1.

Construction of immediate early expression plasmids. This figure shows the strategy used to construct pDIEHR1 (see Materials and Methods).
Antibiotic Sensitivity Experiments
Tn-5B1-4 cells were seeded into six-well culture plates (Corning) in complete TNM-FH at a density of 0.25 × 106cells per well and allowed to adhere for 1 h at 28°C. The media were then removed and duplicate wells were treated with various concentrations of neomycin (G418; Gibco-BRL Life Sciences, Grand Island, NY) or hygromycin (Sigma) dissolved in complete TNM-FH. At various time-points after treatment, cell viability was determined using the trypan blue exclusion method and the results were plotted as the average number of viable cells per milliliter versus treatment time.
Transient Expression Assays
Transient expression assays were performed using a modified calcium phosphate transfection method, as described previously (Summers and Smith, 1987). Tn-5B1-4 cells were seeded into 25-cm2 T-flasks at a density of 2.5 × 106, then transfected with 20 μg of pAcIE1, 10 μg of pAcIE1 plus 10 μg of pIE1HRGalT, 10 of μg pAcIE1 plus 10 μg of pIE1HRST6, or 10 of μg pAcIE1 plus 10 μg of pDIE1GalT/ST6. Each plasmid had been gradient-purified from large-scale bacterial cultures, as described earlier. After a 2-h transfection period at 28°C, the cells were washed twice with complete TNM-FH and incubated for another 22 h at 28°C. The cells were then harvested, washed with Tris-buffered saline (TBS; Tris-HCl [pH 7.4], 1.4 M NaCl), resuspended in TBS, and divided into two equal aliquots. The cells in each aliquot were pelleted and one pellet was resuspended in β1,4GalT buffer (10 mM HEPES [pH 7.4], 140 mM NaCl, 5 mM MnCl2, and 0.5% [v/v] Nonidet P-40 [Calbiochem; San Diego, CA]), whereas the other was resuspended in α2,6SiaT buffer (50 mM sodium phosphate [pH 7.5], 100 mM NaCl, 10 mM MgCl2, and 1.5% [v/v] Triton CF-54 [Sigma]). Both were freeze-thawed, the cell extracts were clarified at top speed in a microcentrifuge for 15 min at 4°C, and the supernatants were harvested. Duplicate samples of the clarified cell extracts were then assayed for β1,4GalT or α2,6SiaT activity. The β1,4GalT assays were done in a total volume of 110 μL of β1,4GalT buffer containing 0.3 μCi of UDP-[3H]-galactose (12.8 Ci/mmol; New England Nuclear, Boston, MA) and 500 μg/mL of ovalbumin (Grade V, Sigma). The α2,6SiaT assays were done in a total volume of 60 μL of α2,6SiaT buffer, containing 50 μg of asialofetuin (Type I, Sigma), 50 μg of bovine serum albumin, and 0.3 μCi of CMP-[3H]-sialic acid (32.8 Ci/mmol; New England Nuclear). Each reaction mixture was incubated at 37°C for 1 h, quenched by the addition of 0.2 mL of the appropriate assay buffer, and spotted onto a glass fiber filter (GF/D Whatman; Hillsboro, OR). The filters were then dried, washed successively with 10% TCA, 5% TCA (twice), 95% ethanol, and absolute ethanol. The filters were redried, then radioactivity was quantified using Ultima Gold F scintillation cocktail (Packard, Meriden, CT) and a liquid scintillation spectrometer (Model LS1801, Beckman, Palo Alto, CA).
Insect Cell Transformation
A method developed for genetic transformation of Sf9 cells (Jarvis et al., 1990; Jarvis and Guarino, 1995) was used to isolate the transformed Tn-5B1-4 cells described in this study. Tn-5B1-4 cells were seeded into 25-cm2 T-flasks at a density of 1.0 × 106 per flask and allowed to attach for 1 h at 28°C. The cells were then cotransfected as described earlier with 20 μg of pIE1HRGalT plus 1 μg of pIE1Hygro, or with 20 μg of pDIE1GalT/ST6 plus 1 μg of pIE1Hygro. At 24 h posttransfection, the cells were removed from the flasks and diluted to a concentration of 2 × 105 cells/mL in complete TNM-FH. Various numbers of cells were then seeded into 60-cm2 culture plates (Corning) and incubated at 28°C for 24 h. The growth media were replaced with complete TNM-FH containing 0.5 mg/mL of hygromycin and the cells were returned to the incubator for 1 week. The media were replaced with fresh selective media and the cells were incubated until colonies of hygromycin-resistant cells began to form. Multiple, well-isolated colonies were picked using cloning cylinders (Bellco), transferred into individual wells of a 96-well plate, and amplified in a stepwise fashion through increasingly larger culture vessels.
Hygromycin-resistant clones isolated and amplified in this way were assayed for expression of the nonselected glycosyltransferase gene(s), as described earlier, except protein concentrations in the lysates were determined and equal amounts of total protein were used for these assays. Total protein concentrations were determined using the bicinchoninic acid assay (Smith et al., 1985) (Pierce Chemical Co., Rockford, IL), with bovine serum albumin as the standard, and 100 μg of total protein was used for each enzyme activity assay. The hygromycin-resistant clones with the highest levels of β1,4GalT and α2,6SiaT activities were designated Tn5β4GalT and Tn5β4GalT/ST6.
Baculovirus Growth Curves
Tn-5B1-4, Tn5β4AGalT, or Tn5β4GalT/ST6 cells were pelleted, resuspended in complete TNM-FH, and infected with AcMNPV at a multiplicity of 10 pfu per cell. The virus was allowed to absorb for 1 h at 28°C, then the inocula were removed and the cells gently washed three times with complete TNM-FH. The cells were resuspended in complete TNM-FH and equal aliquots were dispensed into six-well culture plates. After various incubation times at 28°C, the media were harvested from triplicate wells containing each virus-infected cell type, the triplicates were pooled, and the amounts of infectious viral progeny produced by each cell type were measured by plaque assays on Sf9 cells, as described previously (Summers and Smith, 1987).
Protein Glycosylation Assay
Tn-5B1-4, Tn5β4GalT, or Tn5β4GalT/ST6 cells were grown to 1.0 × 106 cells/mL in spinner flasks with complete TNM-FH and infected with AcMNPV at a multiplicity of 2 pfu/cell. The cells were incubated for 1 h at 28°C, resuspended in 50 mL of complete TNM-FH, and returned to the spinner flasks. Following a 6-h incubation period, the cells were pelleted by low-speed centrifugation, the cell-free media were transferred into ultracentrifuge tubes, and budded virus progeny were pelleted through a 25% (w/v) sucrose solution by centrifugation at 24,000 rpm in a Type 45 (Beckman) rotor for 75 min at 4°C. The viral pellets were resuspended in 0.1 × TE and the major envelope glycoprotein, gp64, was extracted by treating samples with 50 mM Tris-HCl (pH 8.0) containing 140 mM NaCl and 1% (v/v) Nonidet P-40. The viral nucleocapsids were pelleted by centrifugation at top speed in a microcentrifuge for 20 min at 4°C, then the supernatants were harvested and gp64 was immunoprecipitated with a mouse monoclonal antibody (AcV1; Hohmann and Faulkner, 1983), as described previously (Jarvis and Garcia, 1994).
Immune complexes were washed, disrupted, and resolved by discontinuous sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS-PAGE; Laemmli, 1970), then transferred to Immobilon filters (Millipore Corp., Bedford, MA) using an electrophoretic transfer method (Towbin et al., 1979). The membranes were cut into panels corresponding to sets of lanes containing equal amounts of gp64 from each cell line. These panels were then used for antibody or lectin-binding assays, as described previously (Hollister et al., 1998; Jarvis and Finn, 1996), except biotinylated lectins (Vector Labs, Burlingame, CA) were used instead of di-goxigenylated lectins because the latter are no longer commercially available. The lectins used in this study were Ricinus communis agglutinin (RCA) and Sambucus nigra agglutinin (SNA), which are specific for β-linked galactose and terminal, preferably α2,6-linked sialic acid residues, respectively. Secondary reactions were performed using alkaline phosphatase, conjugated streptavidin or goat anti-rabbit immunoglobulin G (IgG; Promega, Inc,, Madison, WI) and binding of the secondary reagents was detected using a standard color reaction (Blake et al., 1984).
For some control experiments designed to demonstrate the specificity of the lectin-binding assays, RCA was pre-incubated with 0.7 M D(+)-galactose and SNA was preincubated with 0.2 M α-lactose for at least 24 h prior to use. For other specificity control experiments, gp64 was pre-treated with endoglycosidases to remove the carbohydrate required for specific lectin binding prior to the lectin binding assays. Immune complexes containing gp64 were treated for 5 h at 37°C with buffer alone, Arthrobacter urefaciens neuraminidase (Calbiochem, San Diego, CA) in 200 mM Na2HPO4 (pH 5.3) plus 7.5% Nonidet P-40, or Diplococcus pneumoniae β-galactosidase (Boehringer Mannheim) in 50 mM Na2HPO4 (pH 7.0) plus 7.5% Noni det P-40. It also is worth noting that lectin-blotting results obtained by our lab have been verified by more sophisticated methods, including high pH anion-exchange chromatography and exoglycosidase sequencing (Hollister and Jarvis, 2000).
RESULTS
Transient Expression of Mammalian Glycosyltransferases in Tn-5B1-4 Cells
The first requirement was to assemble the appropriate immediate early expression plasmids and determine if they could be used to induce the production of functional mammalian glycosyltransferases in Tn-5B1-4 cells. As detailed in the Materials and Methods section, pIE1HRGalT and pIE1HRST6 are immediate early expression plasmids encoding bovine β1,4GalT or rat α2,6SiaT, whereas pIE1GalT/ST6 is a dual immediate early expression plasmid that encodes both of these mammalian glycosyltransferase cDNAs under the control of back-to-back ie1 promoters. Each plasmid was used to transiently transfect Tn-5B1-4 cells in combination with pAcIE1, which encodes the Ac-MNPV transactivator protein, IE1. Tn-5B1-4 cells transected with pAcIE1 alone were used as negative controls. At 24 h posttransfection, cell extracts were prepared and β1,4GalT or α2,6SiaT activity assays were performed, as described earlier. The results showed that β1,4GalT (Fig. 2A) and α2,6SiaT (Fig. 2B) activities were higher in Tn-5B1-4 cells transfected with the immediate early expression plasmids than in cells transfected with pAcIE1 alone. The levels of β1,4GalT activity observed in Tn-5B1-4 cells transfected with pAcIE1 alone were above background in this assay, as determined using boiled cell lysates (data not shown). This observation confirms a previous report that Tn-5B1-4 cells have low levels of endogenous β1,4GalT activity (van Die et al., 1996). In contrast, α2,6SiaT activity was not above background in the pAcIE1-transfected cell lysates and endogenous α2,6SiaT activity has never been reported in Tn-5B1-4 or any other insect cell line. The dual immediate early expression plasmid induced higher levels of both activities than the single immediate early expression plasmids in these transient expression assays, but the reason for this was not investigated. The salient result was that each of the immediate early expression plasmids, pIE1HRGalT, pIE1HRST6, and pDIEGalT/ST6, induced the production of functional glycosyltransferases in Tn-5B1-4 cells.
Figure 2.

Expression of glycosyltransferase activities by transiently transfected Tn-5B1-4 cells. Tn-5B1-4 cells were transfected with the immediate early expression plasmids indicated on the abscissas and assayed for (A) β1,4GalT or (B) α2,6SiaT activity.
Antibiotic Sensitivity of Tn-5B1-4 Cells
In previous experiments, transformed Sf9 cells were selected by cotransfection with a neomycin resistance marker, followed by treatment with neomycin (Hollister et al., 1998; Jarvis et al., 1990). This strategy was based on the finding that Sf9 cells could not survive in growth medium containing 1 mg/mL neomycin unless they were transfected with pIE1Neo, which encodes neomycin phosphotransferase under ie1 control. Prior to undertaking efforts to produce transgenic Tn-5B1-4 cells, it was necessary to establish analogous selection conditions for this cell line. Therefore, we examined the antibiotic sensitivity of Tn-5B1-4 cells. Tn-5B1-4 cells that had been maintained in the absence of common cell culture antibiotics were seeded into culture plates, treated with various concentrations of neomycin or hygromycin, and cell viability was monitored at 24-h intervals. Surprisingly, the results showed that Tn-5B1-4 cells treated with 1 mg/mL of neomycin survived and grew almost as well as the untreated cells during a 4-day observation period (Fig. 3A). In contrast, 0.5 to 1.0 mg/mL of hygromycin arrested cell growth and decreased viability during this time period (Fig. 3B). Thus, hygromycin was the better of the two antibiotics examined for selection of transformed Tn-5B1-4 cell lines. This was confirmed in a subsequent experiment, which showed that Tn-5B1-4 cells were effectively killed after 20-day treatment with 0.5 mg/mL of hygromycin (data not shown).
Figure 3.

Antibiotic sensitivity of Tn-5B1-4 cells. (A) Tn-5B1-4 cells were seeded into six-well culture plates at a density of 0.25 × 106 cells/well and treated with complete TNM-FH (□), or complete TNM-FH containing 0.5 (○), 1.0 (△), or 2.0 (■) mg/mL of neomycin. (B) Tn-5B1-4 cells were seeded as just described and treated with complete TNM-FH (□), or complete TNM-FH containing 0.25 (○), 0.5 (△), or 1.0 (■) mg/mL of hygromycin.
Isolation of Tn5β4GalT and Tn5β4GalT/ST6 Cells
The antibiotic sensitivity experiments set the stage for our attempts to produce transgenic Tn-5B1-4 cell lines that would constitutively express mammalian β1,4GalT or β1,4GalT and α2,6SiaT cDNAs. To produce the first cell line, Tn-5B1-4 cells were cotransfected with pIE1HRGalT plus pIE1Hygro. Hygromycin-resistant colonies were isolated, amplified, and the resulting clones screened for β1,4GalT activity, as described in the Materials and Methods section. The results demonstrate that each of nine independent hygromycin-resistant clones had higher levels of β1,4GalT activity than untransformed Tn-5B1-4 cells (Fig. 4A). Different clones had different levels of activity, ranging from about 1.5- to 3-fold higher than the untransformed cells, and there was about a 2-fold difference between the transformants with the lowest and highest activities. Clone 9 had the highest level of β1,4GalT activity and was designated Tn5β4GalT.
Figure 4.
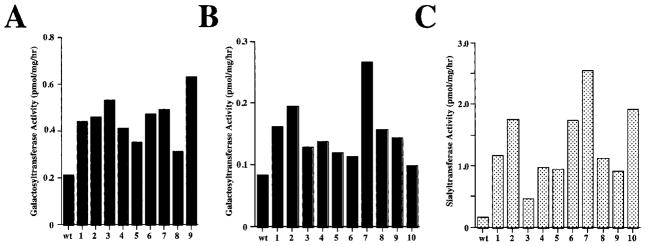
Glycosyltransferase activities in hygromycin-resistant Trichoplusia ni cell clones. (A) Nine hygromycin-resistant clones (1–9) were isolated after cotransfection of Tn-5B1-4 cells with pIE1GalT plus pIE1Hygro (see Materials and Methods). Samples of each clone were extracted and the extracts were assayed in duplicate for β1,4GalT activity (see Materials and Methods). (B, C) In a totally separate transformation experiment, ten independent hygromycin-resistant clones (1–10) were isolated after cotransfection of Tn-5B1-4 cells with pDIE1HRGalT/ST plus pIE1Hygro (see Materials and Methods). Samples of each clone were extracted and the extracts were assayed in duplicate for (B) β1,4GalT and (C) α2,6ST activity (see Materials and Methods). In each case, untransformed Tn-5B1-4 cells (wt) were used as controls.
To produce the second cell line, Tn-5B1-4 cells were cotransfected with pIE1GalT/ST6 plus pIE1Hygro and hygromycin-resistant colonies were isolated, amplified, and screened for β1,4GalT and α2,6SiaT activities. The results of these assays showed that each of ten independent hygromycin-resistant clones had higher levels of both β1,4GalT and α2,6SiaT activities than the untransformed Tn-5B1-4 cells (Fig. 4B, C). The levels of β1,4GalT activity ranged from just slightly above to about three-fold higher than the endogenous level found in untransformed Tn-5B1-4 cells, whereas the levels of α2,6SiaT activity ranged from about two- to tenfold higher than the background level measured using untransformed cells. There was not a perfect correlation between the levels of β1,4GalT and α2,6SiaT activities in the different hygromycin-resistant clones. However, clone 7 had the highest levels of both activities and was designated Tn5β4GalT/ST6.
General Properties of Tn-5B1-4, Tn5β4GalT, and Tn5β4GalT/ST6 Cells
To determine if genetic transformation had altered their basic growth properties, we compared the growth curves of Tn-5B1-4, Tn5β4GalT, and Tn5β4GalT/ST6 cells. The results showed that Tn5β4GalT cells grew to slightly higher densities and Tn5β4GalT/ST6 cells grew to slightly lower densities than Tn-5B1-4 cells (Fig. 5A). However, these differences were minor and the results of this experiment indicated that transformation had no significant effect on the growth properties of Tn5β4GalT or Tn5β4GalT/ST6 cells.
Figure 5.
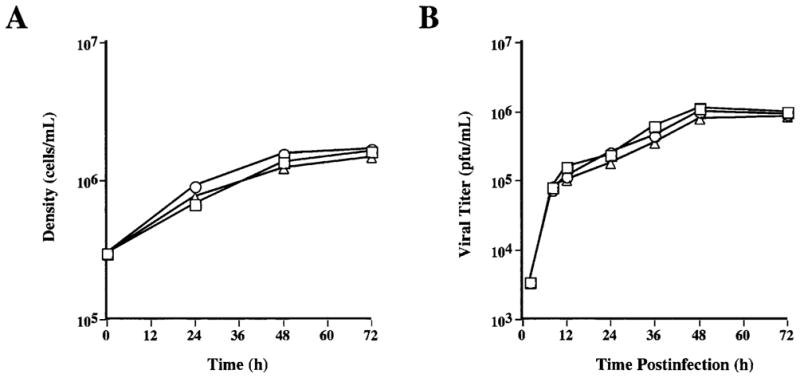
Comparison of parental and transgenic Trichoplusia ni cell properties. (A) The growth curves of Tn-5B1-4 (□), Tn5β4GalT (○), and Tn5β4GalT/ST6 (△) cells were derived by performing viable cell counts in triplicate at various timepoints after seeding spinner flasks at a density of 0.3 × 106 cells/mL. (B) Baculovirus growth curves were derived by measuring viral progeny production at various timepoints after infecting triplicate cultures of Tn-5B1-4 (□), Tn5β4GalT (○), and Tn5β4GalT/ST6 (△) cells with AcMNPV (see Materials and Methods).
Viral growth curve experiments were performed to determine if transformation had altered the ability of Tn5β4GalT or Tn5β4GalT/ST6 cells to serve as hosts for baculoviruses. The results showed that there was little or no difference in the amounts of infectious progeny produced by these cell lines or the parental Tn-5B1-4 cell line (Fig. 5B). Thus, transformation had no detectable effect on the ability of Tn5β4GalT or Tn5β4GalT/ST6 cells to serve as hosts for baculovirus replication.
Genomic Southern blot analyses were performed using Hirt preparations of high-molecular-weight DNA (Hirt, 1967) isolated from Tn-5B1-4, Tn5β4GalT, and Tn5β4GalT/ST6 cells, with fragments of the cloned β1,4GalT and α2,6SiaT cDNAs as probes. The results indicate that multiple copies of the intact immediate early expression plasmids had been integrated as tandem arrays at single sites in the Tn5β4GalT and Tn5β4GalT/ST6 cell genomes (data not shown). This genetic arrangement was the same as has been observed previously for stably transformed Sf9 cell lines (Hollister et al., 1998; Jarvis et al., 1990). Despite this observation, however, we observed that Tn5β4GalT/ST6 cells lost β1,4GalT and α2,6SiaT activities with increasing passage number when they were maintained in the absence of selective pressure (data not shown). Thus, Tn5β4GalT/ST6 cells were not as genetically stable as Tn5β4GalT cells or any of the transformed Sf9 cell lines produced by our lab, as the latter have never lost the ability to express their transgenes, even when maintained in the absence of selective pressure (Hollister et al., 1998; Jarvis et al., 1990; and unpublished observations). Based on this result, we decided to routinely maintain both of the transgenic Tn-5B1-4 cell lines under constant selective pressure, as described in the Materials and Methods section. However, hygromycin was not included in the growth medium of any cultures used for terminal experiments.
Previous studies have shown that baculovirus infection of transformed Sf9 cells initially induces, then represses, expression of integrated transgenes at the transcriptional level (Hollister et al., 1998; Jarvis, 1993). Thus, we examined the effects of baculovirus infection on β1,4GalT and α2,6SiaT activity levels in the transgenic Tn-5B1-4 cell lines. The results showed that there was a transient induction, followed by a slow decay of the β1,4GalT and α2,6SiaT activity in baculovirus-infected Tn5β4GalT and Tn5β4GalT/ST6 cells, as has been observed previously for transformed Sf9 cells (Fig. 6). However, transferase activities in both transgenic cell lines were about the same as or higher than the levels in uninfected cells until at least 48 h postinfection, indicating that these activities remained available well after the peak time of recombinant glycoprotein biosynthesis by conventional baculovirus expression vectors.
Figure 6.
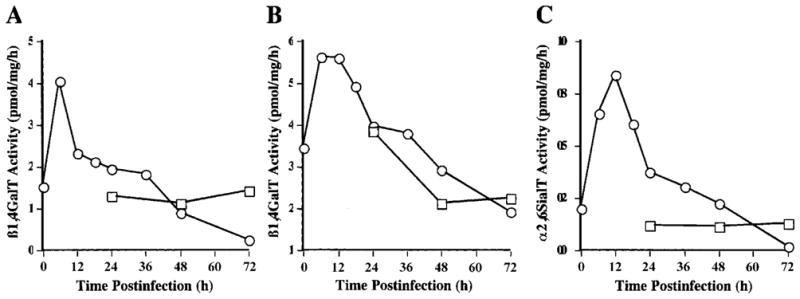
Effects of baculovirus infection on glycosyltransferase activities in transgenic Trichoplusia ni cells. Tn5β4GalT or Tn5β4GalT/ST6 cells were mock-infected (□) or AcMNPV-infected (○), then lysates were prepared at various times after infection and glycosyltransferase assays were performed on duplicate samples (see Materials and Methods). (A) β1,4GalT activity in Tn5β4GalT cells. (B) β1,4GalT activity in Tn5β4GalT/ST6 cells. (C) α2,6SialT activity in Tn5β4GalT/ST6 cells.
Improved Glycosylation of a Foreign Protein by Tn5β4GalT and Tn5β4GalT/ST6 Cells
The major AcMNPV budded virus envelope glycoprotein, gp64, is expressed during the late phase of infection (Miller, 1997; Volkman, 1986). Previous lectin blotting analyses have shown that the gp64 produced by Tn-5B1-4 cells contains no detectable galactose or sialic acids (Jarvis and Finn, 1995). Yet, this same protein acquires both of these sugars when produced by COS cells, which have the requisite glycosyltransferase activities. Thus, we used gp64 as a model to determine if the transgenic Tn-5B1-4 cell lines described in this study could produce a galactosylated and sialylated foreign glycoprotein. gp64 was partially purified from baculovirus-infected Tn-5B1-4, Tn5β4GalT, or Tn5β4GalT/ST6 cells and analyzed by lectin blotting assays, as described in the Materials and Methods section. The results showed that both transgenic cell lines produced differentially glycosylated forms of gp64 (Fig. 7). RCA, which is specific for β-linked galactose, bound only to the gp64 produced by Tn5β4GalT or Tn5β4GalT/ST6 cells, not to the gp64 from untransformed Tn-5B1-4 cells. RCA also bound to the internal positive control, mammalian IgG heavy chain, in each lane. SNA, which is specific for terminal sialic acids and has a preference for α2,6-linked sialic acids, bound only to the gp64 from Tn5β4GalT/ST6 cells and to the IgG heavy chain internal controls. Preincubation of RCA or SNA with competing sugars eliminated or significantly reduced lectin binding, indicating that lectin binding was carbohydrate-specific and not due to protein–protein or other nonspecific interactions.
Figure 7.

Glycosylation of gp64 by parental and transgenic Trichoplusia ni cell lines. gp64 was isolated from AcMNPV-infected Tn-5B1-4 (lanes 1), Tn5β4GalT (lanes 2), and Tn5β4GalT/ST6 (lanes 3) cells and equal aliquots were resolved by SDS-PAGE. The proteins were transferred to an immobilon filter; the filter was cut into strips containing gp64 from each source; and the strips were probed with anti-gp64 (Ab), RCA, or SNA. The specificity of the lectin blots was evaluated by comparing the results obtained after preincubating the lectins in the absence (−) or presence (+) of competing sugars. The arrows indicate the positions of gp64 and the IgG heavy chain.
The specificity of the lectin blotting assay was further examined by pretreating the gp64 from Tn5β4GalT and Tn5β4GalT/ST6 cells with Diplococcus pneumoniae β-galactosidase, which specifically removes terminal sialic acid residues, or Arthrobacter urefaciens neuraminidase, which specifically removes terminal sialic acid residues. The results show that RCA binding was precluded by β-galactosidase pretreatment (Fig. 8A) and SNA binding was precluded by neuraminidase pretreatment (Fig. 8B). The results further confirm that the lectin blotting assays were carbohydrate-specific. It is noteworthy that previous lectin blotting results from our lab have been verified by other more sophisticated methods, including high pH anion-exchange chromatography and exoglycosidase sequencing (Hollister and Jarvis, 2000). Thus, the lectin blotting assays presented in this study clearly show that Tn5β4GalT and Tn5β4GalT/ST6 cells can produce β1,4-galactosylated or α2,6-sialylated forms of gp64, respectively, during baculovirus infection.
Figure 8.

Glycan specificity of RCA and SNA binding to gp64. (A, B) gp64 was isolated from Tn5β4GalT or Tn5β4GalT/ST6 cells and treated with nothing (S), buffer alone (C), β-galactosidase (G), or neuraminidase (N), and probed with anti-gp64 (Ab), RCA, or SNA. The arrows indicate the positions of gp64 and the IgG heavy chain.
DISCUSSION
The overall purpose of this study was to use metabolic engineering to extend the N-glycan processing capabilities of Tn-5B1-4 cells and to produce new cell lines that could serve as improved hosts for foreign glycoprotein production by baculovirus expression vectors. This goal was achieved by using immediate early expression plasmids to introduce chimeric genes encoding constitutively expressable β1,4GalT and/or α2,6SiaT cDNAs into these cells. Integration of these plasmids into the chromosomal DNA of Tn-5B1-4 cells resulted in the isolation of two new, transgenic cell lines called Tn5β4GalT and Tn5β4GalT/ST6. Characterization of these cell lines revealed that they had essentially the same growth properties as the untransformed, parental cell line. Furthermore, Tn-5B1-4, Tn5β4GalT, and Tn5β4GalT/ST6 cells served equally well as hosts for baculovirus replication. Both transgenic cell lines had higher levels of β1,4GalT activity than Tn-5B1-4 cells, and Tn5β4GalT/ST6 cells had α2,6SiaT activity as well, which was not detectable in Tn-5B1-4 cells. Unlike the parental Tn-5B1-4 cell line, both transgenic cell lines were able to add galactose to the N-glycan(s) on a foreign glycoprotein, gp64, at levels that could be detected by RCA lectin blotting. In addition, Tn5β4GalT/ST6 cells were able to produce gp64 with sialylated N-glycans, as evidenced by SNA binding assays accompanied by multiple controls. Together, these findings demonstrated, for the first time, that the Tn-5B1-4 N-glycosylation pathway can be engineered by using genetic transformation methods designed to increase endogenous glycosyltransferase activity levels and/or to introduce new activities into these cells. Interestingly, Tn-5B1-4 cells were found to be resistant to neomycin, which had been used to select stably transformed Sf9 lines in previous studies (Jarvis et at., 1990), and the present study showed that it is necessary to use a different antibiotic to select Tn-5B1-4 transformants. Finally, and most importantly, this study showed that the new, transgenic insect cell lines produced in this way are improved hosts for foreign glycoprotein production by baculovirus expression vectors.
Future studies will be designed to evaluate the precise structures of the glycans on foreign glycoproteins produced by Tn5β4GalT and Tn5β4GalT/ST6 cells, as well as the efficiencies of N-glycan galactosylation and sialylation by these cells. In addition, it will be of interest to determine why the Tn5β4GalT/ST6 cells lost both glycosyltransferase activities with increasing passage number when maintained in the absence of selective pressure. As mentioned in the Results section, this was not true of the Tn5β4GalT cells or any transformed Sf9 cell line produced by our lab in the past. It is possible that the loss of glycosyltransferase activities reflects genetic instability of the single Tn5β4GalT/ST6 cell clone described in detail in this study. This possibility could be examined directly by measuring the glycosyltransferase activities and performing genomic Southern blot analyses with samples isolated from Tn5β4GalT/ST6 cells at different passage levels. These experiments were not pursued in the current study because the apparent instability of Tn5β4GalT/ST6 cells was overcome by maintaining these cells in the presence of hygromycin. However, this might not be practical for large-scale applications and it will be important to determine more precisely how quickly the glycosyltransferase activities of Tn5β4GalT/ST6 cells are lost in the absence of selective pressure, which will reveal whether or not selective pressure is necessary during scale-up.
Acknowledgments
Contract grant sponsors: National Institutes of Health; National Science Foundation
Contract grant numbers: GM49734; BES-9814157; BES-9818001
The authors thank Drs. Joel and Nancy Shaper of Johns Hopkins University School of Medicine for providing the bovine β1,4-galactosyltransferase cDNA, and Dr. James C. Paulson of the Scripps Research Institute for providing the rat α2,6-sialyltransferase cDNA.
References
- Altmann F, Kornfeld G, Dalik T, Staudacher E, Glossl J. Processing of asparagine-linked oligosaccharides in insect cells. N-acetylgluco-saminyltransferase I and II activities in cultured lepidopteran cells. Glycobiology. 1993;3:619–625. doi: 10.1093/glycob/3.6.619. [DOI] [PubMed] [Google Scholar]
- Altmann F, Staudacher E, Wilson IB, Marz L. Insect cells as hosts for the expression of recombinant glycoproteins. Glycoconj J. 1999;16:109–123. doi: 10.1023/a:1026488408951. [DOI] [PubMed] [Google Scholar]
- Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucl Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake MS, Johnston KH, Russell-Jones GJ, Gotschlich EC. A rapid, sensitive method for detection of alkaline phosphatase conjugated anti-antibody on western blot. Anal Biochem. 1984;36:175–179. doi: 10.1016/0003-2697(84)90320-8. [DOI] [PubMed] [Google Scholar]
- Butters TD, Hughes RC, Vischer P. Steps in the biosynthesis of mosquito cell membrane glycoproteins and the effects of tunicamycin. Biochim Biophys Acta. 1981;640:672–686. doi: 10.1016/0005-2736(81)90097-3. [DOI] [PubMed] [Google Scholar]
- Davis TR, Trotter KM, Granados RR, Wood HA. Baculovirus expression of alkaline phosphatase as a reporter gene for evaluation of production, glycosylation and secretion. Bio/Technology. 1992;10:1148–1150. doi: 10.1038/nbt1092-1148. [DOI] [PubMed] [Google Scholar]
- Guarino LA, Gonzalez MA, Summers MD. Complete sequence and enhancer function of the homologous DNA regions of Autographa californica nuclear polyhedrosis virus. J Virol. 1986;60:224–229. doi: 10.1128/jvi.60.1.224-229.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino LA, Summers MD. Nucleotide sequence and temporal expression of a baculovirus regulatory gene. J Virol. 1987;61:2091–2099. doi: 10.1128/jvi.61.7.2091-2099.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hink F. Established insect cell line from the cabbage looper, Trichoplusia ni. Nature. 1970;226:466–467. doi: 10.1038/226466b0. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Hohmann AW, Faulkner P. Monoclonal antibodies to baculovirus structural proteins: determination of specificities by Western blot analysis. Virology. 1983;125:432–444. doi: 10.1016/0042-6822(83)90214-3. [DOI] [PubMed] [Google Scholar]
- Hollister J, Jarvis DL. Engineering lepidopteran insect cells for sialoglycoprotein production by genetic transformation with mammalian β1,4-galactosyltransferase and α2,6-sialyltransferase genes. Glycobiology. 2001;11:1–9. doi: 10.1093/glycob/11.1.1. [DOI] [PubMed] [Google Scholar]
- Hollister JR, Shaper JH, Jarvis DL. Stable expression of mammalian beta 1,4-galactosyltransferase extends the N-glycosylation pathway in insect cells. Glycobiology. 1998;8:473–480. doi: 10.1093/glycob/8.5.473. [DOI] [PubMed] [Google Scholar]
- Jarvis DL. Effects of baculovirus infection on IE1-mediated foreign gene expression in stably transformed insect cells. J Virol. 1993;67:2583–2591. doi: 10.1128/jvi.67.5.2583-2591.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis DL. Baculovirus expression vectors. In: Miller LK, editor. The baculoviruses. New York: Plenum Press; 1997. pp. 389–431. [Google Scholar]
- Jarvis DL, Finn EE. Biochemical analysis of the N-glycosylation pathway in baculovirus-infected lepidopteran insect cells. Virology. 1995;212:500–511. doi: 10.1006/viro.1995.1508. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Finn EE. Modifying the insect cell N-glycosylation pathway with immediate early baculovirus expression vectors. Nat Biotechnol. 1996;14:1288–1292. doi: 10.1038/nbt1096-1288. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Fleming JA, Kovacs GR, Summers MD, Guarino LA. Use of early baculovirus promoters for continuous expression and efficient processing of foreign gene products in stably transformed lepidopteran cells. Bio/Technology. 1990;8:950–955. doi: 10.1038/nbt1090-950. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Garcia A., Jr Biosynthesis and processing of the Autographa californica nuclear polyhedrosis virus gp64 protein. Virology. 1994;205:300–313. doi: 10.1006/viro.1994.1646. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Guarino LA. Continuous foreign gene expression in transformed lepidopteran insect cells. In: Richardson CD, editor. Baculovirus expression protocols. Clifton, NJ: Humana Press; 1995. pp. 187–2O2. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Kawar ZS, Hollister JR. Engineering N-glycosylation pathways in the baculovirus-insect cell system. Curr Opin Biotechnol. 1998;9:528–533. doi: 10.1016/s0958-1669(98)80041-4. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Weinkauf C, Guarino LA. Immediate early baculovirus vectors for foreign gene expression in transformed or infected insect cells. Prot Expr Pur. 1996;8:191–203. doi: 10.1006/prep.1996.0092. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Ann Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Marchal I, Jarvis DL, Cacan R, Verbert A. Glycoproteins from insect cells: Sialylated or not? Biol Chem. 2001;382:151–159. doi: 10.1515/BC.2001.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marz L, Altmann F, Staudacher E, Kubelka V. Protein glycosylation in insects. In: Montreuil J, Vliegenthart JFG, Schachter H, editors. Glycoproteins. Amsterdam: Elsevier; 1995. pp. 543–563. [Google Scholar]
- Miller LK. The baculoviruses. In: Fraenkel-Conrat H, Wagner R, editors. The viruses. New York: Plenum Press; 1997. [Google Scholar]
- Montreuil J, Vliegenthart JFG, Schachter H. In: Glycoproteins. Neuberger A, Van Deenen LLM, editors. Amsterdam: Elsevier; 1995. p. 29a. New comprehensive biochemistry. [Google Scholar]
- Murhammer DW, Goochee CF. Scaleup of insect cell cultures: Protective effects of Pluronic F-68. Bio/Technology. 1988;6:1411–1418. [Google Scholar]
- O’Reilly DR, Miller LK, Luckow VA. Baculovirus expression vectors. New York: WH Freeman; 1992. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1989. [Google Scholar]
- Shaper NL, Shaper JH, Meuth JL, Fox JL, Chang H, Kirsch IR, Hollis GF. Bovine galactosyltransferase: Identification of a clone by direct immunological screening of a cDNA expression library. Proc Natl Acad Sci USA. 1986;83:1573–1577. doi: 10.1073/pnas.83.6.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Stollar V, Stollar BD, Koo R, Harrap KA, Schlesinger RW. Sialic acid contents of sindbis virus from vertebrate and mosquito cells. Equivalence of biological and immunological viral properties. Virology. 1976;69:104–115. doi: 10.1016/0042-6822(76)90198-7. [DOI] [PubMed] [Google Scholar]
- Summers MD, Smith GE. Baculovirus structural polypeptides. Virology. 1978;84:390–402. doi: 10.1016/0042-6822(78)90257-x. [DOI] [PubMed] [Google Scholar]
- Summers MD, Smith GE. Texas Agricultural Experiment Station Bulletin No 1555. 1987. A manual of methods for baculovirus vectors and insect cell culture procedures. [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Die I, van Tetering A, Bakker H, van den Eijnden DH, Joziasse DH. Glycosylation in lepidopteran insect cells: Identification of a β1,4-N-acetylgalactosaminyltransferase involved in the synthesis of complex-type oligosaccharide chains. Glycobiology. 1996;6:157–164. doi: 10.1093/glycob/6.2.157. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J. Essentials of glycobiology. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1999. [PubMed] [Google Scholar]
- Vaughn JL, Goodwin RH, Thompkins GJ, McCawley P. The establishment of two insect cell lines from the insect Spodoptera frugiperda (Lepidoptera:Noctuidae) In Vitro. 1977;13:213–217. doi: 10.1007/BF02615077. [DOI] [PubMed] [Google Scholar]
- Volkman LE. The 64K envelope protein of budded Autographa californica nuclear polyhedrosis virus. Curr Top Microb Immunol. 1986;131:103–118. doi: 10.1007/978-3-642-71589-1_6. [DOI] [PubMed] [Google Scholar]
- Weinstein J, Lee EU, McEntee K, Lai P-H, Paulson JC. Primary structure of β-galactoside a2,6-sialyltransferase. Conversion of membrane-bound enzyme to soluble forms by cleavage of the NH2-terminal signal anchor. J Biol Chem. 1987;262:17735–17743. [PubMed] [Google Scholar]
- Wickham TJ, Davis T, Granados RR, Shuler ML, Wood HA. Screening of insect cell lines for the production of recombinant proteins and infectious virus in the baculovirus expression system. Biotechnol Progr. 1992;8:391–396. doi: 10.1021/bp00017a003. [DOI] [PubMed] [Google Scholar]
- Yates JL, Warren N, Sugden B. Stable replication of plasmids derived from Epstein–Barr virus in various mammalian cells. Nature (London) 1985;313:812–815. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]