

Summary
Endoglin (Eng) is an auxiliary receptor for transforming growth factor-β (TGFβ), with important roles in vascular function. TGFβ regulates angiogenesis through balancing the pro-proliferative and pro-differentiation signaling pathways of endothelial cells (EC). However, the contribution of endoglin to these TGFβ activities, and more specifically modulation of EC phenotype, remains elusive. Mutations in endoglin cause hereditary hemorrhagic telangiectasia-1 in humans. The Eng+/− mice are viable and exhibit some of the vascular defects seen in humans with endoglin haploinsufficiency. In the present study we show that haploinsufficiency of endoglin results in attenuation of retinal neovascularization during oxygen-induced ischemic retinopathy. Although the importance of endoglin expression in angiogenesis and vascular development has been demonstrated, the underlying mechanisms remain obscure. To gain detailed insight into the cell autonomous regulatory mechanisms that affect angiogenic properties of EC, we prepared retinal EC from Eng+/+ and Eng+/− Immorto mice. The Eng+/− EC were more adherent, less migratory, and failed to undergo capillary morphogenesis. Aortic sprouting angiogenesis was similarly attenuated in aortas from Eng+/− mice. In addition, Eng+/− EC expressed increased levels of VEGF but reduced expression of endothelial NO synthase and NO production. Mechanistically, these changes were consistent with sustained activation of mitogen-activated protein kinase (MAPK) pathways, and aberrant Smad-dependent signaling pathways in Eng+/− EC. Taken together, our results underscore the importance of endoglin in both canonical and non-canonical TGFβ signaling pathways modulating both the activation and quiescence of the endothelium during angiogenesis.
Key words: Angiogenesis, Endothelial cells, Extracellular matrix, MAP kinase pathways, Thrombospondins
Introduction
Endoglin/CD105 is a 180 kDa homodimeric transmembrane glycoprotein predominantly expressed on vascular endothelial cells (EC) and functions as an auxiliary receptor for transforming growth factor-β (TGFβ) (Bernabeu et al., 2007; Gougos and Letarte, 1990; Miller et al., 1999). Endoglin expression level in the vasculature increases during angiogenesis. Its decreased expression leads to altered angiogenesis in vitro and aberrant vascular development and function in vivo (Arthur et al., 2000; Bourdeau et al., 1999; Bourdeau et al., 2000; Li et al., 1999). Mutations in a single allele of endoglin gene results in decreased level of the protein and are associated with hereditary hemorrhagic telangiectasia-1 (HHT-1) and vascular malformations in humans. Endoglin deficient (Eng −/−) mice die at E10.5–11.5 from cardiovascular defects due to inappropriate remodeling of mature vascular network (Bourdeau et al., 1999; Li et al., 1999). While endoglin heterozygote (Eng+/−) mice are viable, they exhibit some of the vascular defects seen in humans with endoglin haploinsufficiency (Mahmoud et al., 2010). However, the underlying molecular and cellular mechanisms for endoglin function are not fully understood.
Various angiogenic processes including regulation of EC proliferation and migration, induction of pericyte differentiation, and production of basement membrane depend on TGFβ activity (Antonelli-Orlidge et al., 1989; Hirschi et al., 1998; Neubauer et al., 1999). TGFβ regulates angiogenesis through balancing the pro-proliferative and pro-differentiation signaling pathways of EC. TGFβ modulate EC properties through TGFβ-receptor II (TGFβRII) and two distinct TGFβRI, activin-receptor like kinase-1(ALK1) and ALK5, by balancing signals from these two pathways (Lu, 2008; Lutty et al., 1993; Oh et al., 2000; Seki et al., 2003). Phosphorylated ALKs (ALK1 and ALK5) phosphorylate Smad proteins which mediate the downstream events. In addition, activation of ALK1 requires the presence of both ALK5 and TGFβRII (Goumans et al., 2003). Although the importance of Smad-dependent signaling pathways have been previously considered in aberrant canonical TGFβ signaling pathways, the impact of endoglin on non-canonical TGFβ signaling pathways, including the mitogen-activated protein (MAPK) pathways, remain elusive.
ALK1 activation by TGFβ promotes EC activation during angiogenesis while ALK5 activation is growth inhibitory and is thought to mediate vessel maturation and stability (Lebrin et al., 2005). Lack of TGFβRII or ALK5 induces embryonic lethality at E10.5 due to vascular development defect in the yolk sack (Larsson et al., 2001; Oshima et al., 1996), and EC from ALK5 deficient mice show altered fibronectin production and migration supporting abrogated vascular development (Larsson et al., 2001). ALK1 knockout mice also die at E10.5–11.5 and exhibit failure of capillary network and dilated blood vessels (Oh et al., 2000). Effect of ALK1 mutation is minimal on EC differentiation and vascular development in mice. However, ALK1 mutants exhibit defects in angiogenesis and vascular smooth muscle cell development. Although TGFβ has been generally regarded as an anti-proliferation factor, TGFβ shows both stimulatory and inhibitory effects in angiogenesis (Krupinski et al., 1996; Pepper, 1997). However, the exact contribution of endoglin to TGFβ-mediated angiogenesis activities, and more specifically cell autonomous modulation of EC phenotype, remains poorly understood.
Oxygen-induced ischemic retinopathy (OIR) in the mouse is a highly reproducible model of angiogenesis in vivo and recapitulates the human retinopathy of prematurity condition (Smith et al., 1994). In this model, postnatal day 7 (P7) mice are exposed to 75% oxygen for 5 days. During this time, the developing retinal vasculature is highly sensitive to high oxygen levels. The high oxygen impedes further growth of blood vessels and causes obliteration of the existing vessels. At postnatal day 12, the mice are returned to room air (20% oxygen) for 5 days. During this stage, the lack of sufficient blood vessels causes the retina to become ischemic, upregulating VEGF expression and inducing angiogenesis. Unfortunately, these vessels are fragile and leaky, sprout into the vitreous, and hemorrhage, causing further damage to the retina. How endoglin haploinsufficiency contributes to retinal neovascularization and EC function needs further investigation.
Here we showed that haploinsufficiency of endoglin resulted in attenuation of retinal neovascularization during OIR. We also showed Eng+/− EC are less migratory and fail to undergo capillary morphogenesis in Matrigel. These results were consistent with alterations in Eng+/− endothelial cell-cell and cell-matrix interactions concomitant with changes in production of various ECM proteins. However, the Eng+/− EC proliferated at a faster rate, in part, due to increased VEGF expression and a reduced rate of apoptosis. Mechanistically, the Eng+/− EC exhibited sustained activation of JNK, ERK and p38 MAPKs, while the levels of phosphorylated Akt and Src were modestly affected. A significant decrease in nitric oxide (NO) level, consistent with decreased expression of endothelial nitric oxide synthase (eNOS) and inducible NOS (iNOS) in Eng+/− EC, was also observed. Furthermore, the Eng+/− EC were resistant to the anti-proliferation effect of TGFβ, expressed lower levels of phosphorylated Smad1/5, and did not respond to TGFβ by enhanced Smad1/5 phosphorylation consistent with the reduced expression of TGFβRII in these cells. However, the response of Smad2/3 to TGFβ was not affected in Eng+/− EC. Collectively our results demonstrate that appropriate expression of endoglin is essential for both activation and quiescence state of EC during angiogenesis perhaps through engagement of both canonical and non-canonical TGFβ signaling pathways.
Results
Attenuation of retinal neovascularization in Eng+/− mice
Endoglin is highly expressed during angiogenesis and may play a role in regulating growth of new blood vessels (Duff et al., 2003; Düwel et al., 2007; Jerkic et al., 2006b; ten Dijke et al., 2008). To gain further insight into the physiological role of endoglin during vascular development and neovascularization, we compared retinal neovascularization during OIR in Eng+/+ and Eng+/− mice. As shown in Fig. 1A,B,E Eng+/+ and Eng+/− mice exhibited similar sensitivity to high oxygen-mediated vessel obliteration. Fig. 1C,D show that after return to room air, Eng+/+ mice have numerous vascular tufts sprouting from the retina into the vitreous. This is consistent with significant upregulation of endoglin expression in retinal vasculature and enhanced retinal neovascularization in Eng+/+ mice during OIR (supplementary material Fig. S1). In contrast, Eng+/− mice exhibited little or no neovascularization. The quantitative assessment of the data are shown in Fig. 1F. Thus, consistent with previous work, endoglin expression is essential during angiogenesis.
Fig. 1.
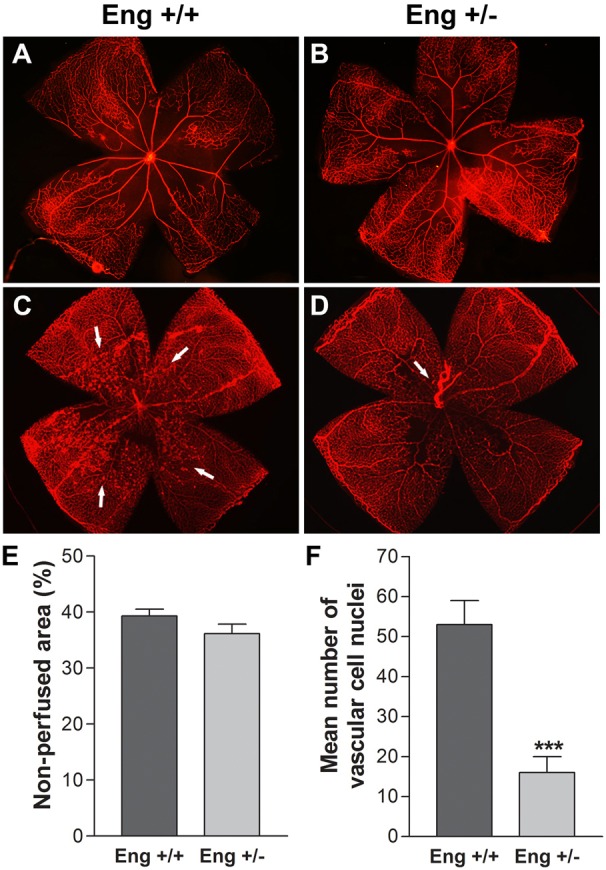
Attenuation of retinal neovascularization in Eng+/− mice. Retinal wholemounts were prepared from 12-day-old Eng+/+ (A) and Eng+/− mice (B) exposed to high oxygen (75%) for 5 days, or postnatal day 17 (P17) following exposure to 75% oxygen for 5 days and room air (20% oxygen) for 5 days (C) and (D). The retinas were stained with collagen IV to visualize the retinal vasculature (magnification ×25). (E) Quantitative assessments of retinal non-perfused areas in response to hypoxia. Data in each bar are the mean non-perfused area relative to whole area of each retina in eights eyes of four mice. (F) Quantitative assessment of retinal neovascularization performed as described in the Materials and Methods section. Data in bars represent the mean number of vascular cell nuclei present on the vitreous side of the retina penetrating the inner limiting membrane (***P<0.001; n = 5). Arrows show neovascular tuffs.
Defective postnatal retinal vascularization in Eng+/− mice
We next examined the retinal vasculature of Eng+/− mice at different postnatal stages of development. The retinal vessels of Eng+/− mice sprouted from the optic nerve and extended to the periphery of the retina at a similar rate as Eng+/+ mice (supplementary material Fig. S2A–D). The Eng+/− mice had similar vascular density as Eng+/+ mice, without a significant impact on vessel diameters and number of secondary and tertiary branches. However, we observed increased number of proliferating vascular cells in retinas of Eng+/− mice compared with Eng+/+ mice (supplementary material Fig. S3).
Endoglin haploinsufficiency alters EC morphology and expression of cell specific markers
We prepared retinal EC from Eng+/+ and Eng+/− Immorto mice using a novel method described by us (Su et al., 2003) and detailed in Material and Methods. EC from Eng+/+ and Eng+/− mice were cultured on gelatin-coated culture dishes. Cell morphology was examined at sub-confluent and confluent states, and we observed a significant change in morphology of Eng+/− EC compared with Eng+/+ EC (Fig. 2A). While Eng+/+ EC showed closely packed cobblestone shape morphology, Eng+/− EC exhibited a more elongated morphology with diminished cell-cell interaction which became more obvious at confluence.
Fig. 2.

Isolation and characterization of mouse retinal EC. (A) The Eng+/+ and Eng+/− EC were isolated and cultured on gelatin-coated plates. Cells were photographed in digital format under subconfluence and confluence conditions. (B) Expression of vascular endothelial cell markers. Retinal EC were examined for expression of PECAM-1, endoglin, B4-lectin, VCAM-1, ICAM-1, ICAM-2, VEGFR1, and VEGFR2 by FACS analysis. The shaded areas show control IgG staining. These experiments were repeated twice with two different isolations of EC with similar results. Representative geometrical means of the expression are noted.
To determine the properties of Eng+/− EC, we first analyzed the expression level of various EC markers including PECAM-1 (CD31), endoglin, and B4-lectin by FACS analysis. As expected, endoglin expression levels were decreased in Eng+/− EC compared with Eng+/+ EC. Moreover PECAM-1, one of the essential membrane proteins for sustained EC integrity and angiogenesis (DiMaio et al., 2008), was downregulated in Eng+/− EC (Fig. 2B). These cells were also less positive for B4-lectin expression compared with Eng+/+ EC. We also determined the expression levels of other EC markers, including ICAM-1, ICAM-2, VCAM-1, VEGFR1, and VEGFR2 (Fig. 2B). We observed a decrease in expression of ICAM-2 and VEGFR1, two proteins with important roles in angiogenesis, in Eng+/− EC. These results suggested a significant role for endoglin expression in modulation of proangiogenic properties of EC. A similar decrease in expression of EC markers including PECAM-1 and ICAM-2 was observed in retinal lysates prepared from Eng+/− mice (supplementary material Fig. S5A,B).
Attenuation of Eng+/− EC capillary morphogenesis and aortic sprouting
Capillary morphogenesis is one of the pivotal properties of EC during angiogenesis and can be assessed by plating EC in Matrigel. The Eng+/− EC showed significantly reduced capillary morphogenesis compared with Eng+/+ EC (Fig. 3A,B). In Eng+/+ EC, we observed well-organized capillary-like networks, however, Eng+/− EC did not form a network and instead aggregated in Matrigel. A quantitative assessment of the data are shown in Fig. 3E. The impact of endoglin haploinsufficiency on vascular sprouting was further investigated using the ex vivo aortic sprouting assay. Aortic rings were isolated from 3-week-old Eng+/+ and Eng+/− mice, and the sprouting was analyzed after 5 days of incubation in Matrigel. Fig. 3C,D show significant attenuation of sprouting angiogenesis in aortic ring from Eng+/− mice. A quantitative assessment of the data are shown in Fig. 3F. Thus, EC expression of endoglin plays an important role in capillary morphogenesis in vitro and sprouting angiogenesis ex vivo, consistent with attenuation of retinal neovascularization in vivo.
Fig. 3.
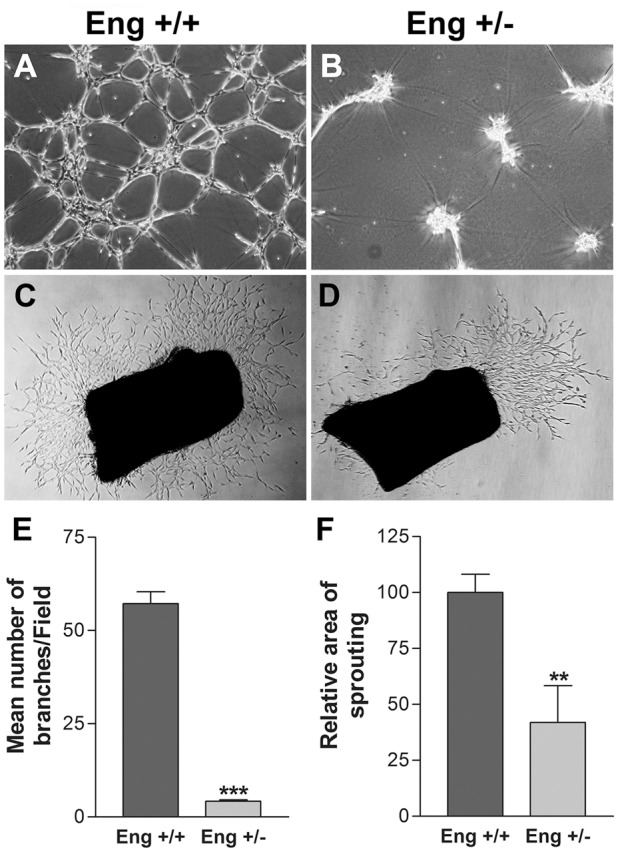
Abrogated capillary morphogenesis of retinal EC and ex vivo sprouting of aortas from Eng+/− mice. Retinal EC from Eng+/+ (A) and Eng+/− (B) were plated in Matrigel-coated plates and incubated for 18 h. The tube-like networks were photographed in digital format. (C,D) The aortas from Eng+/+ and Eng+/− mice were cultured in Matrigel for 5 days and photographed in digital format (magnification ×40). (E) Quantitative assessment of results from A and B. Data are the mean number of branch points from 5 high-power fields (magnification ×100). Note the significant decrease in tube formation by Eng+/− EC compared with Eng+/+ EC (***P<0.001; n = 3). (F) Quantitative assessment of the results from C and D was performed as described in the Materials and methods section. Note the significant decrease in sprouting of aortas from Eng+/− mice compared with Eng+/+ mice (**P<0.01; n = 5). These experiments were repeated with two different isolations of retinal EC and aortas from 3 different mice, with similar results.
Attenuation of NO production in Eng+/− EC
Nitric oxide (NO), produced by nitric oxide synthase (NOS), is an important regulator of angiogenesis. In EC, eNOS is a major source of NO in response to stimulatory factors and shear stress. We observed significantly reduced NO levels in Eng+/− EC compared with Eng+/+ EC (Fig. 4A). To determine the impact of endoglin haploinsufficiency on the expression of NO producing system in EC, eNOS, HSP90, and iNOS expression were analyzed by western blotting. While eNOS expression was dramatically decreased, HSP90, a chaperon protein involved in eNOS activation, expression was not affected in Eng+/− EC (Fig. 4B). Although it is not a major NO-producing enzyme in EC, iNOS, a member of NOS family, was also significantly downregulated in Eng+/− EC (Fig. 4B). The reduced eNOS expression was also observed in retina extracts from 4-week-old Eng+/− mice (Fig. 4C), which showed a significant decrease in endoglin expression (Fig. 4D). These results revealed abrogated NO production in Eng+/− EC, mainly through decreased expression of NO-producing enzymes. However, we observed a comparable eNOS levels in aortas from Eng+/+ and Eng+/− mice (Fig. 4E). This may be attributed, at least in part, to the differences in tissue of origin, and macrovascular versus microvascular endothelium in the aorta and retina, respectively.
Fig. 4.

Attenuation of NOS expression and NO production in Eng+/− EC. (A) Intracellular nitric oxide (NO) level in EC was measured using DAF-FM as described in the Materials and Methods section. Note a significant decrease in the level of intracellular NO in Eng+/− EC compared with Eng+/+ EC (**P<0.01; n = 3). (B) The expression of eNOS, HSP90 and iNOS in cell lysates were analyzed by western blotting. The eNOS (C) and endoglin (D) expression in retina lysates from 4-week-old Eng+/+ and Eng+/− mice was analyzed by western blotting. (E) The expression of eNOS in aorta lysates was analyzed. β-Actin was used as a loading control and relative intensity is shown (*P<0.05, n = 3; **P<0.01, n = 3).
Eng+/− EC were less migratory
The ability of EC to migrate is essential during angiogenesis. To examine the migratory property of EC, we conducted a scratch wound assay. After 48 h, wound in Eng+/+ EC were completely closed, while 60% of wounds in Eng+/− EC monolayer still remained uncovered (Fig. 5A). A quantitative assessment of the data are shown in Fig. 5B. Similar results were observed using the transwell migration assay (Fig. 5C). Formations of actin stress fibers and organization of focal adhesions are pivotal in cell migration. We observed distinct differences between Eng+/+ and Eng+/− EC actin organization and focal adhesions following phalloidin (actin filament) and vinculin (focal adhesion) staining. Compared with Eng+/+, Eng+/− EC actin stress fibers were located on the cell periphery (Fig. 5D). While focal adhesions of Eng+/+ EC were mainly located at the edge of cells with ruffles, Eng+/− EC showed centrally localized focal adhesions (Fig. 5D). Together these results demonstrate an important role for endoglin expression in modulation of EC migration.
Fig. 5.

Attenuation migration, enhanced adhesion and altered expression of extracellular matrix (ECM) proteins in Eng+/− EC. (A) Cell migration was determined by scratch wounding of EC monolayers on gelatin-coated plates and wound closure was monitored by photography within 48 h (magnification ×40). (B) Quantitative assessment of results in A (***P<0.001; n = 5). (C) Rate of cell migration through a transwell, determined by counting the number of cells migrated through the filter (**P<0.01, n = 10). (D) Localization of focal adhesions and actin stress fibers in Eng+/+ and Eng+/− EC by staining with phalloidin (green; actin) and vinculin (red; focal adhesions) (magnification ×630). (E) The adhesion of Eng+/+ and Eng+/− EC to fibronectin, vitronectin, collagen I and collagen IV was determined as described in the Materials and Methods section (***P<0.001, n = 3; **P<0.01, n = 3; *P<0.05, n = 3). (F) The presence of various ECM proteins, in conditioned medium and cell lysates, prepared from Eng+/+ and Eng+/− EC, was analyzed by western blotting for tenascin-C, fibronectin, osteopontin, collagen IV, TSP-1 and TSP-2. The relative intensities of the protein bands were determined using Image J software. These experiments were repeated with two different isolations of EC with similar results.
Eng+/− EC were more adhesive
Endothelial cell migration is influenced by their binding ability to, and composition of, their ECM proteins. Here we determined the adhesion of Eng+/+ and Eng+/− EC to various ECM proteins including fibronectin (FN), vitronectin (VN), collagen I, and collagen IV, as previously described (Park et al., 2010). Varying concentrations of ECM proteins were coated on 96-well plates and cells were allowed to adhere. Non-adherent cells were removed by gently washing the plate until no cells were left in wells coated with BSA. The number of adherent cells in each well was quantified by measuring the levels of intracellular acid phosphatase. The Eng+/− EC were more adherent on FN, VN, and collagen IV compared with Eng+/+ EC (Fig. 5E). Moreover, Eng+/− adhered to collagen I while Eng+/+ EC did not. These results were consistent with reduced migration of Eng+/− EC.
Altered production of ECM proteins in Eng+/− EC
Endothelial cell adhere to ECM proteins through specific cell surface receptor integrins. Cell migration is significantly influenced by the production of ECM proteins and expression of their receptors. We next determined the production of various ECM proteins in Eng+/+ and Eng+/− EC. Tenascin-C, fibronectin, osteopontin, collagen IV, thrombospondin-1 (TSP-1), and TSP-2 are the major ECM components secreted by EC and play important roles in cell migration, wound repair, and inflammation (Lund et al., 2009; Sabet and Gordon, 1989; Tucker, 1991). Fig. 5F shows an increase in production of tenascin-C, osteopontin and collagen IV in Eng+/− EC compared with Eng+/+ EC. In contracts, expression of TSP-1, and TSP-2 was reduced in Eng+/− EC compared with Eng+/+ EC. Thus, endoglin haploinsufficiency has a major impact on expression of ECM proteins and EC adhesive properties. We also examined expression of various integrins by FACS analysis including α1, α2, α3, α5, αv, α5β1, αvβ3, β1, β3 and β8. The Eng+/+ and Eng+/− EC expressed similar levels of these integrins (supplementary material Fig. S4). However, these results do not exclude the possibility of changes in affinity and avidity of these integrins.
Abrogation of cell-cell junction in Eng+/− EC
The altered morphology in Eng+/− EC compared with Eng+/+ EC suggested that alteration in cell-cell interactions may also occur. Formation of cell-cell junction between EC is essential to sustain vascular integrity. Junction formation is mediated by interaction of junctional proteins including VE-cadherin, β-catenin and ZO-1 with intercellular proteins. To visualize junction formation, confluent monolayers of Eng+/+ and Eng+/− EC were stained with antibodies to various junctional proteins. Moreover the expression level of junctional proteins was determined by western blot analysis. Eng+/+ EC showed well-organized junctional localization of these proteins (Fig. 6A) with significant expression of junctional proteins (Fig. 6B). In contrast, Eng+/− EC lacked VE-cadherin and ZO-1 at cell-cell junctions (Fig. 6A) and the expression level of both proteins was significantly reduced (Fig. 6B). Although expression of β-catenin was similar in both Eng+/+ and Eng+/− EC (Fig. 6B), its junctional localization was abrogated in Eng+/− retinal EC (Fig. 6A). The decreased VE-cadherin expression was also observed in retinal lysates from Eng+/− mice compared with Eng+/+ mice (supplementary material Fig. S5A,B). However, an increase in ZO-1 level was observed in retinal lysates from Eng+/− mice. This is in contrast to the decreased level of ZO-1 observed in Eng+/− EC, and may represent the limitation of the in vitro cell culture system and/or contributions from other cell types expressing ZO-1 including retina pigment epithelial cells (supplementary material Fig. S5A,B). Thus, endoglin haploinsufficiency was associated with defects in formation of cell-cell junctions.
Fig. 6.
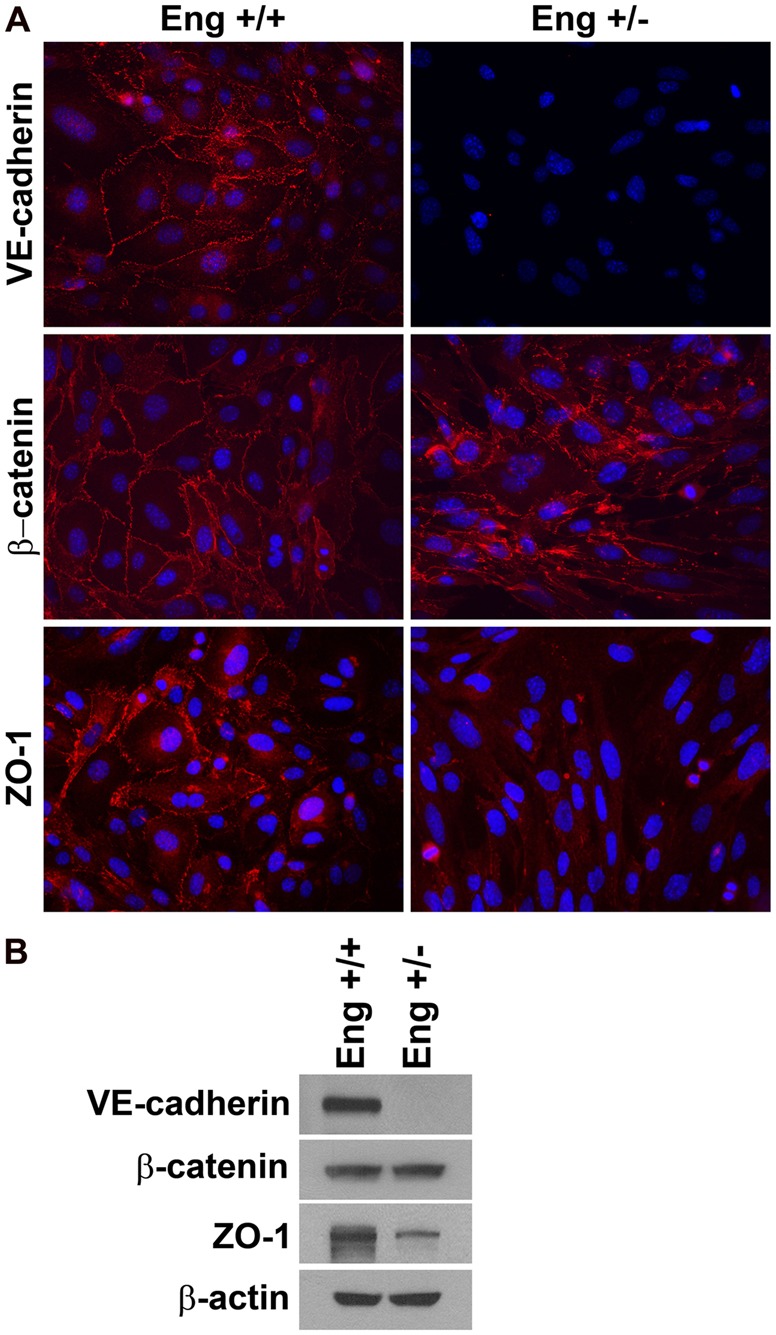
Altered expression and localization of VE-cadherin, β-catenin and ZO-1 in Eng+/− EC. (A) The Eng+/+ and Eng+/− EC were grown on a fibronectin-coated chamber slide to confluence and stained for specific proteins. Note the aberrant localization of these proteins to sites of cell-cell contact in Eng+/− EC compared with Eng+/+ EC (magnification ×400). (B) Cell lysates prepared from Eng+/+ and Eng+/− EC were analyzed by western blotting for VE-cadherin, β-catenin, ZO-1 and β-actin. These experiments were repeated with two different isolations of EC with similar results.
Enhanced proliferation of Eng+/− EC
There are numerous studies investigating the impact of endoglin haploinsufficiency on EC proliferation, and both pro- and anti-proliferative effects have been reported (Pepper, 1997). To examine the effect of endoglin haploinsufficiency on the proliferation of retinal EC, the growth rate of Eng+/+ and Eng+/− EC was determined by counting the number of cells. The rate of Eng+/− EC proliferation was significantly increased compared with Eng+/+ EC, and the number of Eng+/− EC was twice of that of Eng+/+ EC after 12 days (Fig. 7A). To determine whether the increased rate of proliferation was associated with an increase in DNA synthesis, we measured DNA synthesis level in these cells by determining the percent of 5-ethynyl-2′-deoxyuridine (EdU) positive cells. Consistent with increased cell number, DNA synthesis level was significantly higher in Eng+/− compared with Eng+/+ EC (Fig. 7B). Thus, endoglin haploinsufficiency was associated with enhanced proliferation of EC, consistent with enhanced proliferation observed in the retinal vasculature in vivo (supplementary material Fig. S3).
Fig. 7.

Altered proliferation and apoptosis and sustained activation of MAPK signaling pathways in Eng+/− EC. The rate of EC proliferation was determined by counting the number of cells after different times in culture (A) and by analyzing the rate of DNA synthesis (B) (**P<0.01; n = 3). (C) H2O2 toxicity of EC was measured by MTS assay. Note a significantly greater viability in Eng+/− EC compared with Eng+/+ EC (***P<0.001; n = 3). (D) The rate of apoptosis was determined by measuring caspase-3/7 activity. The cell stressors H2O2 and staurosporine were also used in EC growth medium for 8 h (*P<0.05 and ***P<0.001; n = 3). (E) The Eng+/− EC produced a significantly higher level of VEGF compared with Eng+/+ EC (***P<0.001; n = 3). These experiments were repeated with two different isolations of EC with similar results. (F) The active and total level of Src, AKT, JNK, p38, and ERK in Eng+/+ and Eng+/− EC was determined by western blot analysis. These experiments were repeated three times and relative intensity was analyzed by densitometry (*P<0.05, n = 3). (G) Analysis of active and total ERK level in aortas from Eng+/+ and Eng+/− mice (*P<0.05, n = 4).
Eng+/− EC were more resistant to oxidative challenge
Hydrogen peroxide (H2O2) is generally used as an inducer of oxidative stress. We next determined the cytotoxic effect of H2O2 on Eng+/+ and Eng−/− EC using the MTS assay. We observed decreased cell viability by H2O2 (0–2 mM for 2 days) in a concentration-dependent manner (not shown). The incubation with 1.75 mM H2O2 decreased viability of Eng+/+ EC by 30%, while Eng+/− retinal EC showed 50% viability (Fig. 7C). Thus, Eng+/− EC were less susceptible to oxidative stress-induced cytotoxicity.
We next examined whether the endoglin haploinsufficiency impacts EC apoptosis. Staurosporine is a well recognized inducer of apoptosis in many cell types and was used as positive control. Apoptosis was determined by measuring active caspase 3/7 level in EC following incubation with H2O2 (1.75 mM) or staurosporine (2 µM) for 8 h. The medium control, H2O2, or staurosporine-treated Eng+/− EC showed significantly lower levels of active caspase 3/7 compared with Eng+/+ EC (Fig. 7D). Together these results suggest that endoglin haploinsufficiency was associated with protection of EC from apoptotic insults.
Vascular endothelial growth factor (VEGF) is a known survival factor for EC. To further explore the more protective and proliferative feature of Eng+/− EC, we analyzed VEGF levels produced by Eng+/− and Eng+/+ EC. We observed a dramatic increase in the level of VEGF produced by Eng+/− EC compared with Eng+/+ EC (Fig. 7E). These results were consistent with a significant increase in VEGF levels in retinal lysates from Eng+/− mice (supplementary material Fig. S5C). Thus, endoglin haploinsufficiency in EC was associated with increased production of VEGF, resistance to pro-apoptotic stimuli and enhanced proliferation.
Sustained activation of non-canonical TGFβ signaling pathways in Eng+/− EC
The alterations in adhesion and migration of Eng+/− cells, along with increased levels of VEGF suggested alterations in the intracellular signaling pathways. To determine the impact of endoglin on various Smad-independent signaling pathways, we analyzed activation of the Src kinase, AKT1/PKB and MAPK signaling pathways in Eng+/+ and Eng+/− EC (Fig. 7F). AKT activation, downstream of the ECM/integrin and growth factor/receptor signaling pathways, promotes cell survival and Src kinase plays an important role in cell proliferation and migration. The impact of endoglin haploinsufficiency on the expression and activation of AKT and Src kinase was not significant. These results are inconsistent with a recent report suggesting an important role for endoglin in activation of PI3K and Akt (Lee et al., 2012). However, MAPKs, including extracellular-signal-regulated kinases (ERKs), c-Jun N-terminal kinase (JNK) and p38, were more active in Eng+/− EC. MAPKs are involved in regulation of gene expression, differentiation, proliferation, survival and apoptosis of various cell types including EC. The sustained activation of MAPK/ERKs was also observed in aortas from Eng+/− mice compared with Eng+/+ mice (Fig. 7G). Thus, appropriate expression of endoglin may be essential for dampening the activity of MAPK signaling pathways and promoting the mature quiescence phenotype of vascular EC.
The anti-proliferative effect of TGFβ was abrogated in Eng+/− EC
The Eng+/− EC exhibited enhanced proliferation as recently reported in early embryonic EC isolated from Eng−/− mice (Pece-Barbara et al., 2005) and in retinal vessels of mice with conditional deletion of endoglin (Mahmoud et al., 2010). However, the underlying mechanisms remain elusive. The impact of endoglin haploinsufficiency on TGFβRII, ALK1 and ALK5 expression was analyzed by western blotting. Fig. 8A shows that TGFβRII and ALK5 levels were decreased in Eng+/− EC. Consistent with our in vitro data, we observed a decrease in expression of ALK5 in retinal lysates from Eng+/− mice compared with Eng+/+ mice, while expression of TGFβRII was not affected (supplementary material Fig. S5A,B).
Fig. 8.

Decreased expression of TGFβRII and ALK5 and attenuation of TGFβ1-mediated anti-proliferation in Eng+/− EC. (A) Expression of TGFβRII, ALK1 and ALK5 in Eng+/+ and Eng+/− EC was determined by western blot analysis. The effect of TGFβ on the rate of EC proliferation was determined by counting cell numbers after incubating with TGFβ1 (1 ng/ml for 3 days; **P<0.01, n = 3) (B) and by analyzing DNA synthesis rate using FACScan flow cytometry following incubation of EC with TGFβ1 (1 ng/ml for 3 days; *P<0.05; n = 3) (C). (D) Activation of Smad1/5 and Smad 2/3 in Eng+/+ and Eng+/− EC were analyzed by western blotting. Cells were serum starved for 24 h, serum stimulated with and without TGFβ (1 ng/ml) for 30 min and used for analysis. These experiments were repeated three times with two different isolations of EC with similar results.
Although TGFβ is known to downregulate EC proliferation, the role of TGFβ on cell proliferation under various conditions is still unclear (Pepper, 1997). To examine the effect of TGFβ on EC proliferation, Eng+/+ and Eng+/− EC were incubated with TGFβ1 (0.2 or 1 ng/ml) in growth medium for 3 days and the rate of proliferation was determined by counting the number of cells. The proliferation of Eng+/+ EC was inhibited by TGFβ1, 50% of medium control, while Eng+/− EC were not affected (Fig. 8B). Moreover, we observed the attenuation of DNA synthesis in Eng+/+ EC, while that of Eng+/− EC was not affected by TGFβ1 incubation (Fig. 8C). Thus, endoglin haploinsufficiency is associated with impaired TGFβ-mediated inhibition of EC proliferation.
Endoglin haploinsufficiency impacted Smad expression and activation
It is generally accepted that Smad1/5 activation induces EC proliferation and migration, while Smad2/3 activation inhibits these activities. ALK1 and ALK5 are subtypes of TGFβRI. ALK5 is universally expressed on various cells and mediates Smad2/3 activation promoting the quiescence of EC. The ALK1 is exclusively expressed on EC and mediates Smad1/5 activation inducing EC activation. The expression of ALK5 in Eng+/− EC was significantly decreased, while ALK1 expression was comparable with Eng+/+ EC (Fig. 8A). Since our results demonstrated decreased migration and increased proliferation in Eng+/− EC (Figs. 5 and 7), we determined the impact of endoglin haploinsufficiency on Smad expression and activation in response to TGFβ1. Activation of Smad1/5 and Smad2/3 were analyzed through detection of phosphorylated Smad1/5 and Smad2/3 by western blotting.
The total Smad1/5 and Smad2/3 expression levels were modestly increased by endoglin haploinsufficiency. However, basal level of pSmad1/5 was significantly higher in Eng+/+ EC compared with Eng+/− EC under normal growth conditions. Serum starvation resulted in a significant decrease in the level pSmad1/5 in both Eng+/+ and Eng+/− EC (Fig. 8D). Serum stimulation restored basal levels of pSmad1/5, and TGFβ1 minimally affected the level pSmad1/5 in both Eng+/+ and Eng+/− EC. Addition of TGFβ1 had a modest effect on serum induced pSmad1/5 levels. Thus, the activation of Smad1/5 in Eng+/− EC was significantly lower compared with Eng+/+ EC, and TGFβ by itself had a modest effect on Smad1/5 phosphorylation. Similar to pSmad1/5, basal level of pSmad2/3 was downregulated in Eng+/− EC, and serum starvation significantly abrogated Smad2/3 phosphorylation in both Eng+/+ and Eng+/− EC (Fig. 8D). Serum stimulation restored Smad2/3 phosphorylation to the basal level, and addition of TGFβ1 enhanced their phosphorylation. In contrast to Smad1/5 phosphorylation, TGFβ1 itself was sufficient to induce Smad2/3 phosphorylation in both Eng+/+ and Eng+/− EC (Fig. 8D). Thus, Eng+/− EC are less responsive to TGFβ1-mediated Smad1/5 activation, but are similarly responsive to TGFβ1-mediated Smad2/3 activation as Eng+/+ EC.
Discussion
Here we explored the impact of endoglin haploinsufficiency on retinal vascular development and neovascularization and retinal EC function. Using Eng+/− mice we showed that endoglin haploinsufficiency mitigated retinal neovascularization during OIR. We also observed defects in postnatal retinal vascular development, including increased proliferation. Thus, endoglin expression is essential for appropriate retinal vasculature development and angiogenesis. To gain insight into cell autonomous function of endoglin, we isolated retinal EC from Eng+/+ and Eng+/− mice. Our results indicated, for the first time, a significant role for endoglin not only in pro-angiogenic but also in pro-differentiation properties of EC. These activities of endoglin were mediated through appropriate modulation of both canonical and non-canonical TGFβ signaling pathways.
The abrogated migratory features of Eng+/− EC has been previously reported suggesting that downregulation of smad1/5/8 activation is involved (Lee et al., 2008; Ray et al., 2010). EC migration is important in capillary morphogenesis and wound closure, and it is influenced by cell-ECM protein interactions (Dejana et al., 2009). Thus, enhanced adhesion of Eng+/− EC observed here is consistent with their attenuated migration. Although endoglin haploinsufficiency minimally impacted the expression of various integrins, we observed alterations in production of various ECM proteins. These changes in ECM protein composition may have a significant impact on the EC local microenvironment and angiogenesis defects noted with endoglin haploinsufficiency. This notion is further supported by a recent study demonstrating the significant impact of varying matrix rigidity on TGFβ signaling in epithelial cells (Leight et al., 2012), while decreasing rigidity increased TGFβ-induced apoptosis, the increased rigidity resulted in epithelial-mesenchymal transition and enhanced migration.
VE-cadherin expression and junctional localization are required for optimal TGFβ signaling in EC (Rudini et al., 2008). It is suggested that VE-cadherin may participate in the recruitment of TGFβRII and enhanced TGFβRII/TGFβRI assembly into an active signaling receptor. Thus, reduced levels of VE-cadherin in EC may contribute to impaired TGFβ signaling and vascular defects. However, a role for VE-cadherin alterations in pathogenesis of endoglin haploinsufficiency has not been previously reported. Our results indicate that endoglin haploinsufficiency has a significant impact on EC junctional organization and function, and are consistent with an increase in TGFβ-mediated Smad2/3 phosphorylation in Eng+/− EC (Fig. 8D) as previously reported by downregulation of VE-cadherin (Rudini et al., 2008) and/or activation of P38 MAPK (Carta et al., 2009; Mu et al., 2012).
The EC proliferation is an essential component of the angiogenic activation in response to various proangiogenic stimuli, including VEGF, and its attenuation is essential for formation of functional, mature blood vessel. A number of studies have shown that endoglin haploinsufficiency or downregulation results in decreased EC proliferation (Jerkic et al., 2006a; Li et al., 2000). Here we observed increased proliferation in Eng+/− compared with Eng+/+ EC associated with increased rate of DNA synthesis as well as reduced rate of apoptosis. In addition, Eng+/− EC were protected from cytotoxic effects of H2O2 and staurosporine. Thus, while TGFβ promotes apoptosis of EC (Pollman et al., 1999a; Pollman et al., 1999b) appropriate expression of endoglin may contribute to this activity. However, the mechanism by which endoglin signaling contributes to the protection and/or apoptosis of EC remains elusive.
VEGF is a proangiogenic factor with prosurvival activity in EC (Gerber et al., 1998; Leung et al., 1989; Neufeld et al., 1999). We observed a significant increase in VEGF expression in Eng+/− EC, which could protect EC from cytotoxic insults as an autocrine and/or paracrine survival factor. This notion is supported by sustained activation of MAPK signaling pathways in Eng+/− EC promoting cell survival and proliferation. Previous studies have shown activation of MAPK pathways by TGFβ through Smad-independent pathways (Derynck and Zhang, 2003; Lebrin et al., 2004). Although endoglin haploinsufficiency moderately impacted the Akt and Src kinase activation, the sustained activation of MAPK pathways, including ERKs, JNK and p38, were observed in Eng+/− EC. Thus, endoglin haploinsufficiency was associated with sustained activation of MAPK signaling pathways and enhanced cell proliferation and survival. We have previously shown that sustained activation of MAPK/ERKs in EC results in disruption of cadherin-mediated cell-cell interactions, as well as decreased expression of VE-cadherin and PECAM-1 (Sheibani et al., 2000; Wu and Sheibani, 2003). To our knowledge this is the first report that appropriate expression of endoglin is essential for attenuation of pro-proliferative and pro-migratory signaling pathways in EC.
Endoglin modulates eNOS expression through TGFβ signaling (Santibanez et al., 2007). The cellular localization of endoglin in caveolae, along with other proteins with important role in angiogenesis including PECAM-1 and eNOS (Toporsian et al., 2005), suggest an important role for endoglin in coordination of angiogenic activities. Perhaps, the co-localization of endoglin and eNOS in caveolae regulates NO production. Our results indicate that endoglin haploinsufficiency is associated with decreased eNOS expression, NO production and attenuation of EC capillary morphogenesis, as previously demonstrated (Tang et al., 2010). How endoglin haploinsufficiency contributes to aberrant regulation of eNOS expression and/or NO production is subject of current investigation in our laboratory.
TGFβ is generally accepted as an anti-proliferative agent in EC. We showed that the ability of TGFβ to inhibit EC proliferation depends on endoglin expression. TGFβ affects angiogenesis through interactions with its receptors, including TGFβRII, TGFβRI (ALK1 or ALK5) and endoglin. We observed a significant decrease in expression of TGFβRII in Eng+/− EC. Endoglin haploinsufficiency also resulted in decreased ALK5 expression without affecting ALK1 levels (Fig. 8A). The basal Smad1/5 and Smad2/3 expression levels were modestly increased in Eng+/− EC compared with Eng+/+ EC, and their level was relatively constant in response to serum stimulation and/or TGFβ. Although the basal expression of Smad1/5 and Smad2/3 was not affected by endoglin haploinsufficiency, we observed a significant decrease in the level of pSmad1/5 and pSmad2/3 under normal culture conditions. Since TGFβ-bound TGFβRII is prerequisite for ALK1 and ALK5 activation, decreased TGFβRII may alter the activation of its down-stream effectors, Smad1/5 and Smad2/3. Moreover, unchanged ALK1 level and decreased Smad1/5 phosphorylation in Eng+/− retinal EC suggest that Smad1/5 activation is dependent on normal expression of endoglin and TGFβRII. These results are consistent with a recent report indicating that inhibition of ALK1 signaling during postnatal retinal vascularization in mice results in retinal hypervascularization and the appearance of arteriovenous malformations (Larrivée et al., 2012). In addition, they showed that the synergistic signaling from ALK1 and activated Notch is essential for repression of VEGF signaling and endothelial cell sprouting, and their disruption may contribute to HHT vascular lesions.
In summary, consistent with previous observations (Jerkic et al., 2006b; Lebrin et al., 2004), we showed that Eng+/− EC exhibit defects in their angiogenic properties including cell migration and capillary morphogenesis which were associated with attenuation of Smad1/5 activation, increased survival and proliferation. Thus, increased proliferation of Eng+/− EC cannot be simply explained by enhanced proliferative signaling through Smad1/5 activation and/or reduced anti-proliferative signaling through Smad2/3. We observed increased VEGF expression and sustained activation of MAPK pathways in Eng+/− EC supporting the increased proliferation and survival of these cells, and perhaps decreased expression of eNOS, PECAM-1, ICAM-2, VEGFRI, and VE-cadherin, all of which are important in angiogenesis. Cumulatively our results imply that endoglin-mediated Smad-independent signaling pathways play a key role in regulation of EC growth and survival with significant impact on angiogenesis and vascular development. However, signaling through ALK1/Smad1/5 and perhaps its synergism with activated Notch signaling, is essential for migration and capillary morphogenesis of EC (Larrivée et al., 2012; Lee et al., 2012).
Materials and Methods
Experimental animals
The mice used for these studies were maintained and treated according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Wisconsin. Endoglin heterozygous (Eng+/−) mice were provided by Dr Dean Lee (University of Utah, St Lake City, UT) and the mice were screened as previously described (Li et al., 1999). These mice were crossed to Immorto mice expressing a temperature-sensitive SV40 large T antigen (Charles River Laboratories, Wilmington, MA) to generate Eng+/− mice that carry the Immorto transgene. The Immorto/Eng+/− mice were identified by PCR analysis of DNA isolated from tail biopsies. The PCR primer sequences were as follows: Immorto-forward: 5′-CCTCTGAGCTATTCCAGAAGTAGTG-3′, Immorto-reverse: 5′-TTAGAGCTTTAAATCTCTGTAGGTAG-3′; Neo-forward: 5′-TGCTCTCCATCTGCACGAGACTAG-3′, Neo-reverse: 5′-GAGTTGCTTGTGGTGAACGCTCAG-3′; Endoglin-forward: 5′-CACAGCTGTAATCTCAGCACTTG-3′, and Endoglin-reverse: 5′-GATTGGATCCATTGTGGTAGCTG-3′.
Oxygen-induced ischemic retinopathy
The 7-day-old (P7) pups and their mother were placed in an airtight incubator and exposed to an atmosphere of 75±0.5% oxygen for 5 days. Incubator temperature was maintained at 23±2°C, and oxygen was continuously monitored with a PROOX model 110 oxygen controller (Reming Bioinstruments CO., Redfield, NY). Mice were brought to room air for 5 days, and then pups were sacrificed for retinal whole mount preparations and neovascularization analysis as described below.
Examination of retinal vasculature
Vessel obliteration and the retinal vascular pattern were analyzed using retinal wholemount stained with anti-collagen IV antibody as described previously (Wang et al., 2003). Mouse eyes were enucleated and briefly fixed in 4% paraformaldehyde (10 min on ice). The eyeballs were fixed in 70% ethanol for 24 h at −20°C. Retinas were dissected in PBS and then washed with PBS three times, 10 min each. Following incubation in blocking buffer (50% fetal calf serum, 20% normal goat serum in PBS) for 2 h, the retinas were incubated with rabbit anti-collagen IV antibody (Millipore, Billerica, MA; diluted 1∶500 in blocking buffer) at 4°C overnight. Retinas were then washed three times with PBS, 10 min each, incubated with secondary antibody Alexa Fluor 594 goat-anti-rabbit (Molecular probes, Eugene, OR; 1∶500 dilution prepared in blocking buffer) for 2 h at room temperature, washed four times with PBS, 30 min each, and mounted on a slide with PBS/glycerol (2vol/vol). The staining was viewed by fluorescence microscopy, and images were captured in digital format using a Zeiss microscope (Axio-Phot) and Axio-vision software (Carl Zeiss, Chester, VA).
Quantification of neovascular proliferative retinopathy
Quantification of vitreous neovascularization on P17 was performed as previously described (Wang et al., 2003). Briefly, mouse eyes were enucleated, fixed in formalin for 24 h and embedded in paraffin. Serial sections (6 µm thick), each separated by at least 40 µm, were taken from around the region of the optic nerve. The hematoxylin- and PAS-stained sections were examined in masked fashion for the presence of neovascular tufts projecting into the vitreous from the retina. The neovascular score was defined as the mean number of neovascular nuclei per section found in eight sections (four on each side of the optic nerve) per eye.
BrdU and collagen IV staining of wholemount retinas
The detection of cell proliferation on the retinal blood vessels was assessed by immunohistochemistry for 5-bromo-2′-deoxyuridine (BrdU) incorporation and type IV collagen (staining blood vessels). Mice were injected intraperitoneally with BrdU (Sigma, St. Louis, MO) 0.12 g/kg of body mass dissolved in water. One and a half hours later the animals were sacrificed, their eyes were removed, and fixed immediately in 4% paraformaldehyde for 3 min on ice. Eyes were then transferred to 70% ethanol (v/v) and stored at −20°C for at least 2 h. Retinas were dissected in PBS, washed for 30 min in PBS containing 1% Triton X-100 to permeabilize cell membranes and placed in 2 M HCl at 37°C for 1 h. Each retina was then washed in 0.1 M sodium borate for 30 min to neutralize the HCl. Retinas were then washed in PBS containing 1% Triton X-100 for 15 min and incubated with a monoclonal antibody to BrdU (Roche, Indianapolis, IN; diluted 1∶250 in PBS with 1% BSA, BSA) at 4°C overnight or room temperature for 2 h. Following incubation, retinas were washed for 10 min in PBS containing 1% Triton X-100 and incubated with anti-mouse CY2 antibody (Jackson's Laboratory) diluted 1∶500 in PBS containing 1% BSA for 2 h. Retinas were then washed with PBS and stained with anti-collagen IV antibody (Millipore) as described above. The collagen IV and secondary antibodies were used at 1∶1000 and 1∶500, respectively. After a final wash in PBS for 30 min, the retinas were mounted in PBS∶glycerol (2∶1, v/v). Retinas were viewed by fluorescence microscopy and images were captured in digital format using a Zeiss microscope. Results are expressed as the mean number of positive staining cells per whole retina from five eyes of five mice.
Immunohistochemical staining of the frozen sections
Mouse eyes were enucleated and embedded in optimal cutting temperature (OCT) compound at −80°C. Sections (9 µm) were cut on cryostat, placed on glass slides, and allowed to dry for 2 h. For fluorescence microscopy, sections were fixed in cold acetone (4°C) on ice for 10 min, followed by three washes with PBS, 5 min each. Sections were incubated in blocking solution (1% BSA, 0.2% skim milk, and 0.3% Triton X-100 in PBS) for 15 min at room temperature. Sections were then incubated with rat anti-mouse endoglin (BD Biosciences, 1∶500 dilution prepared in blocking solution) overnight at 4°C in a humid environment. After three washes in PBS, 5 min each, sections were incubated with secondary antibody Alexa Fluor 594-goat-anti-rat (Molecular probes, 1∶500 dilution prepared in blocking solution). Sections were washed three times in PBS, covered with PBS∶glycerol (2vol/1vol), and mounted with a coverslip. Retina sections were viewed by fluorescence microscopy, and images were captured in digital format using a Zeiss microscope.
Tissue preparation, isolation, and culture of retinal endothelial cells
Eyes from one litter (6–7 pups) of 4-week-old wild type and Eng+/− Immorto mice were enucleated and hemisected. Retinal EC were isolated by colleting retinas under dissecting microscope, rinsed with serum free Dulbecco's modified Eagle's medium (DMEM), minced into small pieces with a sterilized razor blade in a 60 mm dish and digested with 5 ml of collagenase type I (1 mg/ml in serum free DMEM, Worthington, Lakewood, NJ) at 37°C for 45 min. Cells were next rinsed with 5 ml of DMEM containing 10% fetal bovine serum (FBS), centrifuged for 5 minutes at 400 g, resuspended in 5 ml of DMEM with 10% FBS, and filtrated through a double layer of sterile 40 µm nylon mesh (Sefar America Inc., Fisher Scientific, Hanover Park, IL). Cells were centrifuged at 400 g for 10 min and rinsed twice with DMEM containing 10% FBS. The cell pellet was resuspended in 1 ml of DMEM with 10% FBS and incubated with 10 µl of sheep anti-rat magnetic beads (Invitrogen) precoated with anti-platelet endothelial cell adhesion molecule-1 (PECAM-1) antibody (MEC 13.3, BD Biosciences, Bedford, MA) at 4°C for 1 h on a rocker. Following incubation, the magnetic beads were collected using a magnetic tube holder and washed six times with 1 ml of DMEM containing 10% FBS. The bound cells were plated on fibronectin (BD Biosciences) coated 24 well plates (2 µg/ml in serum free DMEM) in 1 ml of endothelial cell growth medium in a single well, and incubated in tissue culture incubator at 33°C and 5% CO2. Endothelial cells were grown in DMEM containing 10% FBS, 2 mM L-glutamine, 2 mM sodium pyrovate, 20 mM HEPES, 1% non-essential amino acids, 100 µg streptomycin, 100 U/ml penicillin, 55 U/ml heparin (Sigma), endothelial growth supplement 100 µg/ml (Sigma), and murine recombinant interferon-γ 44 U/ml (R&D Systems, Minneapolis, MN). Cells were incubated at 33°C with 5% CO2 and progressively passaged to larger plates and maintained on 1% gelatin-coated 60 mm tissue culture dishes.
FACS analysis
Retinal EC on gelatin-coated 60 mm tissue culture dishes were washed once with PBS containing 0.04% EDTA, and incubated with 3 ml of cell dissociation solution (TBS containing 2 mM EDTA and 0.05% BSA). The cells were removed and washed once with DMEM containing 10% FBS. Cells were then fixed in 0.5 ml of 2% paraformaldehyde for 30 min on ice, and incubated with primary antibody. To detect vascular EC markers, anti-PECAM-1 (R&D Systems), anti-VE-cadherin (Santa Cruz, Santa Cruz, CA), anti-Endoglin, anti-ICAM-1, and anti-ICAM-2 (BD Bioscience), anti-VCAM-1 (Chemicon), anti-VEGF receptors (VEGFR1 and VEGFR2; R&D Systems) antibodies, or FITC conjugated B4-lectin (Sigma) were prepared in 0.5 ml of TBS with 1% BSA, 0.1% Triton X-100 and incubated for 30 min on ice. Following incubation with primary antibody, cells were washed twice with TBS, and then incubated with appropriate FITC-conjugated secondary antibody (Pierce, Rockford, IL; 1∶200 dilution in 0.5 ml of TBS with 1% BSA for 30 min on ice. The cells were washed twice with TBS and resuspended in 0.5 ml of PBS, and analyzed by FACScan caliber flow cytometer (Becton Dickinson).
Capillary morphogenesis assay
For capillary morphogenesis, 35 mm culture dishes were coated with 0.5 ml of Matrigel (10 mg/ml, BD Bioscience) and allowed to harden by incubation for 30 min at 37°C. Cells were removed by trypsinization, washed with DMEM containing 10% FBS, and resuspended at 1×105 cells/ml in EC growth medium. Cells (2×105) in 2 ml were gently added to Matrigel coated plates, incubated at 37°C for 18 h, and capillary morphogenesis was monitored and photographed in digital format. For quantitative assessment of the data, the mean numbers of branch points were determined by counting the number of branch points in five high-power fields (×100).
Ex vivo aortic sprouting assays
Aortas were isolated from CO2-euthanized 3-week-old Eng+/+ or Eng+/− mice. The isolated aortas were washed with serum-free DMEM three times on ice and adipose tissues around aortas were carefully removed with microdissecting forceps and iridectomy scissors. Next, the aortic rings (1–1.5 mm long, 5–6 per aorta) were prepared and embedded on 0.5 ml of Matrigel gel coated 35 mm culture dishes, and DMEM containing 1% FBS, 100 µg streptomycin, 100 U/ml penicillin was applied for 2 days at 37°C. Then medium was changed to EC growth medium, and EC sprouting was photographed after 4 days using a Nikon Microscope (×40; Nikon Eclipse; Chicago, IL). For quantitative assessment of sprouting, the areas of sprouting of tissue were assessed using ImageJ software (NIH; http://rsb.info.nih.gov/ij). Lysates were also prepared from aortas for western blot analysis as described below.
Scratch wound assays
EC (4×105) were plated in 60 mm tissue culture dishes and allowed to reach confluence (2–3 days). Monolayered confluent retinal EC were wounded using a 1 ml micropipette tip and floating cells were removed by rinsing with DMEM containing 10% FBS twice. To exclude the potential contribution of cell proliferation to wound closure, cells were fed with EC growth medium containing 1 µM 5-fluorouracil (Sigma) (Wu and Sheibani, 2003). The wound closure by cell migration was monitored every 24 h and photographed in digital format. For quantitative assessment, the distances migrated as percent of total distance were determined as previously described (Wu and Sheibani, 2003).
Transwell migration assays
The transwell migration assay was performed as previously described (Kondo et al., 2007). Briefly, 8 µm-pore Costar transwells (Corning, Corning, NY), were coated with fibronectin (FN; 10 µg/ml in PBS) at 4°C overnight. The FN-coated transwells were rinsed with PBS and blocked with 2% BSA in PBS for 1 h at room temperature. After trypsinization, retinal EC were resuspended in serum-free DMEM, counted, and 1×105 cells in 0.1 ml was added to top of the transwell membrane, and incubated for 3 h at 37°C. Following the incubation, the membranes were fixed with 2% paraformaldehyde (PFA) for 10 min at room temperature. Following hematoxylin/eosin staining, the stained membrane was mounted on a glass slide. The number of migrated cells through the membrane was analyzed by counting 10 high-power fields (×200).
Cell adhesion assays
Cell adhesion was determined using 96 well flat-bottom plates (Nunc Immunoplate Maxisorp, Fisher Scientific) coated with various concentrations of extracellular matrix proteins, including fibronectin, vitronectin, collagen I, and collagen IV (BD Biosciences), or BSA as control. The proteins were serially diluted in TBS containing 2 mM CaCl2 and 2 mM MgCl2 (50 µl/well; TBS with Ca/Mg) and allowed to coat overnight at 4°C. After washed with 200 µl of TBS with Ca/Mg, the plates were blocked with TBS with Ca/Mg containing 1% BSA (200 µl) at room temperature for 1 h. Cells were incubated with 3 ml of cell dissociation buffer (2 mM EDTA and 0.05% BSA in TBS), removed from the tissue culture plates, washed with TBS and resuspended in cell binding buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 4 mg/ml BSA. The plate were washed once with TBS with Ca/Mg and incubated with 50 µl of TBS with Ca/Mg and 50 µl of cell suspension (3×104 cells/well) at 37°C in a humidified tissue culture incubator for 2 h. Following incubation, the plates were gently washed with 200 µl of TBS with Ca/Mg until no cells were left in the wells coated with BSA. The adherent cells were lysed in100 µl of lysis buffer (50 mM sodium acetate pH 5.0, 1% Triton X-100, 6 mg/ml p-nitrophenyl phosphate) and incubated overnight at 4°C. The next day, the reaction was neutralized by adding 50 µl of 1 M NaOH and the absorbance at 405 nm was determined using a microplate reader (Thermomax, Molecular Devices, Sunnyvale, CA).
Western blot analysis
Cells (2×105) were plated in 60 mm tissue culture dishes and allowed to reach ∼90% confluence. After rinsed once with serum free DMEM, cells were incubated with EC growth medium without FBS for 2 days. Conditioned medium (CM) was collected and cell debris were removed by centrifugation. The cells were lysed in 0.1 ml of lysis buffer [50 mM HEPES pH 7.5, 100 mM NaCl, 0.1 mM EDTA, 1 mM CaCl2, 1 mM MgCl2, 1% Triton X-100, 1% NP-40 and protease inhibitor cocktail (Roche Biochemicals, Mannheim, Germany)]. The amount of CM for loading was adjusted according to total protein concentration of cell lysates. To detect phospho-eNOS and phospho-Smads, cells were serum starved for 24 h with no serum containing EC growth medium, incubated with EC growth medium containing serum for 30 min, rinsed the cells with PBS containing 1 mM Na3OV4 twice and lysed in 0.1 ml of 3 mM Na3OV4 and 5 mM NaF containing lysis buffer. Retinas from one mouse was lysed in the lysis buffer (2 eyes in 0.1 ml of lysis buffer: 10 mM HEPES, 142.5 mM KCl, 5 mM MgCl2, 1% NP-40, 1 mM Na3VO4, 1 mM NaF and protease inhibitor cocktail). The protein concentration was determined using by BCA protein assay (Bio-Rad, Hercules, CA) and samples were adjusted for protein content, mixed with appropriate volume of 6×SDS loading buffer and analyzed by SDS-PAGE (4–20% Tris-glycine gels, Invitrogen). The proteins were transferred to a nitrocellulose membrane and incubated in blocking buffer (5% skim milk in TBS containing 0.05% Tween 20) for at least 1 h and then incubated with appropriate primary antibody. Anti-tenascin-C, -Collagen IV (Chemicon), -fibronectin, -c-Src, -eNOS (Santa Cruz), -osteopontin, -TSP-2, -JNK and -phospho-JNK (R&D Systems), -TSP-1 (A6.1, Neo Markers, Fremont, CA), -phospho-eNOS, -Smad1, -Smad2/3, -phospho-Smad1/5, -phospho-Smad2/3, -HSP90, -AKT, -phospho-AKT, -ERK, -phospho-ERK, -p38, -phospho-p38, -phospho-Src (Cell Signaling), -iNOS (BD Science) and -β-actin (Sigma) antibodies were diluted to 1∶1000 ratio in blocking buffer for 2 h at room temperature. The membranes were then incubated with appropriate secondary HRP-conjugated antibody for 1.5 h, washed and developed using ECL (GE Healthcare, Buckinghamshire, UK). The β-actin was used to control for loading.
Indirect immunofluorescence staining
To visualize cell-cell junction formation, retinal EC (1×105) were plated on fibronectin-coated glass multi chamber slides and allowed to reach confluence (1–2 days). Cells were washed in cold PBS, fixed and permeabilized with methanol for 15 min on the ice and blocked with 1% BSA in TBS at 37°C for 20 min. Slides were washed with TBS and incubated with anti-VE-cadherin, β-catenin (BD Bioscience) and ZO-1 (Zymed, Carlsbad, CA) antibodies in TBS containing 1% BSA at 37°C for 40 min. For visualization of focal adhesions and actin stress fibers cells were stained with anti-vinculin antibody and FITC-conjugated phalloidin (Sigma) in TBS containing 1% BSA, respectively. Following washing with TBS, cells were incubated with appropriate Cy3-conjugated secondary antibody (1∶800 dilution in TBS containing 1% BSA) at 37°C for 40 min. Cells were washed five times with TBS, mounted and analyzed using a fluorescent microscope (Carl Zeiss) and images were captured in digital format.
Cell proliferation assays
The cell proliferation assay was performed by plating cells in multiple sets of 60 mm tissue culture dishes. The cell numbers were counted every other day for 2 weeks. 1×104 cells were plated in triplicate on gelatin-coated 60 mm tissue culture dishes and the cells were fed every other day. The cell number was determined by counting on days not fed.
The DNA synthesis rate was measured by Click-iTTM EdU Alexa Fluor 488 cell proliferation assay (Invitrogen) according to manufacturer's instruction. The assay measures the amount of incorporated EdU (5-ethynyl-2′-deoxyuridine), a nucleoside analogue of thymidine, during cell proliferation. For assay, Eng+/+ and Eng+/− retinal EC (5×105) were plated in 60 mm tissue culture dishes and incubated with 30 µM EdU in culture medium for 2 h at 33°C. The DNA synthesis levels were determined by determining the percent of EdU positive cells using FACScan caliber flow cytometer (Becton Dickinson).
Apoptosis and cell viability assays
Apoptosis level was determined by measuring active caspase 3/7. Caspase activation was analyzed using Caspase-Glo 3/7 Assay Systems (Promega, Madison, WI) according to the manufacturer's instruction. The assay provides caspase-3/7 DEVD-aminoluciferin substrate and the caspase 3/7 activity was determined by measuring luminescence. Eng+/+ and Eng+/− retinal EC (5×103) were plated on white 96 well plates (Fisher) and incubated with 1.75 mM hydrogen peroxide (H2O2, Fisher Scientific, Fair Lawn, NJ) or 2 µM staurosporine (Invitrogen) in culture medium as apoptotic inducer for 8 h at 33°C. The caspase 3/7 activity was detected by a luminescence microplate reader (Victa2 1420 Multilabel Counter, PerkinElmer, Waltham, MA).
Cellular viability of EC was determined by MTS viability assay (Promega). After plated on 96 well plate, retinal EC (4×103) were incubated with 1.75 mM H2O2 in culture medium for 2 days at 33°C and incubated with MTS solution for 3 h. Cellular viability was determined through measuring absorbance (490 nm) using a microplate reader (Thermomax, Molecular Devices, Sunnyvale, CA).
VEGF measurements
The amount of secreted vascular endothelial growth factor (VEGF) from Eng+/+ and Eng+/− EC was determined using mouse VEGF Immuno assay kit (R&D Systems). Cells (2×105) were plated in 60 mm culture dishes and allowed to reach ∼90% confluence. The EC were then rinsed once with serum free DMEM and incubated in EC growth medium without serum for 2 days. The CM was collected, centrifuged to remove cell debris and the level of secreted VEGF was determined by ELISA. For determination of retinal VEGF levels, retinas from one mouse were placed in 50 µl of PBS, sonicated, centrifuged and total protein content was determined by using BCA protein assay. Then lysates were adjusted for total protein content and the VEGF levels were determined by ELISA. To determine the amount of VEGF, a standard VEGF curve was prepared along with the samples and used to determine VEGF concentration.
Intracellular NO level
The intracellular nitric oxide (NO) production in Eng+/+ and Eng+/− retinal EC was determined using DAF-FM diacetate (Invitrogen). DAF-FM diacetate is a cell-permeable molecule and non-fluorescent until reacts with NO. By the interaction with NO, DAF-FM diacetate changes its structure to cell membrane-impermeable and yields fluorescent. Retinal EC (5×103 cells) were plated on gelatin-coated 96 well black/clear bottom plates (BD Falcon) and incubated for 1 day. Cells were then incubated with 30 µM DAF-FM (0.1 ml/well) in fresh EC culture medium for 40 min. After the incubation, the medium was changed with fresh EC growth medium (0.1 ml/well), incubated for an additional 30 min and cells were washed with TBS (0.1 ml/well) twice. DAF-FM fluorescence was detected (excitation 485 nm/emission 535 nm) using a fluorescent microplate reader (Victa2 1420 Multilabel Counter, PerkinElmer). Assays were conducted three times in triplicate and normalized to cell numbers.
Statistical analysis
Statistical differences between control and treated samples were evaluated with student's unpaired t-test (2-tailed) or two-way ANOVA with Bonferroni correction for multiple comparisons when appropriate. Means±s.e.m. are shown. P values ≤0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank Dr Dean Li for proving the Eng+/− mice.
Footnotes
Author contributions
S.P. performed the majority of the experiments, data collection and analysis, and wrote the manuscript; T.A.D., W.L. and S.W. performed the in vivo retinal vasculature development, OIR, and retinal staining; C.M.S. generated the mice and edited the manuscript; N.S. conceived the study, and contributed to the interpretation, manuscript writing and editing.
Funding
This work was supported by the National Institutes of Health [grant numbers EY016995, EY021357 (to N.S.) and P30-EY016665]; a University of Wisconsin Paul P. Carbone Cancer Center Support Grant from the National Institutes of Health [grant number P30 CA014520]; and an unrestricted departmental award from Research to Prevent Blindness. N.S. is a recipient of a Research Award from the American Diabetes Association [grant number 1-10-BS-160] and the Retina Research Foundation. C.M.S. is supported by a grant from American Heart Association [grant number 0950057G]. S.P. was supported by a Predoctoral Award from AstraZeneca. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.117275/-/DC1
References
- Antonelli-Orlidge A., Saunders K. B., Smith S. R., D'Amore P. A. (1989). An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc. Natl. Acad. Sci. USA 86, 4544–4548 10.1073/pnas.86.12.4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur H. M., Ure J., Smith A. J., Renforth G., Wilson D. I., Torsney E., Charlton R., Parums D. V., Jowett T., Marchuk D. A. et al. (2000). Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 217, 42–53 10.1006/dbio.1999.9534 [DOI] [PubMed] [Google Scholar]
- Bernabeu C., Conley B. A., Vary C. P. (2007). Novel biochemical pathways of endoglin in vascular cell physiology. J. Cell. Biochem. 102, 1375–1388 10.1002/jcb.21594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau A., Dumont D. J., Letarte M. (1999). A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 104, 1343–1351 10.1172/JCI8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau A., Faughnan M. E., Letarte M. (2000). Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc. Med. 10, 279–285 [DOI] [PubMed] [Google Scholar]
- Carta L., Smaldone S., Zilberberg L., Loch D., Dietz H. C., Rifkin D. B., Ramirez F. (2009). p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J. Biol. Chem. 284, 5630–5636 10.1074/jbc.M806962200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E., Tournier-Lasserve E., Weinstein B. M. (2009). The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev. Cell 16, 209–221 10.1016/j.devcel.2009.01.004 [DOI] [PubMed] [Google Scholar]
- Derynck R., Zhang Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584 10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- DiMaio T. A., Wang S., Huang Q., Scheef E. A., Sorenson C. M., Sheibani N. (2008). Attenuation of retinal vascular development and neovascularization in PECAM-1-deficient mice. Dev. Biol. 315, 72–88 10.1016/j.ydbio.2007.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff S. E., Li C., Garland J. M., Kumar S. (2003). CD105 is important for angiogenesis: evidence and potential applications. FASEB J. 17, 984–992 10.1096/fj.02-0634rev [DOI] [PubMed] [Google Scholar]
- Düwel A., Eleno N., Jerkic M., Arevalo M., Bolaños J. P., Bernabeu C., López-Novoa J. M. (2007). Reduced tumor growth and angiogenesis in endoglin-haploinsufficient mice. Tumour Biol. 28, 1–8 10.1159/000097040 [DOI] [PubMed] [Google Scholar]
- Gerber H. P., McMurtrey A., Kowalski J., Yan M., Keyt B. A., Dixit V., Ferrara N. (1998). Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 273, 30336–30343 10.1074/jbc.273.46.30336 [DOI] [PubMed] [Google Scholar]
- Gougos A., Letarte M. (1990). Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 265, 8361–8364 [PubMed] [Google Scholar]
- Goumans M. J., Valdimarsdottir G., Itoh S., Lebrin F., Larsson J., Mummery C., Karlsson S., ten Dijke P. (2003). Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 12, 817–828 10.1016/S1097-2765(03)00386-1 [DOI] [PubMed] [Google Scholar]
- Hirschi K. K., Rohovsky S. A., D'Amore P. A. (1998). PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol. 141, 805–814 10.1083/jcb.141.3.805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerkic M., Rivas-Elena J. V., Santibanez J. F., Prieto M., Rodríguez-Barbero A., Perez-Barriocanal F., Pericacho M., Arévalo M., Vary C. P., Letarte M. et al. (2006a). Endoglin regulates cyclooxygenase-2 expression and activity. Circ. Res. 99, 248–256 10.1161/01.RES.0000236755.98627.69 [DOI] [PubMed] [Google Scholar]
- Jerkic M., Rodríguez-Barbero A., Prieto M., Toporsian M., Pericacho M., Rivas-Elena J. V., Obreo J., Wang A., Pérez-Barriocanal F., Arévalo M. et al. (2006b). Reduced angiogenic responses in adult Endoglin heterozygous mice. Cardiovasc. Res. 69, 845–854 10.1016/j.cardiores.2005.11.020 [DOI] [PubMed] [Google Scholar]
- Kondo S., Scheef E. A., Sheibani N., Sorenson C. M. (2007). PECAM-1 isoform-specific regulation of kidney endothelial cell migration and capillary morphogenesis. Am. J. Physiol. 292, C2070–C2083 10.1152/ajpcell.00489.2006 [DOI] [PubMed] [Google Scholar]
- Krupinski J., Kumar P., Kumar S., Kaluza J. (1996). Increased expression of TGF-β 1 in brain tissue after ischemic stroke in humans. Stroke 27, 852–857 10.1161/01.STR.27.5.852 [DOI] [PubMed] [Google Scholar]
- Larrivée B., Prahst C., Gordon E., del Toro R., Mathivet T., Duarte A., Simons M., Eichmann A. (2012). ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 22, 489–500 10.1016/j.devcel.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson J., Goumans M-J., Sjöstrand L. J., van Rooijen M. A., Ward D., Levéen P., Xu X., ten Dijke P., Mummery C. L., Karlsson S. (2001). Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 20, 1663–1673 10.1093/emboj/20.7.1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F., Goumans M. J., Jonker L., Carvalho R. L., Valdimarsdottir G., Thorikay M., Mummery C., Arthur H. M., ten Dijke P. (2004). Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 23, 4018–4028 10.1038/sj.emboj.7600386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F., Deckers M., Bertolino P., Ten Dijke P. (2005). TGF-beta receptor function in the endothelium. Cardiovasc. Res. 65, 599–608 10.1016/j.cardiores.2004.10.036 [DOI] [PubMed] [Google Scholar]
- Lee N. Y., Ray B., How T., Blobe G. C. (2008). Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J. Biol. Chem. 283, 32527–32533 10.1074/jbc.M803059200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N. Y., Golzio C., Gatza C. E., Sharma A., Katsanis N., Blobe G. C. (2012). Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Mol. Biol. Cell 23, 2412–2423 10.1091/mbc.E11-12-0993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leight J. L., Wozniak M. A., Chen S., Lynch M. L., Chen C. S. (2012). Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 23, 781–791 10.1091/mbc.E11-06-0537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D. W., Cachianes G., Kuang W. J., Goeddel D. V., Ferrara N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309 10.1126/science.2479986 [DOI] [PubMed] [Google Scholar]
- Li D. Y., Sorensen L. K., Brooke B. S., Urness L. D., Davis E. C., Taylor D. G., Boak B. B., Wendel D. P. (1999). Defective angiogenesis in mice lacking endoglin. Science 284, 1534–1537 10.1126/science.284.5419.1534 [DOI] [PubMed] [Google Scholar]
- Li C., Hampson I. N., Hampson L., Kumar P., Bernabeu C., Kumar S. LI(2000). CD105 antagonizes the inhibitory signaling of transforming growth factor β1 on human vascular endothelial cells. FASEB J. 14, 55–64 [DOI] [PubMed] [Google Scholar]
- Lu Q. (2008). Transforming growth factor-β1 protects against pulmonary artery endothelial cell apoptosis via ALK5. Am. J. Physiol. 295, L123–L133 10.1152/ajplung.00402.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund S. A., Giachelli C. M., Scatena M. (2009). The role of osteopontin in inflammatory processes. J. Cell Commun. Signal 3, 311–322 10.1007/s12079-009-0068-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutty G. A., Merges C., Threlkeld A. B., Crone S., McLeod D. S. (1993). Heterogeneity in localization of isoforms of TGF-beta in human retina, vitreous, and choroid. Invest. Ophthalmol. Vis. Sci. 34, 477–487 [PubMed] [Google Scholar]
- Mahmoud M., Allinson K. R., Zhai Z., Oakenfull R., Ghandi P., Adams R. H., Fruttiger M., Arthur H. M. (2010). Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 106, 1425–1433 10.1161/CIRCRESAHA.109.211037 [DOI] [PubMed] [Google Scholar]
- Miller D. W., Graulich W., Karges B., Stahl S., Ernst M., Ramaswamy A., Sedlacek H. H., Müller R., Adamkiewicz J. (1999). Elevated expression of endoglin, a component of the TGF-beta-receptor complex, correlates with proliferation of tumor endothelial cells. Int. J. Cancer 81, 568–572 [DOI] [PubMed] [Google Scholar]
- Mu Y., Gudey S. K., Landström M. (2012). Non-Smad signaling pathways. Cell Tissue Res. 347, 11–20 10.1007/s00441-011-1201-y [DOI] [PubMed] [Google Scholar]
- Neubauer K., Krüger M., Quondamatteo F., Knittel T., Saile B., Ramadori G. (1999). Transforming growth factor-beta1 stimulates the synthesis of basement membrane proteins laminin, collagen type IV and entactin in rat liver sinusoidal endothelial cells. J. Hepatol. 31, 692–702 10.1016/S0168-8278(99)80350-X [DOI] [PubMed] [Google Scholar]
- Neufeld G., Cohen T., Gengrinovitch S., Poltorak Z. (1999). Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 13, 9–22 [PubMed] [Google Scholar]
- Oh S. P., Seki T., Goss K. A., Imamura T., Yi Y., Donahoe P. K., Li L., Miyazono K., ten Dijke P., Kim S. et al. (2000). Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 97, 2626–2631 10.1073/pnas.97.6.2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M., Oshima H., Taketo M. M. (1996). TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 179, 297–302 10.1006/dbio.1996.0259 [DOI] [PubMed] [Google Scholar]
- Park S., DiMaio T. A., Scheef E. A., Sorenson C. M., Sheibani N. (2010). PECAM-1 regulates proangiogenic properties of endothelial cells through modulation of cell-cell and cell-matrix interactions. Am. J. Physiol. 299, C1468–C1484 10.1152/ajpcell.00246.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece-Barbara N., Vera S., Kathirkamathamby K., Liebner S., Di Guglielmo G. M., Dejana E., Wrana J. L., Letarte M. (2005). Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J. Biol. Chem. 280, 27800–27808 10.1074/jbc.M503471200 [DOI] [PubMed] [Google Scholar]
- Pepper M. S. (1997). Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 8, 21–43 10.1016/S1359-6101(96)00048-2 [DOI] [PubMed] [Google Scholar]
- Pollman M. J., Naumovski L., Gibbons G. H. (1999a). Endothelial cell apoptosis in capillary network remodeling. J. Cell. Physiol. 178, 359–370 [DOI] [PubMed] [Google Scholar]
- Pollman M. J., Naumovski L., Gibbons G. H. (1999b). Vascular cell apoptosis: cell type-specific modulation by transforming growth factor-beta1 in endothelial cells versus smooth muscle cells. Circulation 99, 2019–2026 10.1161/01.CIR.99.15.2019 [DOI] [PubMed] [Google Scholar]
- Ray B. N., Lee N. Y., How T., Blobe G. C. (2010). ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 31, 435–441 10.1093/carcin/bgp327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudini N., Felici A., Giampietro C., Lampugnani M., Corada M., Swirsding K., Garrè M., Liebner S., Letarte M., ten Dijke P. et al. (2008). VE-cadherin is a critical endothelial regulator of TGF-beta signalling. EMBO J. 27, 993–1004 10.1038/emboj.2008.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabet M. D., Gordon S. R. (1989). Ultrastructural immunocytochemical localization of fibronectin deposition during corneal endothelial wound repair. Evidence for cytoskeletal involvement. Biol. Cell 65, 171–179 [PubMed] [Google Scholar]
- Santibanez J. F., Letamendia A., Perez-Barriocanal F., Silvestri C., Saura M., Vary C. P., Lopez-Novoa J. M., Attisano L., Bernabeu C. (2007). Endoglin increases eNOS expression by modulating Smad2 protein levels and Smad2-dependent TGF-beta signaling. J. Cell. Physiol. 210, 456–468 10.1002/jcp.20878 [DOI] [PubMed] [Google Scholar]
- Seki T., Yun J., Oh S. P. (2003). Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ. Res. 93, 682–689 10.1161/01.RES.0000095246.40391.3B [DOI] [PubMed] [Google Scholar]
- Sheibani N., Sorenson C. M., Frazier W. A. (2000). Differential modulation of cadherin-mediated cell-cell adhesion by platelet endothelial cell adhesion molecule-1 isoforms through activation of extracellular regulated kinases. Mol. Biol. Cell 11, 2793–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. E., Wesolowski E., McLellan A., Kostyk S. K., D'Amato R., Sullivan R., D'Amore P. A. (1994). Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 [PubMed] [Google Scholar]
- Su X., Sorenson C. M., Sheibani N. (2003). Isolation and characterization of murine retinal endothelial cells. Mol. Vis. 9, 171–178 [PubMed] [Google Scholar]
- Tang Y., Scheef E. A., Gurel Z., Sorenson C. M., Jefcoate C. R., Sheibani N. (2010). CYP1B1 and endothelial nitric oxide synthase combine to sustain proangiogenic functions of endothelial cells under hyperoxic stress. Am. J. Physiol. 298, C665–C678 10.1152/ajpcell.00153.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P., Goumans M. J., Pardali E. (2008). Endoglin in angiogenesis and vascular diseases. Angiogenesis 11, 79–89 10.1007/s10456-008-9101-9 [DOI] [PubMed] [Google Scholar]
- Toporsian M., Gros R., Kabir M. G., Vera S., Govindaraju K., Eidelman D. H., Husain M., Letarte M. (2005). A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ. Res. 96, 684–692 10.1161/01.RES.0000159936.38601.22 [DOI] [PubMed] [Google Scholar]
- Tucker R. P. (1991). The distribution of J1/tenascin and its transcript during the development of the avian cornea. Differentiation 48, 59–66 10.1111/j.1432-0436.1991.tb00243.x [DOI] [PubMed] [Google Scholar]
- Wang S., Wu Z., Sorenson C. M., Lawler J., Sheibani N. (2003). Thrombospondin-1-deficient mice exhibit increased vascular density during retinal vascular development and are less sensitive to hyperoxia-mediated vessel obliteration. Dev. Dyn. 228, 630–642 10.1002/dvdy.10412 [DOI] [PubMed] [Google Scholar]
- Wu J., Sheibani N. (2003). Modulation of VE-cadherin and PECAM-1 mediated cell-cell adhesions by mitogen-activated protein kinases. J. Cell. Biochem. 90, 121–137 10.1002/jcb.10600 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.