

Summary
Phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α) by the kinase GCN2 attenuates protein synthesis during amino acid starvation in yeast, whereas in mammals a family of related eIF2α kinases regulate translation in response to a variety of stresses. Unlike single-celled eukaryotes, mammals also possess two specific eIF2α phosphatases, PPP1R15a and PPP1R15b, whose combined deletion leads to a poorly understood early embryonic lethality. We report the characterisation of the first non-mammalian eIF2α phosphatase and the use of Drosophila to dissect its role during development. The Drosophila protein demonstrates features of both mammalian proteins, including limited sequence homology and association with the endoplasmic reticulum. Of note, although this protein is not transcriptionally regulated, its expression is controlled by the presence of upstream open reading frames in its 5′UTR, enabling induction in response to eIF2α phosphorylation. Moreover, we show that its expression is necessary for embryonic and larval development and that this is to oppose the inhibitory effects of GCN2 on anabolic growth.
Key words: CREP, GADD34, GCN2, Development, eIF2α
Introduction
Early in eukaryotic evolution, cells gained the capacity to regulate protein synthesis in response to stress through phosphorylation of eukaryotic initiation factor 2 on its alpha subunit (eIF2α) (Wek et al., 2006; Williams et al., 1989). The eIF2 complex delivers a methionyl-tRNA necessary for the initiation of protein synthesis by ribosomes. This process requires the hydrolysis of GTP and so during its catalytic cycle eIF2 interacts with a GTP exchange factor, eIF2B. Phosphorylated eIF2α binds with high affinity to eIF2B inhibiting further nucleotide exchange and thus attenuating the rate of global protein synthesis; however, a subset of mRNAs is translated more efficiently under these conditions owing to the presence of small open reading frames within their 5′ untranslated regions, so-called uORFs (Abastado et al., 1991; Dever et al., 1992). In the case of the mammalian stress-responsive transcription factor ATF4, the final uORF overlaps out of frame with the true coding sequence (Lu et al., 2004a; Vattem and Wek, 2004). This arrangement acts to prevent eIF2-GTP-Met ternary complex-bound ribosomes from arriving at the true start codon in the correct frame to allow translation of the protein. In contrast, when ternary complex is limiting a proportion of scanning ribosomes skip the inhibitory uORFs allowing more efficient translation of the protein mRNA. In yeast, this process governs the general control response (GCN) to amino acid starvation in which the kinase GCN2 phosphorylates eIF2α (Sui2p) both reducing the demand for scarce amino acids while inducing a transcriptional programme through enhanced translation of the transcription factor GCN4 (Abastado et al., 1991; Dever et al., 1992).
Whereas unicellular eukaryotes possess only a single eIF2α kinase, GCN2 has given rise in metazoans to a family of kinases that respond to a variety of stresses. In mammals, GCN2 retains its role as the master regulator of the response to amino acid insufficiency (Wek et al., 2006), but is joined by PERK, responding to endoplasmic reticulum (ER) stress (Harding et al., 1999; Shi et al., 1998); HRI, responding to deficiency of haem (Chen et al., 1991); and PKR, which responds to viral double-stranded RNA (Berry et al., 1985). This family evolved incrementally, since insects and worms have only GCN2 and PERK while vertebrates possess all four family members. In mammals, the activity of these kinases is antagonised by two highly selective protein phosphatases comprising the PP1 catalytic subunit complexed either with the constitutively expressed PPP1R15b subunit (previously named CReP) (Jousse et al., 2003) or the stress-inducible isoform PPP1R15a (GADD34) (Novoa et al., 2001). Curiously, no orthologues of the PPP1R15 proteins have been identified either in yeast or in nematodes, which have been used extensively to study the consequences of eIF2α phosphorylation. In mice, deletion of the PPP1R15a isoform is well tolerated (Marciniak et al., 2004). These animals are outwardly healthy and fertile. In contrast, although PPP1R15b deficient mice survive gestation, they exhibit severe growth retardation and impaired erythropoiesis (Harding et al., 2009). However, the double knockout is embryonic lethal at a preimplantation stage prior to formation of the blastocyst cavity and hatching from the zona pellucida, demonstrating the essential nature of eIF2α dephosphorylation during embryogenesis but preventing its detailed study in mammals (Harding et al., 2009).
We set out to characterise a putative PPP1R15 orthologue in Drosophila melanogaster in order to shed light on the evolutionary relevance of this family of phosphatases. Herein, we show that Drosophila PPP1R15 (dPPP1R15; previously known as CG3825) is a functional eIF2α phosphatase that shares 59% homology over a 61-residue stretch with the mammalian PPP1R15 proteins. Although dPPP1R15 is expressed constitutively with no evidence of transcriptional regulation in response to endoplasmic reticulum stress, we show that translation of its mRNA is tightly regulated by the presence of uORFs within the 5′ untranslated region. Finally, we demonstrate that regulated dephosphorylation of eIF2α by dPPP1R15 is essential for anabolic larval growth and that it principally antagonises GCN2 during development. This represents the first description of a non-mammalian eIF2α phosphatase and suggests its evolution occurred to enable anabolic growth in metazoans.
Results
dPPP1R15 is widely expressed during embryogenesis
We have previously described the generation of a Drosophila model system that allows us to study the consequences of eIF2α phosphorylation in vivo (Malzer et al., 2010). The kinase PERK, which phosphorylates eIF2α in response to ER stress, is highly conserved in all metozoans. When we expressed Drosophila PERK in the eye imaginal disc, we observed a profound impairment of eye development due to delayed cell cycle progression and enhanced apoptotic cell death. Rescue of this phenotype by ectopic expression of the murine ER stress-inducible eIF2α phosphatase PPP1R15a confirmed these effects to be mediated by selective phosphorylation of eIF2α. Sequence comparisons revealed a single putative PPP1R15 homologue in Drosophila, originally labeled CG3825, with 59% homology over a 61 amino acid region (Fig. 1A). Previously, this gene product has been annotated as dGadd34 on the basis of this homology, although its function had not been studied (Carra et al., 2010). We therefore tested its ability to antagonise PERK in our in vivo model. Overexpression of dPPP1R15 alone had no detectable effect on eye development but efficiently rescued the developmental defect induced by ectopic expression of PERK (Fig. 1B). This closely resembled the effect of murine PPP1R15a supporting a role for dPPP1R15 in eIF2α dephosphorylation in Drosophila.
Fig. 1.

dPPP1R15 shares sequence and functional homology with other PPP1R15 family members. (A) Extract of protein alignment of C-termini of PPP1R15 proteins and dPPP1R15 shows high similarity. Conservations are marked with boxes and the conserved PP1α binding KVxF-motif is marked with an asterisk. (B) Representative light and electron micrographs of flies expressing GMR-Gal4>UAS-dPERK alone or with GMR-Gal4>UAS-mGADD34 or GMR-Gal4>UAS-dPPP1R15. (C) dPPP1R15 mRNA was detected by in situ hybridisation of OregonR whole embryos using DIG-labelled dPPP1R15 anti-sense probe. Embryo orientation: anterior to the left, dorsal to the top. Representative images are shown for stages 2, 5, 9, 13 and 17; anti-sense (upper panels) and sense probes (lower panels). Images have been cropped for clarity.
Information from the public modEncode database indicates that dPPP1R15 is abundant during all stages of Drosophila development with peak expression during embryogenesis and pupal stages (Graveley et al., 2011), while the FlyAtlas expression database indicates that dPPP1R15 transcript is abundant in larvae and adults especially in larval secretory tissues and adult neuronal tissue (Chintapalli et al., 2007). To address the lack of detailed expression data during embryogenesis we performed in situ hybridisation for dPPP1R15 transcripts. This revealed dPPP1R15 mRNA to be a widely expressed throughout embryogenesis (Fig. 1C). The high abundance of dPPP1R15 mRNA in the central yolk sac of stage 5 embryos before cellularisation at the initiation of zygotic transcription suggested a significant maternal contribution (Fig. 1C). During gastrulation, uniform dPPP1R15 expression was replaced by more tissue-specific expression in the invaginating endoderm and the surrounding ectoderm. High signal was also seen in the epidermis, the brain and central nervous system, the Malpighian tubules and the primordia of the mid- and hindgut (Fig. 1C). At later stages of embryogenesis, expression of dPPP1R15 mRNA persisted with high signal in the CNS.
dPPP1R15 and PP1 form an ER-associated eIF2α phosphatase
In mammalian cells PPP1R15 subunits form stable complexes with protein phosphatase 1 (PP1) that direct the catalytic activity towards eIF2α (Brush et al., 2003; Novoa et al., 2001). Both PPP1R15a and PPP1R15b have been shown to associate with endomembranes, at least partially directed by an amphipathic helix within their N-termini (Brush et al., 2003; Kloft et al., 2012; Zhou et al., 2011). This association with the ER is more pronounced for PPP1R15b than for PPP1R15a. The function of this membrane association is unclear, but has been suggested to encourage eIF2α dephosphorylation at the ER or to play a role in regulating the degradation of PPP1R15 proteins (Brush et al., 2003; Zhou et al., 2011). We set out to determine the subcellular localisation of dPPP1R15 by generating fusion proteins tagged with GFP. In the case of PPP1R15a, the addition of tags to its N-terminus significantly stabilises the protein, although both N- and C-terminally tagged mammalian PPP1R15a can associate with the endoplasmic reticulum (Brush and Shenolikar, 2008; Zhou et al., 2011). We observed that both N- and C-terminally tagged dPPP1R15 expressed well in mammalian cells, although the C-terminally tagged protein consistently showed multiple degradation products (Fig. 2A). Similar instability was seen for C-terminally His6-tagged dPPP1R15 expressed in Drosophila Schneider S2 cells, suggesting that this was not the result of the heterologous expression system (data not shown). When transgenic protein was expressed in mammalian HEK293T cells and purified using GFP-TRAP® beads, both GFP-dPPP1R15 and dPPP1R15-GFP co-purified with endogenous mammalian PP1 catalytic subunit (Fig. 2A). To test the specificity of the interaction of dPPP1R15 and PP1, we mutated valine 249 to glutamic acid, since equivalent mutations of the KVxF domains of other PP1 regulatory subunits are know to abolish interaction with the catalytic PP1 subunit (Brush et al., 2003). Affinity purification confirmed that the V249E mutant failed to bind PP1 (Fig. 2B). Interestingly, only the C-terminally tagged protein demonstrated association with endomembranes (Fig. 2C). Co-localisation with the marker protein mCherry-ER confirmed these membranes to be the ER (Fig. 2D). The failure of N-terminally tagged dPPP1R15 to bind ER membranes is likely to reflect steric hindrance from the tag with the N-terminal amphipathic helix. Comparison of mammalian and Drosophila PPP1R15 sequences revealed a much shorter distance between the N-terminus and the potential amphipathic helix in the insect protein: only 4 residues in dPPP1R15 compared with 26 residues in PPP1R15a and 75 residues in PPP1R15b.
Fig. 2.
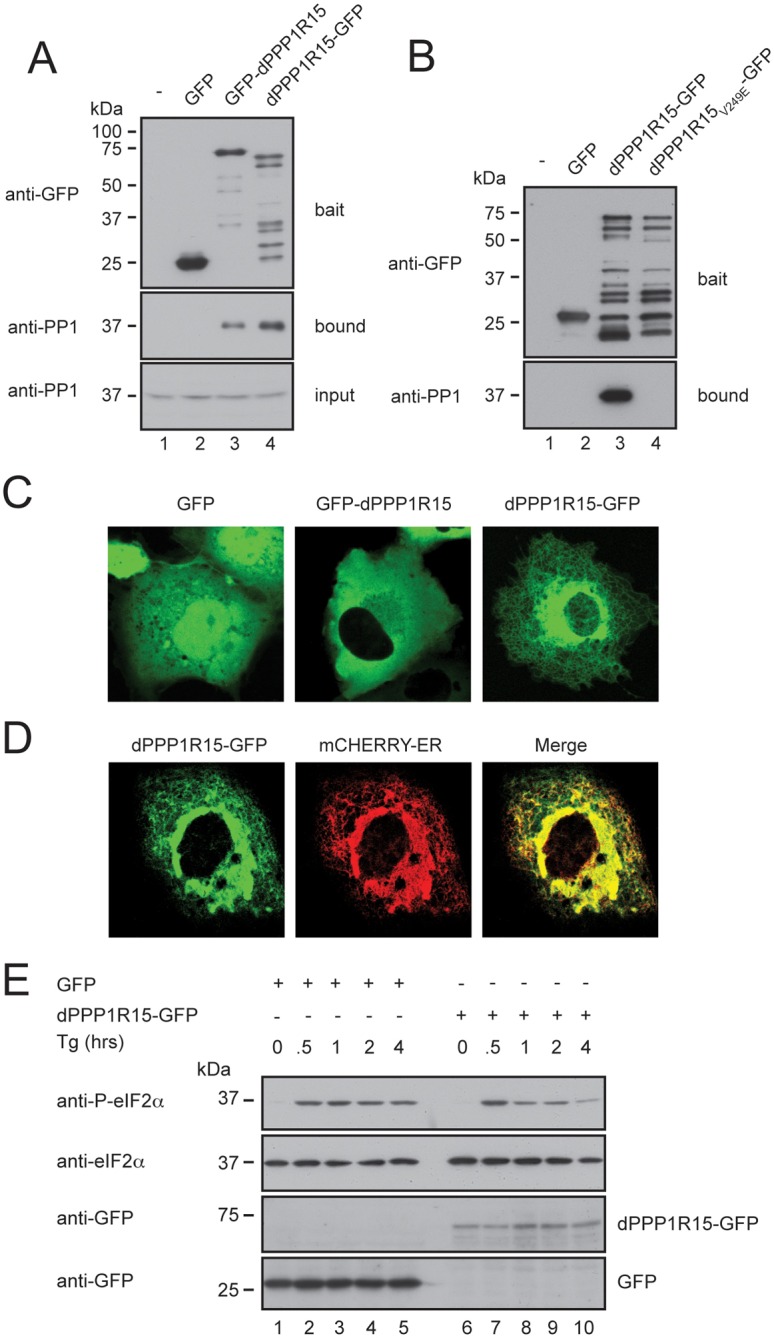
dPPP1R15 and PP1 form an ER-associated eIF2α phosphatase when expressed in mammalian cells. (A) Immunoblots of GFP-Trap affinity purification of lysates from HEK 293T cells expressing no transgene (lane 1), GFP alone (lane 2), GFP-dPPP1R15 (lane 3) or dPPP1R15-GFP (lane 4). Lysates were subjected to GFP-Trap affinity-purification and analysed by 10% (w/v) SDS-PAGE and probed for GFP and PP1. Input samples were probed for PP1. (B) Immunoblots of GFP-Trap affinity purification of lysates from HEK293T cells expressing no transgene (lane 1), GFP alone (lane 2), dPPP1R15-GFP (lane 3) or dPPP1R15V249E-GFP (lane 4). Lysates were subjected to GFP-Trap affinity-purification and analysed by 10% (w/v) SDS-PAGE and probed for GFP and PP1. Note large number of degradation products with C-terminally tagged dPPP1R15, as also seen in A, lane 4. (C) Representative fluorescence confocal images of COS-7 cells expressing GFP, GFP-dPPP1R15 or dPPP1R15-GFP. (D) Representative fluorescence confocal image of a COS-7 cell expressing dPPP1R15-GFP and the ER marker ER-mCherry. (E) Immunoblot of lysates from HEK 293T cells expressing either dPPP1R15-GFP (lanes 6–10) or GFP alone (lanes 1–5), followed by treatment with 500 nM thapsigargin for the indicated times. Lysates were analysed by 10% (w/v) SDS-PAGE followed by immunoblot for phosphorylation of eIF2α (P-eIF2α), total eIF2α and GFP.
Next, we assessed the effect of dPPP1R15-GFP expression on eIF2α phosphorylation. Treatment with the SERCA calcium pump inhibitor thapsigargin causes rapid depletion of ER calcium stores and is associated with the activation of ER stress. This triggers the phosphorylation of eIF2α by PERK followed by gradual dephosphorylation by PPP1R15a and PPP1R15b (Jousse et al., 2003; Novoa et al., 2001). In cells expressing dPPP1R15-GFP, we observed hastening of eIF2α dephosphorylation compared with cells expressing GFP alone (Fig. 2E). Taken together, these data indicate that dPPP1R15 is a regulatory subunit of PP1 able to direct the dephosphorylation of eIF2α at the ER membrane.
dPPP1R15 is functionally homologous to mammalian PPP1R15b
In mammals eIF2α dephosphorylation is achieved by either of two PP1 regulatory subunits: PPP1R15a (Novoa et al., 2001) and PPP1R15b (Jousse et al., 2003). Deletion of the inducible PPP1R15a isoform is well tolerated in mice (Marciniak et al., 2004). Indeed, PPP1R15a deficient mice are fertile and demonstrate increased tolerance to ER stress-induced tissue damage. In contrast, deletion of the constitutive PPP1R15b isoform is associated with growth retardation and defects of erythropoiesis (Harding et al., 2009). Although dPPP1R15 has previously been annotated dGadd34 on the basis of sequence homology, in fact it shares equal homology with both PPP1R15a (also known as GADD34) and PPP1R15b (also known as CReP) (Fig. 1A). To test if ER stress could induce dPPP1R15 transcriptionally we next treated Schneider S2 insect cells with 2 mM dithiothreitol (DTT) for 6 hours and extracted RNA for qPCR analysis (Fig. 3A). The induction of ER stress was confirmed by the transcription induction of both hsc3 (Drosophila BiP) and of Ero1l, which are both targets of the unfolded protein response (Fig. 3A). In contrast, the level of dPPP1R15 mRNA failed to increase significantly during ER stress. This and the tight ER localisation seen with dPPP1R15-GFP suggest that at a functional level the behavior of dPPP1R15 more closely resembles that of PPP1R15b.
Fig. 3.
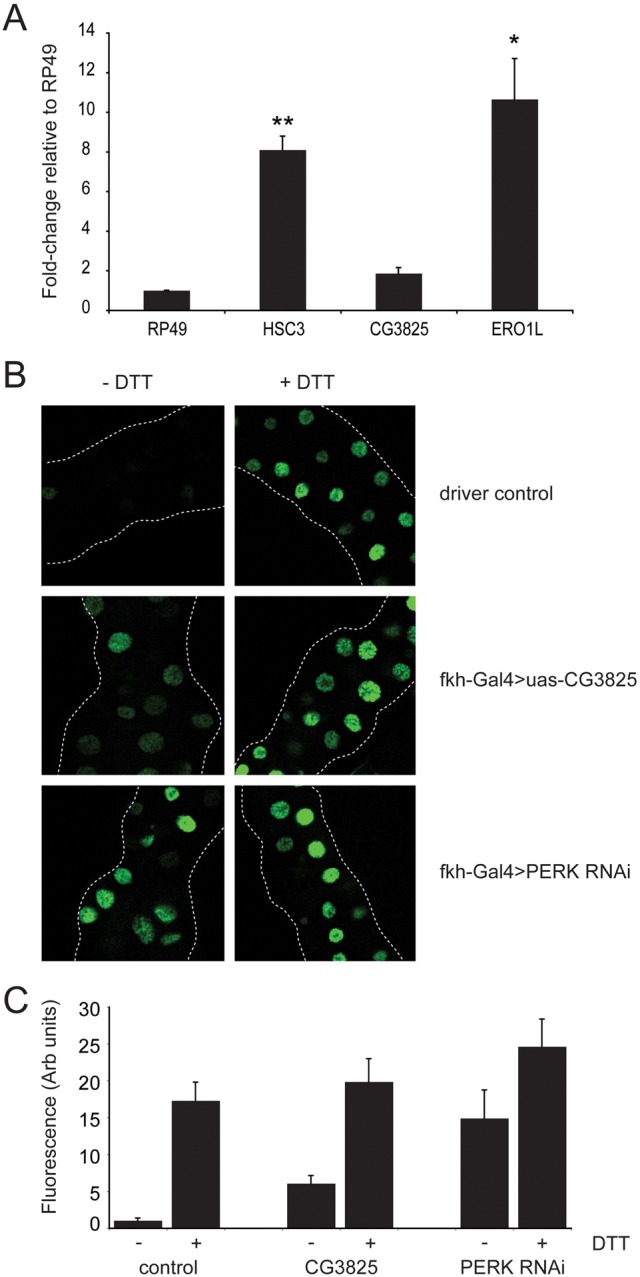
dPPP1R15 is not regulated transcriptionally by ER stress. (A) Transcriptional fold-changes of dPPP1R15, HSC3 and ERO1L mRNA measured by qPCR after 6 hours of treatment with 2 mM DTT and expressed relative to RP49. Means + s.e.m. are shown; n = 3 independent repeats, *P<0.05, **P<0.01 compared with untreated control. (B) Representative fluorescence images of salivary glands of early 3rd instar larvae. Salivary gland specific expression of either XBP1-eGFP reporter alone or coexpressed with Fkh-Gal4>UAS-dPPP1R15 Fkh-Gal4>UAS-dPERK RNAi (v16427). Salivary glands were dissected from early 3rd instar larvae and tissue further incubated at room temperature in Express Five® SFM media with or without addition of DTT (final 2 mM) for 3 hours. After tissue fixation, and mounting, the appearance of GFP signal was analysed using a LSM510 META (Zeiss) fluorescence confocal microscope. Flies were reared at 25°C. (C) Quantification of data from B.
We wished to test in vivo if dPPP1R15 serves to oppose the effects of PERK. Levels of endoplasmic reticulum stress can be assayed in Drosophila tissues using a reporter based on the splicing of the mRNA of the transcription factor XBP1 (Ryoo et al., 2007). Under basal conditions XBP1u protein levels are low owing to the presence of an inhibitory intron within its mRNA and the protein itself is unable to transactivate target genes. Upon the induction of ER stress, the intron is excised by activated IRE1 (Calfon et al., 2002). This alters the frame of the mRNA 3′ to the excision event and thus generates a new active C-terminal XBP1s domain. By fusing the coding sequence of GFP with the 3′ end of XBP1, this change of frame can be used to regulate the expression of GFP. The reporter was expressed in the salivary glands of third instar larvae, which were then dissected and a subset were treated ex vivo with DTT to induce endoplasmic reticulum stress (Fig. 3B,C). This caused the accumulation of GFP within cell nuclei indicating the activation of IRE1. We observed that in this tissue, regulation of protein synthesis by PERK is necessary to prevent ER stress in developing larvae, since RNAi depletion of PERK caused basal activation of the XBP1-GFP reporter (Fig. 3B,C). Overexpressed dPPP1R15 in the salivary gland similarly induced XBP1 splicing in otherwise unstressed cells, consistent with induction of ER stress.
Translation of dPPP1R15 mRNA is regulated by uORFs within its 5′UTR
An inspection of the mRNA sequence of dPPP1R15 cDNA clone AY051450 revealed the presence of two uORFs within its 5′UTR (Fig. 4A). The presence of these features and the finding that uORF2 overlapped out-of-frame with the dPPP1R15 coding sequence suggested that translation of this mRNA might be responsive to eIF2α phosphorylation. To test this, we fused the 5′UTR of dPPP1R15 cDNA clone AY051450 (GH117127) including the first three amino acid residues of dPPP1R15 with firefly luciferase (5′UTR-Luciferase) and cloned this into pcDNA3.1, so that synthesis of the reporter mRNA would be driven by a strong heterologous CMV promoter (Fig. 4B). Transfection of HEK293T cells with 5′UTR-Luciferase or pcDNA3.1 encoding firefly luciferase alone (Luc) demonstrated that the 5′UTR of dPPP1R15 repressed basal expression; pEGFP served as a transfection control (Fig. 4C).
Fig. 4.

dPPP1R15 expression is regulated translationally. (A) Representation of the structure of dPPP1R15 mRNA; the coding sequence is shown in purple and the two uORFs in orange. (B) Schematic of reporter mRNAs: firefly luciferase alone (Luc), uORFs 1 and 2 (5′UTR-Luc), uORF1 mutated (uORF2 only 5′UTR-Luc) and uORF2 mutated (uORF1 only 5′UTR-Luc). (C) HEK 293T cells were transfected for 16 hours with GFP and either firefly Luc or 5′UTR-Luc expressing vectors. Lysates were analysed by 10% (w/v) SDS-PAGE followed by immunoblot for dPPP1R15 and GFP. (D) Graph of fold-changes of firefly luminescence signal from HEK 293T cells transiently transfected with either firefly Luc reporters or Renilla luciferase transfection control. After 16 hours of tunicamycin treatments (concentration 0–2.5 µg/ml) firefly and Renilla luciferase activities were assayed and expressed as firefly/Renilla luciferase activity normalised to activity of untreated controls. (E) Dmel cells were treated with 2 mM DTT for indicated times and the lysates separated by 12% (w/v) SDS-PAGE followed by immunoblot analysis anti-dPPP1R15 (SMAR-2) antibody and HSC3. (F) HEK 293T cells were transiently transfected with Luc-pcDNA3.1 or 5′UTR-Luc-pcDNA3.1 and after 16 hours were treated with or without 2.5 µg/ml tunicamycin. Luciferase mRNA levels were determined by qPCR and expressed relative to β-actin mRNA.
Next, to test if phosphorylation of eIF2α would derepress translation of mRNA possessing the 5′UTR of dPPP1R15, we transfected HEK293T cells with each construct and after 6 hours treated with increasing concentrations of tunicamycin. The cells were co-transfected with pRL-TK encoding Renilla luciferase to control for transfection efficiency. Firefly and Renilla luciferase activities were determined 16 hours thereafter using a luminescence assay and firefly luciferase activity was normalised to Renilla luciferase activity (Fig. 4D). Tunicamycin treatment caused a dose-dependent increase of firefly luciferase activity in cells transfected with the 5′UTR-Luciferase reporter (Fig. 4D, red line). In contrast, a modest decrease in translation of the luciferase signal was found with the reporter construct lacking the 5′UTR (Fig. 4D, blue line). To test if the regulation of translation imparted by the 5′UTR of dPPP1R15 was dependent upon uORF1 or uORF2 we next mutated the start codon of each uORF by site-directed mutagenesis. Both uORF1 and uORF2 in fact possess two ATGs each. When both ATGs of uORF1 were mutated to AGG (uORF2 only 5′UTR-Luciferase) no significant difference in reporter activity was detected on treatment with tunicamycin (Fig. 4D, green line). However, when both ATG codons were mutated to AGG in the uORF2 (uORF1 only 5′UTR-Luciferase), a marked reduction in responsiveness to tunicamycin was seen (Fig. 4D, purple line). Of note, when each ATG of uORF2 was mutated to AGG individually, the loss of tunicamycin sensitivity was very modest, suggesting that there may be a degree of redundancy in the site of uORF2 initiation (data not shown). These data indicate that uORF2, which overlaps out-of-frame with the coding sequence of dPPP1R15, is responsible for most of the translational regulation imparted by the 5′UTR.
To confirm that expression of dPPP1R15 is regulated in insect cells despite the lack of a significant induction of its mRNA, we treated Schneider S2 cells for up to 6 hours with 2 mM DTT and immunoblotted for dPPP1R15 protein expression using an antiserum raised against the dPPP1R15 peptide sequence LTPVHRDRVYQARFLHED (CRB Ltd, UK). In unstressed cells, little dPPP1R15 protein could be detected, but following treatment with DTT a dPPP1R15-immunoreactive band was induced (Fig. 4E). This contrasted with the failure to induce dPPP1R15 mRNA even at 6 hours in this cell line (Fig. 3A). To test whether the increased synthesis of luciferase observed during ER stress solely represented enhanced translation initiation we determined the level of luciferase transcript by qPCR under basal conditions and following treatment with tunicamycin (2.5 µg/ml) (Fig. 4F). When normalised to β-actin, control mRNA lacking the 5′UTR (Luc) was expressed 2.7-fold more efficiently than that of the UTR-Luc construct; however, following the induction of ER stress, the level of luciferase mRNA fell in cells transfected with the Luc control vector yet remained stable in UTR-Luc transfected cells. This suggests that the 5′UTR of dPPP1R15 may additionally confer stability during ER stress. Taken together, these results suggest that dPPP1R15 is regulated at the translational level through increased translation initiation and enhanced mRNA stability during ER stress.
Embryonic and larval expression of dPPP1R15 is required for development
Deletion of both PPP1R15 paralogues in mice has been shown to cause early embryonic lethality that could be rescued with a phosphorylation-resistant mutant of eIF2α (Harding et al., 2009). This indicated that regulation of protein synthesis through changes in eIF2 phosphorylation status plays a critical role during embryogenesis. However, the nature of this regulation could not be elucidated further in mice owing to the severity of the phenotype and the technical challenges inherent in the study of embryogenesis in placental mammals. We therefore took advantage of our Drosophila system to study the effects of PPP1R15 activity on development. Expression of RNAi against dPPP1R15 in all cells under the control of the strong tubulin-Gal4 driver caused significant embryonic lethality associated with failure of dorsal closure in some cases (Fig. 5A). Similar effects were observed using an epithelial selective driver (e22c-Gal4) (Fig. 5A). However, when a weaker ubiquitous driver, daughterless-Gal4, was used, embryos survived to eclosure but showed a delayed larval development (Fig. 5B-F).
Fig. 5.

Embryonic and larval expression of dPPP1R15 is required for development. (A) Knockdown of dPPP1R15 using tubulin-Gal4 driver or epithelial e22c-Gal4 leads to embryonic lethality. Light microscopy, phase contrast images are shown of cuticle preparations of wild-type, tub-Gal4>dPPP1R15 RNAi and e22c-Gal4>dPPP1R15 RNAi embryos. All flies were reared at 25°C. (B) Graph illustrates eclosion of embryos expressing RNAi to dPPP1R15 (red), dPERK (green) or GCN2 (light blue) or combinations of dPERK and dPPP1R15 (violet) and of GCN2 and dPPP1R155 (orange) at 25°C. RNAi expression was driven by daughterless-Gal4. Flies with only daughterless-Gal4 are referred to as No RNAi (dark blue, obscured by red CG3825 line). (C) Graph illustrating the developmental stage distribution of flies with impaired eIF2α phosphorylation assessed 6 days after egg laying at 25°C. Gene silencing was achieved using daughterless-Gal4 driver. Dark orange, pupa; orange, 3rd instar larva; yellow, 1st and 2nd instar larva. (D) Graph illustrates the developmental stage distribution of flies with impaired eIF2α phosphorylation assessed 14 days after egg lying at 25°C. Purple, adult; dark orange, pupa; orange, 3rd instar larva. (E) Images of mouth hook preparations of larvae on day 6 after egg lying. 3rd instar wild-type larvae (upper left) display typical 3rd instar mouth hook. In contrast, retained 2nd instar mouth hook (arrowhead) was found in Da-Gal4>dPPP1R15 RNAi larvae (upper right). Moulting defect was completely rescued by the co-silencing of dPERK or GCN2 RNAi by dPPP1R15 RNAi (middle right and lower right). (F) Representative images of flies obtained after 14 days at 25°C.
When either of the two eIF2α kinases expressed in flies, GCN2 and PERK, were depleted in the embryo by daughterless-Gal4>UAS-RNAi, the majority of embryos eclosed successfully. However, knockdown of both GCN2 and dPPP1R15 in the same animals showed a strong reduction in the rate of eclosion, with only 20% eclosing within the first 48 hours (Fig. 5B). This indicates that precise regulation of eIF2α phosphorylation is necessary for efficient embryogenesis and that both dPPP1R15 and GCN2 play important roles in this. While the developing animal can tolerate partial depletion of either dPPP1R15 or GCN2, perhaps due to compensatory changes in the other’s activity, loss of both results in marked impairment of development (Fig. 5B).
When animals surviving to eclosion were then followed, it became apparent that depletion of dPPP1R15 caused a marked developmental delay (Fig. 5C–F). By day 6, more than half of the control animals were at the pupal stage compared with none of the dPPP1R15 RNAi larvae (Fig. 5C). After 6 days, most daughterless-Gal4>UAS-dPPP1R15 RNAi animals reached the third instar stage but were smaller than controls, appeared sluggish and often displayed molting defects including retention of previous instar mouth hooks and cuticle shedding (Fig. 5E). At 14 days after egg laying, the majority of animals expressing dPPP1R15 RNAi were third instar larvae in contrast to controls, most of which were adult flies (Fig. 5D,F). At 20 days, most dPPP1R15 RNAi animals were alive but remained arrested at the third instar stage (data not shown). Depletion of PERK was well tolerated during development and animals appeared phenotypically healthy (Fig. 5C–F); however, there was a subtle loss of fitness since after several generations daughterless-Gal4>UAS-PERK RNAi lines consistently died out. When both PERK and dPPP1R15 were depleted in the same animals, a modest rescue of the dPPP1R15 phenotype was observed (Fig. 5C–F). However, these animals often died during eclosion from the pupal case and those animals surviving to adulthood showed severe mobility defects and strongly reduced longevity (data not shown). Depletion of GCN2 was lethal during pupariation and this could not be rescued by depletion of dPPP1R15; however, knockdown of GCN2 did appear partially to rescue the effect of dPPP1R15 depletion as animals depleted of both GCN2 and dPPP1R15 developed further than animals depleted of dPPP1R15 alone (Fig. 5C–F).
Taken together, these observations reveal an important role for dPPP1R15 during larval development, principally to antagonise the function of GCN2.
Discussion
Dephosphorylation of eIF2α by PPP1R15 in mammals is important in the response to many forms of cellular stress, in haematopoiesis and in mammalian embryonic development (Dalton et al., 2012). However, studies of its role in development have been hampered by functional redundancy between the two mammalian isoforms and by the severity of the double knockout phenotype (Harding et al., 2009). Moreover, until now, no invertebrate PPP1R15 had been identified to enable its study during development and, by extension, a better understanding of its evolution. In this study we have determined that dPPP1R15 (formerly CG3825) shares sequence homology with both mammalian PPP1R15 proteins, has a similar localisation at the ER and functionally antagonises the eIF2α kinases both in vivo and in cultured cells. Its tight ER association and lack of significant transcriptional regulation both suggest that it shares more functional homology with the PPP1R15b isoform than PPP1R15a.
The presence of regulatory uORFs upstream of the dPPP1R15 start codon is reminiscent of both PPP1R15a and PPP1R15b. It is noteworthy that the 5′UTR found within the mRNA of both mouse and human PPP1R15a contain two uORFs (Lee et al., 2009). In each case, uORF2 is necessary for translational regulation of the true coding sequence, while uORF1 appears to play a less important role. This contrasts with the mRNA of mammalian ATF4 in which uORF1 is required for the regulatory function since its translation renders the scanning ribosome depleted of active eIF2, thus reducing the likelihood of translating the inhibitory uORF2 (Lu et al., 2004b; Vattem and Wek, 2004). Our findings suggest that the regulation of dPPP1R15 shares a uORF2-dominant mechanism with mammalian PPP1R15a. Loss of uORF1 from the 5′UTR of dPPP1R15 had little effect on translation of the true coding sequence following treatment with tunicamycin, while loss of uORF2 markedly reduced translation during ER stress. The mRNA of human PPP1R15b (NM_032833.3) also contains two non-overlapping uORFs but it remains unclear whether these play a regulatory function.
Mice lacking both Ppp1r15 genes (Harding et al., 2009) and Drosophila depleted of dPPP1R15 by RNAi driven by tubulin-Gal4 or e22c-Gal4 (this study) show markedly impaired embryonic development. Use of the weaker ubiquitous driver daughterless-Gal4 allowed for a less dramatic depletion of dPPP1R15 enabling us to study of the role of eIF2α phosphorylation at later stages of development, which revealed a genetic interaction between dPPP1R15 and the nutrient sensing kinase GCN2. The biphasic effects of genetic manipulation of eIF2α phosphorylation, with toxicity caused by the loss of the kinase GCN2 or the phosphatase dPPP1R15 is striking. While lack of dPPP1R15 may lead to excessive phosphorylation of eIF2α and thus a global impairment of protein synthesis, it is less clear why loss of the kinase GCN2 under conditions of adequate nutrient availability should impair development. A link between dysregulated eIF2α phosphorylation and impaired larval growth has been shown previously by the overexpression of a mutant form of eIF2α in which the phosphorylated serine was mutated to alanine (phosphorylation resistant) or aspartic acid (phosphomimetic) (Qu et al., 1997). The phosphorylation resistant animals grew faster and larger. In contrast, the phosphomimetic mutants experienced developmental delay. This is consistent with our observation of impaired growth of animals depleted of PPP1R15 by RNAi in which eIF2α is likely to remain phosphorylated for longer periods. The defects of growth and molting have also been seen with mutations of the DmATF4 gene, cryptocephal (Hewes et al., 2000). This transcription factor is regulated in many organisms by a translational mechanism similar to that we observed for dPPP1R15 (Lu et al., 2004a). Since loss of GCN2 will be accompanied by reduction of eIF2α phosphorylation it will also have lower ATF4 levels mimicking loss of cryptocephal. Thus, either excessive or deficient phosphorylation of eIF2α is likely to result in impaired larval development.
A previous genetic screen for modifiers of growth in Drosophila identified slimfast, an amino acid transporter (Colombani et al., 2003). Remarkably, when slimfast was depleted selectively in the fatbody, animals suffered a global defect in growth. It is known that the TOR pathway is involved in the balancing protein synthesis with nutrient supply via ribosomal protein S6 kinase and eIF4E-BP and much of the effect of amino acid starvation has been attributed to these pathways (Colombani et al., 2003; Layalle et al., 2008). However, our study suggests that GCN2 may also participate. Moreover, there is evidence for cross talk between the TOR and GCN2 pathways (Cherkasova and Hinnebusch, 2003). Further work is required to determine if such cross talk might play a role in nutrient sensing by the fat body.
While the interactions between dPPP1R15 and its cognate kinases during larval development are readily explained by a simple antagonistic relationship, the apparent synthetic lethal interaction between dPPP1R15 and dGCN2 knockdown during embryogenesis is curious. One might speculate that during embryogenesis the phosphorylation of eIF2α by dGCN2 may serve to promote translation of dPPP1R15. If partial knockdown of dPPP1R15 (by daughterless-Gal4 driven RNAi) is compensated for during embryogenesis by enhanced translation of the residual dPPP1R15 mRNA in response to phosphorylation of eIF2α by GCN2, then reduction in the level of dGCN2 may paradoxically reduce the level of dPPP1R15 still further. If dPPP1R15 protein is required at a late stage of embryogenesis, for example to limit the effects of dPERK signaling triggered by the development of secretory tissues, then the earlier failure to translate dPPP1R15 might lead to failure to eclose.
Our study suggests that coordinate regulation of eIF2α phosphorylation during larval development is important in determining the rate of anabolic growth and, in turn, successful completion of pupariation. We have characterised the first example of an invertebrate eIF2α phosphatase and demonstrated that during development it primarily antagonises the function of GCN2. Further studies will address the mechanism by which alteration of eIF2α phosphorylation selectively within the fat body can impair global tissue growth.
Materials and Methods
Expression plasmids
To generate UAS-dPPP1R15 flies, the coding sequence of dPPP1R15 was amplified from cDNA clone GH11727 (Drosophila Genomics Research Centre, Indiana University, USA) and cloned into pUASTattB. For synthesis of dPPP1R15 RNA hybridisation probes, the coding sequence of dPPP1R15 was amplified and cloned into pCRII-TOPO vector (Invitrogen). To express dPPP1R15 protein in mammalian cells, the coding sequence was cloned into pcDNA3.1(+)-myc-his (Invitrogen), pEGFP-C2 and pEGFP-N3 (Clontech). The coding sequence of firefly luciferase 2 gene was amplified with incorporation of 5′ EcoRI and 3′ XhoI restriction sites. This was then cloned into the mammalian expression vector pcDNA3.1(+) (Invitrogen) to generate Luc-pcDNA3.1. To investigate the regulatory properties of the 5′UTR of dPPP1R15 mRNA, the 5′UTR sequence and the first nine nucleotides of the protein coding sequence were amplified by polymerase chain reaction from cDNA clone GH11727 (AY051450) with incorporation of 5′ Kpn1 and 3′ EcoRI restriction sites. This was cloned upstream of the luciferase 2 coding sequence in Luc-pcDNA3.1 KpnI and EcoRI in the multicloning site to generate 5′UTR-Luc-pcDNA3.1. As a transfection control in the luciferase reporter assay pRL-TK Renilla luciferase plasmid (Promega) was co-transfected.
Cell culture
For studies of dPPP1R15 in mammalian cells, HEK293T or COS-7 cells were maintained in DMEM medium supplemented with 10% heat-inactivated FBS and incubated at 37°C with 5% CO2. COS-7 cells in glass bottom 3.5 cm Petri dishes were transfected with 1 µg of GFP-dPPP1R15 or dPPP1R15-GFP constructs using Lipofectamine LTX (Invitrogen) and imaged 25–35 hours post-transfection. Before live cell imaging, a medium change to DME media: Ham’s F-12 (1∶1) with HEPES buffer, without Phenol Red (Sigma Aldrich) was conducted to avoid interference with Phenol red. To test for PP1 binding, HEK293T cells in 10 cm Petri dishes were transiently transfected with 6 µg of dPPP1R15-GFP construct, lysed 36 hours later and subjected to GFP-trap affinity purification (Chromotek, Germany). To evaluate the effect of dPPP1R15 on eIF2α phosphorylation, HEK293T cells in 10 cm Petri dishes were transiently transfected with 6 µg of vector expressing dPPP1R15-GFP or GFP for 24 hours prior to drug treatment. For studies in Drosophila cells the Drosophila melanogaster late-stage embryo-derived cell line called Dmel (a kind gift from Dr Jenny Hirst, Cambridge, UK) was used. The cells were grown in suspension in serum-free Express Five® SFM medium (Invitrogen) supplemented with 2 mM L-glutamine (Invitrogen) and antibiotics (100 U/ml Penicillin G and 100 µg/ml Streptomycin) and cultured in a cooled incubator at 25°C.
Immunoblot
Mammalian cell lysates were prepared in lysis buffer H [HEPES pH 7.9 10 mM; NaCl 50 mM; sucrose 0.5 M; EDTA 0.1 mM; Triton X-100 0.5% (v/v)] supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitors (10 mM tetrasodium pyrophosphate, 15.5 mM β-glycerophosphate, 100 mM NaF). Dmel cells were lysed in lysis buffer containing 50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1% (v/v) Triton X-100 supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitors. Pull down experiments were performed using GFP-Trap® A beads (Chromotek, Germany) according to the manufacturer’s protocol. Lysates of Drosophila animals or heads were prepared by homogenisation directly in 1× SDS loading buffer containing phosphatase inhibitors. Lysates were boiled for 5 minutes and centrifuged at 15,000 g to clear. Unless stated otherwise, gel loading per lane corresponded to: three fly heads; one adult fly; half of one third-instar larva.
Commercially used primary antibodies used were: mouse anti-GFP antibody (ab1218, Abcam, UK; 1∶1000), rabbit anti-PP1a antibody (no. 2582; Cell signaling; 1∶1000), goat anti-firefly luciferase (Fisher Scientific; 1∶20000), rabbit p-eIF2α (Cell signaling; 1∶1000), anti-β-actin (ab3280, Abcam, Cambridge, UK; 1∶1000), rat Troponin H cross reacts with dBiP/hsc3 MAC 143 (Babraham Institute, Cambridge, UK; 1∶2000). Non-commercial antibodies included: anti-total eIF2α mouse monoclonal (kind gift of D. Ron, Cambridge, UK; 1∶10000); custom rabbit anti-dPPP1R15 peptide antibody raised against C-terminal end peptide[C]-LTPVHRDRVYQARFLHED-acid) (CRB Ltd, Cambridge, UK; 1∶1000).
Fluorescence microscopy
An LSM510 META confocal fluorescence microscope (Zeiss Ltd, Welwyn Garden City, UK) equipped with a 37°C incubation chamber without CO2 supply was used for live cell imaging to determine subcellular localisation of GFP-tagged dPPP1R15 protein in mammalian cells. Images were taken using 63× magnification with oil immersion and post-imaging modifications were performed using the LSM510 imaging and Adobe Photoshop software. For detection of XBP1 splicing in Drosophila larval salivary gland cells using the GFP reporter flies (Ryoo et al., 2007), an LSM510 microscope with 20× magnification with FITC filter (excitation of 488 nm) was used. For post-imaging processing the LSM510 processing package and ImageJ software were used.
Luciferase assays
To analyse the regulatory function of the 5′UTR of the dPPP1R15 mRNA, HEK293T cells were transfected with 200 ng of Luc-pcDNA3.1 or with 1 µg of 5′UTR-Luc.pcDNA3.1 constructs in addition to 50 ng TK-Renilla luciferase plasmid using Lipofectamine LTX. Six hours post-transfection, cells were treated for 16 hours with tunicamycin then firefly and Renilla luciferase luminescence were measured sequentially using the dual-luciferase assay kit (Promega, Southampton, UK) and a Glomax luminometer (Promega). The ratio of firefly/Renilla luminescence was calculated and expressed as fold change compared to untreated samples.
To assess ER stress, HEK293T cells were co-transfected with 2 µg of a plasmid encoding firefly luciferase under the control of a UPR response element (p5×ATF6-Luc) and 50 ng of the TK-Renilla luciferase transfection efficiency control reporter plasmid. The cells were transfected for 6 hours and then induced with tunicamycin for 16 hours. Transfected cells were lysed and assayed for firefly and Renilla luciferase activity using the dual luciferase kit (Promega).
In situ hybridisation
Digoxeginin-labelled sense and anti-sense dPPP1R15 RNA probes were synthesised from linearised dPPP1R15.pCRII-TOPO plasmid using the DIG RNA labelling SP6/T7 kit (Roche Applied Science). Oregon R embryo fixation and whole embryo in situ hybridisation was performed as described previously (Tautz and Pfeifle, 1989). Detection of DIG-labelled probe was achieved using an alkaline phosphatase-conjugated anti-DIG antibody and NBT/X-phosphate catalysation.
Fly husbandry
Crosses were set up and flies maintained at 25°C if not stated otherwise. Crosses expressing dPERK in the eye (GMR-Gal4>UAS-dPERK) were performed at 18°C due to pupal lethality as described previously (Malzer et al., 2010). The UAS-dPPP1R15 transgenic fly line was generated using the PhiC31 system and the 51D-attP landing site in y1w1118; 51DattP;+ embryos (Bestgene, USA). Other lines used were: w[*];P{w[+mC] = Gal4-ninaE.GMR}12 BL1104; w[1118]; P{da-Gal4.w[-]}3 BL8641; w[1118]; +; tub-Gal4/TM6b,tb (a kind gift of S. Imarisio, University of Cambridge); P{fkh-Gal4.H} (III); arm-Gal4 (III) β-catenin (a kind gift of S. Imairsio, University of Cambridge); yw.hs-flp122;e22c-Gal4/Cyo;MKRS/TM6b,tb (a kind gift of Joaquín de Navascués, University of Cambridge). All RNAi lines used were obtained from the Vienna Drosophila RNAi Centre (Vienna, Austria): CG3825/dPPP1R15 RNAi v15238; dPERK RNAi v16427; GCN2 RNAi V103976.
Hatching assay
Twenty mated da-Gal4 driver line female virgins were crossed with each transgenic RNAi line and allowed to lay eggs onto grape juice agar for 4 hours followed by counting of the eggs at time 0 (100% ‘unhatched’). For the following 48 hours, the number of eggs remaining ‘unhatched’ was counted every 12 hours. The grape juice plates were then incubated at 25°C for either 6 or 14 days after egg. The animals were then staged by light microscopy; the distinction between second and third instar larvae was based on the morphology of the anterior spiracles.
qPCR
RNA from a 10 ml suspension of Schneider S2 insect cells each treated with DTT for various time points or untreated control were isolated using the RNAeasy Mini kit (Qiagen, Crawley, UK). One microgram of RNA was treated with 1 U DNase I (Invitrogen, Paisley, UK) in a 10 µl reaction volume then transcribed into cDNA using Oligo-dT primers and the reverse transcription kit (Promega, Southampton, UK). The QuantiTect SYBR® Green PCR kit (Qiagen, Crawley, UK) was used for qPCR reactions in an ABI 7900 HT Fast real-time PCR machine (Applied Biosystems, Carlsbad, UK). All samples were measured in triplicate in 25 µl reaction volumes. Amplification efficiencies for each primer pair were determined using logarithmic standard dilution of an untreated Schneider S2 cell sample from 1∶10–1∶10000 on each PCR plate. Efficiencies of between 80 and 120% were accepted. The PCR program used was: 95°C for 15 minutes; 40 cycles of 94°C for 15 seconds; 54°C for 30 seconds; 72°C for 30 seconds. To calculate changes in expression levels relative to the untreated sample and housekeeper gene RP49 the ΔΔCt method was used.
For measurements of firefly, Renilla luciferase and β-actin mRNA level by qPCR HEK293T cells were transfected and treated as described for the luciferase assay but harvested by trypsinisation. RNA was then purified using the RNAeasy Mini kit (Qiagen, Crawley, UK). Primer sequences were: FLuc.S, 5′-GCCATTCTACCCACTCGAAG-3′; FLuc.AS, 5′-CTCGAAGTACTCGGCGTAGG-3′, product size: 139 bp; RLuc.S, 5′-GAGCATCAAGATAAGATCAAAGCA-3′; RLuc.AS, 5′-CTTCACCTTTCTCTTTGAATGGTT-3′, product size: 241 bp; β-actin.S, 5′-CTCTTCCAGCCTTCCTTCCT-3′; β-actin.AS, 5′-AGCACTGTGTTGGCGTACAG-3′, product size: 116 bp. An annealing temperature of 58°C was used for all mammalian qPCR primers.
Footnotes
Author contributions
E.M., M.S.S., L.E.D., S.E.T., N.H. performed the experiments under the supervision of S.J.M. E.M. and S.J.M. designed the project and wrote the manuscript. H.S., D.A.L. and D.C.C. contributed experimental advice. All authors commented on the final manuscript.
Funding
This work was supported by the UK Medical Research Council (MRC); and the British Society for Cell Biology. S.J.M. is an MRC Senior Clinical Fellow [grant number G1002610]. Deposited in PMC for release after 6 months.
References
- Abastado J. P., Miller P. F., Jackson B. M., Hinnebusch A. G. (1991). Suppression of ribosomal reinitiation at upstream open reading frames in amino acid-starved cells forms the basis for GCN4 translational control. Mol. Cell. Biol. 11, 486–496 10.1128/MCB.11.1.486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M. J., Knutson G. S., Lasky S. R., Munemitsu S. M., Samuel C. E. (1985). Mechanism of interferon action. Purification and substrate specificities of the double-stranded RNA-dependent protein kinase from untreated and interferon-treated mouse fibroblasts. J. Biol. Chem. 260, 11240–11247 [PubMed] [Google Scholar]
- Brush M. H., Shenolikar S. (2008). Control of cellular GADD34 levels by the 26S proteasome. Mol. Cell. Biol. 28, 6989–7000 10.1128/MCB.00724-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush M. H., Weiser D. C., Shenolikar S. (2003). Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 23, 1292–1303 10.1128/MCB.23.4.1292-1303.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- Carra S., Boncoraglio A., Kanon B., Brunsting J. F., Minoia M., Rana A., Vos M. J., Seidel K., Sibon O. C., Kampinga H. H. (2010). Identification of the Drosophila ortholog of HSPB8: implication of HSPB8 loss of function in protein folding diseases. J. Biol. Chem. 285, 37811–37822 10.1074/jbc.M110.127498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. J., Throop M. S., Gehrke L., Kuo I., Pal J. K., Brodsky M., London I. M. (1991). Cloning of the cDNA of the heme-regulated eukaryotic initiation factor 2 alpha (eIF-2 alpha) kinase of rabbit reticulocytes: homology to yeast GCN2 protein kinase and human double-stranded-RNA-dependent eIF-2 alpha kinase. Proc. Natl. Acad. Sci. USA 88, 7729–7733 10.1073/pnas.88.17.7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkasova V. A., Hinnebusch A. G. (2003). Translational control by TOR and TAP42 through dephosphorylation of eIF2alpha kinase GCN2. Genes Dev. 17, 859–872 10.1101/gad.1069003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli V. R., Wang J., Dow J. A. (2007). Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39, 715–720 10.1038/ng2049 [DOI] [PubMed] [Google Scholar]
- Colombani J., Raisin S., Pantalacci S., Radimerski T., Montagne J., Léopold P. (2003). A nutrient sensor mechanism controls Drosophila growth. Cell 114, 739–749 10.1016/S0092-8674(03)00713-X [DOI] [PubMed] [Google Scholar]
- Dalton L. E., Healey E., Irving J., Marciniak S. J. (2012). Phosphoproteins in stress-induced disease. Prog. Mol. Biol. Transl. Sci. 106, 189–221 10.1016/B978-0-12-396456-4.00003-1 [DOI] [PubMed] [Google Scholar]
- Dever T. E., Feng L., Wek R. C., Cigan A. M., Donahue T. F., Hinnebusch A. G. (1992). Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596 10.1016/0092-8674(92)90193-G [DOI] [PubMed] [Google Scholar]
- Graveley B. R., Brooks A. N., Carlson J. W., Duff M. O., Landolin J. M., Yang L., Artieri C. G., van Baren M. J., Boley N., Booth B. W. et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479 10.1038/nature09715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H. P., Zhang Y., Ron D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- Harding H. P., Zhang Y., Scheuner D., Chen J. J., Kaufman R. J., Ron D. (2009). Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc. Natl. Acad. Sci. USA 106, 1832–1837 10.1073/pnas.0809632106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewes R. S., Schaefer A. M., Taghert P. H. (2000). The cryptocephal gene (ATF4) encodes multiple basic-leucine zipper proteins controlling molting and metamorphosis in Drosophila. Genetics 155, 1711–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousse C., Oyadomari S., Novoa I., Lu P. D., Zhang Y., Harding H. P., Ron D. (2003). Inhibition of a constitutive translation initiation factor 2α phosphatase, CReP, promotes survival of stressed cells. J. Cell Biol. 163, 767–775 10.1083/jcb.200308075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloft N., Neukirch C., von Hoven G., Bobkiewicz W., Weis S., Boller K., Husmann M. (2012). A subunit of eukaryotic translation initiation factor 2α-phosphatase (CreP/PPP1R15B) regulates membrane traffic. J. Biol. Chem. 287, 35299–35317 10.1074/jbc.M112.379883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layalle S., Arquier N., Léopold P. (2008). The TOR pathway couples nutrition and developmental timing in Drosophila. Dev. Cell 15, 568–577 10.1016/j.devcel.2008.08.003 [DOI] [PubMed] [Google Scholar]
- Lee Y. Y., Cevallos R. C., Jan E. (2009). An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 284, 6661–6673 10.1074/jbc.M806735200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P. D., Harding H. P., Ron D. (2004a). Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33 10.1083/jcb.200408003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P. D., Jousse C., Marciniak S. J., Zhang Y., Novoa I., Scheuner D., Kaufman R. J., Ron D., Harding H. P. (2004b). Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 23, 169–179 10.1038/sj.emboj.7600030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malzer E., Daly M. L., Moloney A., Sendall T. J., Thomas S. E., Ryder E., Ryoo H. D., Crowther D. C., Lomas D. A., Marciniak S. J. (2010). Impaired tissue growth is mediated by checkpoint kinase 1 (CHK1) in the integrated stress response. J. Cell Sci. 123, 2892–2900 10.1242/jcs.070078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., Ron D. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 10.1101/gad.1250704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I., Zeng H., Harding H. P., Ron D. (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 153, 1011–1022 10.1083/jcb.153.5.1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S., Perlaky S. E., Organ E. L., Crawford D., Cavener D. R. (1997). Mutations at the Ser50 residue of translation factor eIF-2alpha dominantly affect developmental rate, body weight, and viability of Drosophila melanogaster. Gene Expr. 6, 349–360 [PMC free article] [PubMed] [Google Scholar]
- Ryoo H. D., Domingos P. M., Kang M. J., Steller H. (2007). Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 26, 242–252 10.1038/sj.emboj.7601477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Vattem K. M., Sood R., An J., Liang J., Stramm L., Wek R. C. (1998). Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18, 7499–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D., Pfeifle C. (1989). A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 98, 81–85 10.1007/BF00291041 [DOI] [PubMed] [Google Scholar]
- Vattem K. M., Wek R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 101, 11269–11274 10.1073/pnas.0400541101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek R. C., Jiang H. Y., Anthony T. G. (2006). Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11 10.1042/BST0340007 [DOI] [PubMed] [Google Scholar]
- Williams N. P., Hinnebusch A. G., Donahue T. F. (1989). Mutations in the structural genes for eukaryotic initiation factors 2 alpha and 2 beta of Saccharomyces cerevisiae disrupt translational control of GCN4 mRNA. Proc. Natl. Acad. Sci. USA 86, 7515–7519 10.1073/pnas.86.19.7515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Brush M. H., Choy M. S., Shenolikar S. (2011). Association with endoplasmic reticulum promotes proteasomal degradation of GADD34 protein. J. Biol. Chem. 286, 21687–21696 10.1074/jbc.M110.212787 [DOI] [PMC free article] [PubMed] [Google Scholar]