

Summary
Phospholipase D2 (PLD2) is a cell-signaling molecule that bears two activities: a guanine-nucleotide exchange factor (GEF) and a lipase that reside in the PX/PH domains and in two HKD domains, respectively. Upon cell stimulation, the GEF activity yields Rac2-GTP and the lipase activity yields phosphatidic acid (PA). In the present study, we show for the first time that these activities regulate one another. Upon cell stimulation, both GEF and lipase activities are quickly (within ∼3 min) elevated. As soon as it is produced, PA positively feeds back on the GEF and further activates it. Rac2-GTP, on the other hand, is inhibitory to the lipase activity. PLD2 would remain downregulated if it were not for the contribution of the tyrosine kinase Janus kinase 3 (JAK3), which restores lipase action (by phosphorylation at Y415). Conversely, the GEF is inhibited upon phosphorylation by JAK3 and is effectively terminated by this action and by the increasing accumulation of PA at >15 min of cell stimulation. This PA interferes with the ability of the GEF to bind to its substrate (Rac2-GTP). Thus, both temporal inter-regulation and phosphorylation-dependent mechanisms are involved in determining a GEF–lipase switch within the same molecule. Human neutrophils stimulated by interleukin-8 follow a biphasic pattern of GEF and lipase activation that can be explained by such an intramolecular switch. This is the first report of a temporal inter-regulation of two enzymatic activities that reside in the same molecule with profound biological consequences in leukocyte cell migration.
Key words: HL-60 cells, Cell migration, Cell signalling, Chemotaxis, Leukocyte, Tyrosine kinase JAK
Introduction
Small GTPases are guanine nucleotide binding proteins that can switch between the inactive GDP-bound form (GDP-GTPase) and the active GTP-bound form (GTP-GTPase) (Haeusler et al., 2006; Matsuda et al., 1996). The Rho family GTPases, Rho, Rac and Cdc42, regulate cell migration, phagocytosis, cell polarity, neurite extraction/retention and cell survival among other functions (Chae et al., 2008b; Cougoule et al., 2006; Wang et al., 2008; Yamauchi et al., 2004).
Rac2 is abundant in hematopoietic cells and is involved in the activation of a variety of kinases, such as p42/p44 and p38 MAPK, JNK and Akt (Fumagalli et al., 2007; Gu et al., 2003; Guo et al., 2008). In addition, Rac2 regulates actin cytoskeletal machinery during the formation of lamellipodia and membrane ruffles, cell migration and adhesion of immune cells (Gomez-Cambronero, 2011). Abnormal activation of Rho GTPases is implicated in diseases, such as tumorigenesis (Aznar and Lacal, 2001; Benitah et al., 2003; Karlsson et al., 2009), endometriosis (Yotova et al., 2011) and Fragile X syndrome (Chen et al., 2010). Hence, activation of Rho GTPases and the upstream guanine-nucleotide exchange factors (GEFs) is tightly regulated (Pai et al., 2010).
In a recent study, we reported for the first time that phospholipase D2 (PLD2) acts as a GEF for the Rac2 GTPase, which is a novel characterization of a dual activity enzyme with both lipase and GEF activities embedded in the same protein (Gomez-Cambronero, 2011; Mahankali et al., 2011b). PLD2 has a phox homology (PX) domain, a pleckstrin homology (PH) domain and two HKD catalytic domains. HKD domains mediate lipase activity, while the GEF activity site resides in the PX domain and is assisted by the PH domain (Gomez-Cambronero, 2011; Mahankali et al., 2011b).
PLD2 via its lipase activity breaks down phosphatidylcholine to phosphatidic acid (PA) and choline. PLD and its product PA are associated with a wide variety of cellular functions including that of neutrophils, which is the primary defense in the immune system (Jang et al., 2012; Norton et al., 2011). PA is one of the major lipid second messengers that in turn triggers many signaling pathways such as phosphatidylinositol 4-phosphate 5-kinases (type I), mTOR (mammalian target of rapamycin) kinase, sphingosine kinase 1 and Raf-1 protein kinase (Cazzolli et al., 2006; Corrotte et al., 2006; Foster, 2009; Frondorf et al., 2010; Lehman et al., 2007; Rose et al., 1995; Song et al., 1991). PA regulates small GTPases like Ras, Rac1 by changing their membrane localization (Zhang and Du, 2009). PA exerts most of the above effects by binding to its protein targets (Chae et al., 2008a; Cockcroft, 2009). We have shown that Rho GTPases upon activation by upstream receptor tyrosine kinases are specifically implicated in lamellipodia formation and cell movement (Mahankali et al., 2011a). In spite of all this, a good understanding of the connection between the roles of PLD2/PA and Rac GTPases in cell movement is still lacking. Also, it is essential to understand if both lipase and GEF activities of PLD2 are turned on at the same time or at sequential times during cell stimulation.
PLD2 is a newly discovered GEF for Ras and Rac2 (Gomez-Cambronero, 2011; Henkels et al., 2013; Mahankali et al., 2012; Mahankali et al., 2011b). Another study also demonstrated PLD2's GEF activity towards RhoA (Jeon et al., 2011). However, very little is known about how PLD2-GEF activity is regulated. Phosphorylation plays a key role in GEF regulation. Some GEFs are also regulated by autoinhibition, which usually is relieved by phosphorylation or interaction with other proteins. ERK1/2 induces and positively regulates GEFH1 catalytic activity (Fujishiro et al., 2008), whereas two mitotic kinases, auroraA/B and Cdk1/cyclinB, negatively mediate phosphorylation of GEFH1, as does PAR1b (Yamahashi et al., 2011), which reduces the catalytic activity of GEFH1 and, therefore, activation of RhoA (Birkenfeld et al., 2007). PLD2 is a phosphoprotein, regulated by kinases such as EGFR, JAK3 and Src tyrosine kinases (Henkels et al., 2010). Even though phosphorylation is implicated in lipase activity regulation of PLD2, the mechanism by which PLD2's GEF activity is activated is not known.
In the present study, we report a novel mechanism of how the products of both the lipase and GEF activities regulate the alternate activity. We report for the first time a biphasic effect of PA on GEF, as well as a divergent effect of the tyrosine kinase JAK3 on GEF and lipase activities. We also report that the temporal regulation of the dual enzyme PLD2 is also present in human neutrophils, which underlies the progress of chemotaxis, as it likely occurs at a site of injury.
Results
PA has a dual effect on PLD2 GEF activity: time and concentration determines the effect
As PLD2 bears two different catalytic activities, GEF and lipase, in the same molecule, we reasoned that the product of one activity could influence the other activity given the close spatial proximity that exists between the two catalytic sites. We hypothesized that Rac2-GTP could affect the lipase component and that PA could affect the GEF counterpart.
To examine the effect of PA on PLD2-GEF, PLD2-mediated Rac2 activation (Rac2-GTP pulldown) was measured in the absence or presence of 1 or 300 nM PA. Rac2 activation (also known as ‘GTP loading’) was measured by p21 activated kinase binding domain (PBD) pulldown assay as elaborated in the Methods section. PBD has an affinity for Rac2-GTP but not for Rac2-GDP. As shown in Fig. 1A,B, PA augments the effect on the GEF activity at lower concentrations of PA (1 nM), but it is inhibitory at higher concentrations (300 nM). We believe that PA provides an initial positive feedback on the catalytic GEF site and a larger negative feedback on the same activity as more and more PA accumulates in the cell. Similar results (Fig. 1C,D) were observed in an in vitro approach where recombinant PLD2 and Rac2 were used for PBD pulldown assays. The last panels in Fig. 1A and Fig. 1C are loading controls showing increasing amounts of PLD2 expression in lysates and total Rac2 levels, respectively. Also shown are the densitometry line graphs (Fig. 1B,D), indicating that PA affects PLD2-mediated Rac2 activation dually, positively at low concentrations and negatively at higher concentrations.
Fig. 1.

PA influences PLD2-mediated Rac2 activation in vivo and in vitro. Rac2 activation was measured by a p21 activated kinase binding domain (PBD) pulldown assay. PBD has affinity for Rac2-GTP but not for Rac2-GDP. (A,C) PBD pulldown assay in whole cells transfected with myc-PLD2-WT (A) or with purified, baculoviral myc-tagged PLD2 and HA-tagged Rac2 (C) in the presence of 1 or 300 nM PA (DOPA, a membrane-soluble form of PA, was used for the whole cell experiments). Expression of increasing PLD2 and Rac2 equal loading controls are shown in western blots (W.B.) with anti-PLD2 or anti-HA antibodies, respectively. (B,D) Densitometry line graphs corresponding to A and C, respectively. Results shown are representative of three independent experiments performed in duplicate.
PA interferes with a PLD2–Rac2 association
We next intended to understand the mechanism that could explain the negative effect of PA on PLD2-GEF. We hypothesized that PA interferes with a functional PLD2–Rac2 association. A static protein–protein interaction between the lipase and the GTPase has been shown previously by our lab (Peng et al., 2011a; Peng et al., 2011b). Co-immunoprecipitations of PLD2 and Rac2 were performed in the absence or presence of PA in whole cells (Fig. 2A) and with recombinant PLD2 and Rac2 (Fig. 2C). Fig. 2A shows Rac2–PLD2 binding as myc-tagged Rac2 was immunoprecipitated using anti-HA antibodies specific for HA-tagged PLD2, while Fig. 2C also shows Rac2–PLD2 binding using purified proteins as HA-tagged Rac2 was immunoprecipitated using anti-myc antibodies specific for myc-tagged PLD2. In general for both Fig. 2A,C, increasing amounts of PA from 1 nM to 1000 nM affected the binding of Rac2 to PLD2; PA first helped and then impeded with increasing concentrations in an ‘oscillatory’ pattern (Fig. 2B,D). The bottom panels in Fig. 2A,C are loading controls representing total Rac2 or total PLD2 and total recombinant Rac2, respectively.
Fig. 2.

PA interferes with protein–protein interaction between PLD2 and Rac2. (A) In vivo PLD2–Rac2 binding in the absence or presence of increasing PA. COS-7 cells were co-transfected with constant myc-tagged Rac2 and constant HA-tagged PLD2-WT. HA-tagged PLD2-WT was immunoprecipitated using mouse anti-HA-agarose followed by subsequent western blotting (W.B.) using rabbit anti-myc antibody specific for the myc-tagged Rac2. (B,D) Densitometry line graphs of in vivo and in vitro immunoprecipitations shown in A and C, respectively. Representative total cellular Rac2 and PLD2 (20% of total cellular protein) or recombinant Rac2 equal loading controls are shown in the last two panels of A or bottom panel of C, respectively. (C) In vitro PLD2–Rac2 binding in the absence or presence of increasing PA. Purified, baculoviral myc-tagged Rac2 and HA-tagged PLD2-WT proteins were used for in vitro binding assays. Similar to A, myc-tagged PLD2-WT was immunoprecipitated using mouse anti-myc-agarose followed by subsequent western blotting using rabbit anti-HA antibody specific for the HA-tagged Rac2. Results shown are representative of three independent experiments performed in duplicate.
The PX domain of PLD2 preferentially binds to PA over other phospholipids
Interaction of PA with a variety of proteins has been reported in previous studies, which affects the localization and/or activities of these proteins (Stace et al., 2008; Stace and Ktistakis, 2006). For example, by interacting with PKC, PA directs membrane translocation of PKC (Jose Lopez-Andreo et al., 2003). PA via interaction with sphingosine kinase 1 stimulates its kinase activity and thereby aids in the formation of sphingosine-1-phosphate (Delon et al., 2004). PA interacts with the PX domain of p47PHOX protein (Karathanasis and Wilson, 2002). In the present study, as it is evident that PA interferes with PLD2's GEF activity and its interaction with Rac2 (Figs 1,2), we hypothesized that PLD2 has a preference for PA, whereby PLD2 might bind PA at the GEF catalytic site or close by and prevent PLD2–Rac2 interaction. A liposome co-sedimentation assay was performed with purified recombinant PLD2 or GST–PX. Liposomes composed of phosphatidylcholine (PC), both PC and PA, both PC and PI(4,5)P2 (weight ratio 6∶4) or both PC and PI(3)P were incubated with purified full length PLD2 or GST–PX followed by SDS-PAGE and western blot analyses. Fig. 3A indicates that full-length PLD2 binds to all lipids tested. However, PX preferentially binds to PA over PI and PIP2 (Fig. 3A). Since PA is synthesized at the C-terminus of PLD2 at its two lipase HKD domains, it is understandable that if PLD2 binds PA at another site (PX), then this could enable a negative effect on the GEF activity, as in the case of an allosteric regulation.
Fig. 3.
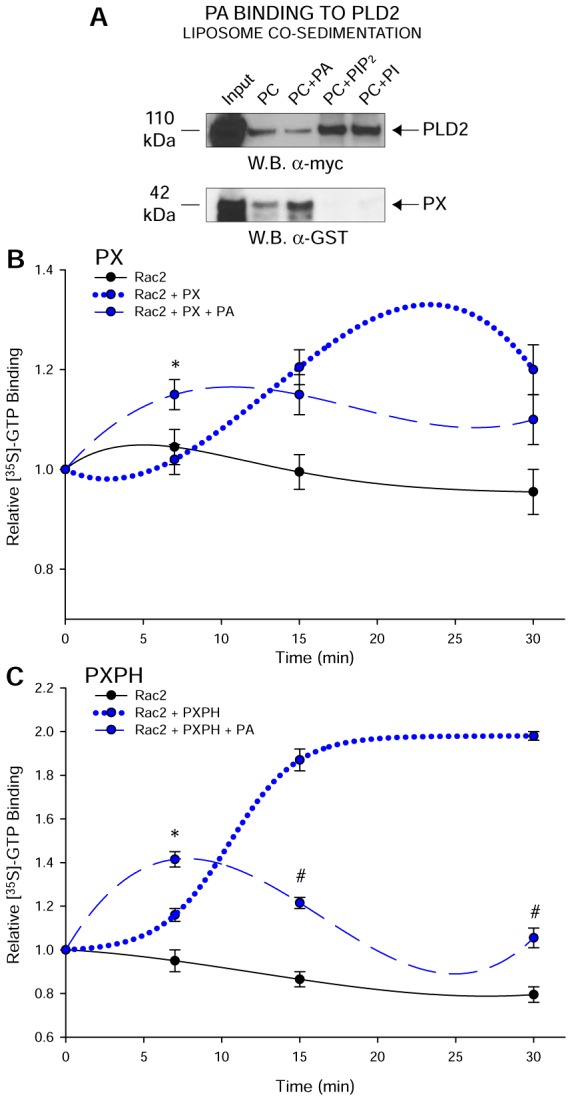
PA binds PLD2 at the PX domain and negatively regulates Rac2 binding. (A) Liposome co-sedimentation assays using purified, recombinant full-length PLD2 or GST–PX (purified). Liposomes composed of phosphatidylcholine (PC), both PC and PA, both PC and PI(3)P or both PC and PI(4,5)P2 (weight ratio 6∶4) were incubated with purified full length PLD2 or GST–PX followed by SDS and western blot (W.B.) analyses. Full length PLD2 interacts with all the four lipids including PA. PX domain binds to PA preferentially but not to PI or PIP2. Lane 1 of all the panels shows protein input in the liposome co-sedimentation assay. (B,C) Negative effect of PA on [35S]-GTP binding of Rac2 mediated by PX (B) and PXPH (C) respectively. GTP binding assays were performed in the absence or presence of 300 nM PA. Triplicate results are means±s.e.m. and are expressed in terms of relative [35S]-GTPγS binding. Curves in B and C were best fit by a polynomial cubic equation. *denotes significant differences (P<0.005) above controls; #denotes significant differences (P<0.005) below controls.
We next determined the ability of GST–PX or GST–PXPH (the slightly larger truncated version of PLD2) to catalyze GTP binding to Rac2 in a temporal fashion. Fig. 3B,C show a transient upregulating effect of PA but an overall negative effect of PA on GEF activity. All of these experiments suggest that the decrease in GEF activity by PA is due to a decreased protein–protein interaction between Rac2 and PLD2 at the PX domain level.
Rac2 alters PLD2-lipase activity
Having established the effect of PA on PLD2-GEF activity, we next asked the inverse question, i.e. if Rac2-GTP (GEF reaction product) could influence PLD-lipase activity. First, using cell lysates, we show that increasing overexpression of Rac2 in COS-7 cells led to a robust inhibition of lipase activity (Fig. 4A). Second, using purified, recombinant proteins, we show in Fig. 4B that Rac2-WT has a negative effect on the lipase activity, which is accentuated by GTP but not by GDP. To further confirm this, we observed that in recombinant Rac2-T17N (constitutively GDP-bound), the negative effect on lipase activity is no longer observed (Fig. 4B). Thus, activated Rac2 (i.e. GTP-bound) has a negative regulatory effect on PLD2-lipase activity.
Fig. 4.
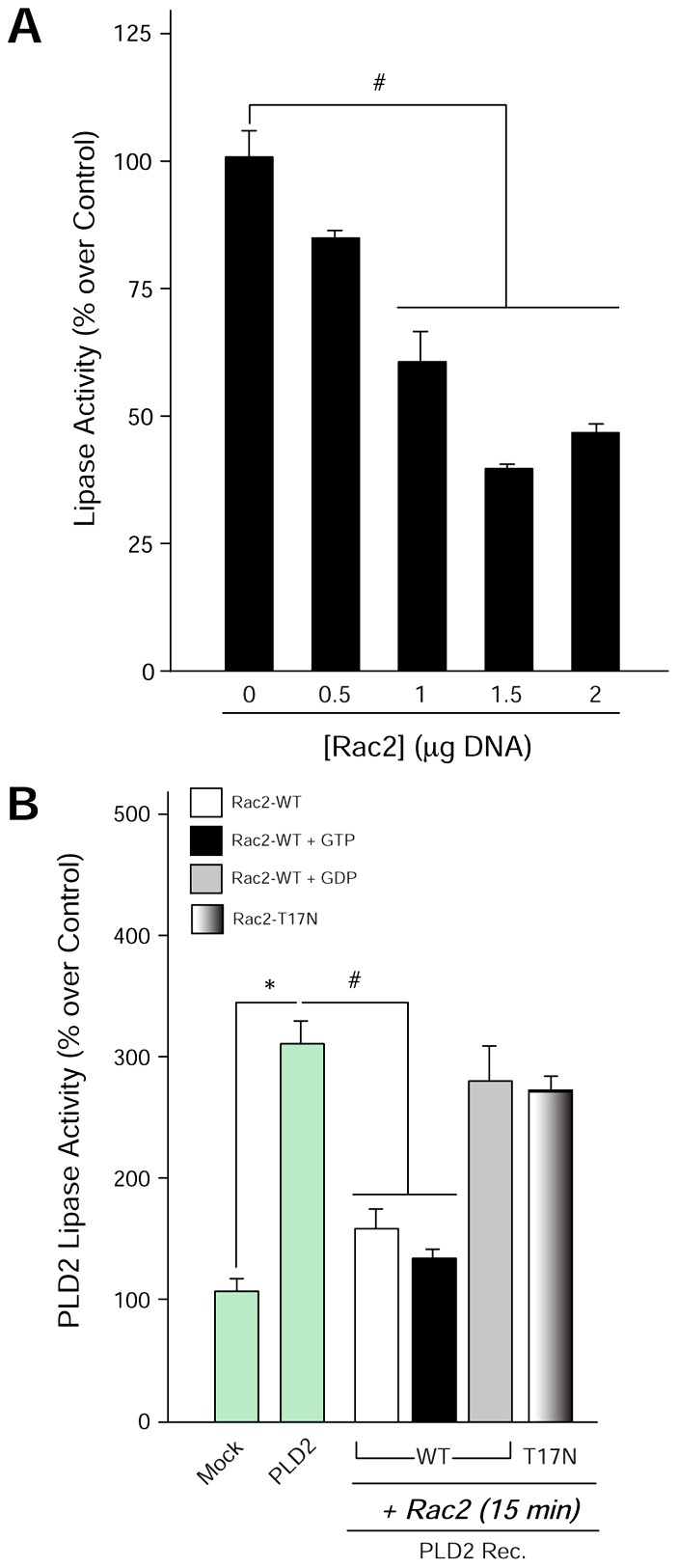
Negative effect of Rac2-GTP on PLD2-lipase activity. (A) PLD2 lipase activity is inhibited by Rac2 in a dose-dependent fashion. COS-7 cells were overexpressed with a constant amount of PLD2 and increasing concentrations of Rac2, and post-transfection PLD2 lipase activity was measured. (B) PLD2 lipase activity (using recombinant PLD2) in the presence of recombinant Rac2, GTP-bound Rac2-WT, GDP-bound Rac2-WT or Rac2-T17N at late times. Rac2-WT when GTP bound inhibits PLD2-lipase, whereas the effect is negated in the presence of GDP-bound Rac2-WT or constitutively inactive Rac2-T17N. Triplicate results are means±s.e.m. and are expressed in terms of either activity (% over control). *denotes significant differences (P<0.005) above controls; #denotes significant differences (P<0.005) below controls.
Phosphorylation dependent mechanism in a PLD2 GEF–lipase switch: role of tyrosine kinase JAK3
Since the previous figures indicate that the products of the lipase and GEF reactions (PA and Rac2-GTP, respectively) regulate the opposite activity, we next investigated (1) whether a GEF–lipase activity switch exists and, if so, (2) whether this switch is external to the PLD2 molecule. As PLD2 is a phosphoprotein and its lipase activity is regulated by phosphorylation/dephosphorylation mechanisms, we asked if PLD2-GEF is also regulated by phosphorylation/dephosphorylation using two different approaches. First, we performed GEF assays using purified, recombinant proteins. Of the four candidate tyrosine kinases tested, Src, EGFR, Fes and JAK3, only JAK3 showed a negative effect on the GEF activity of PLD2 (Fig. 5A), which is represented by a ∼50% decrease in GEF activity. JAK3 is a non-receptor tyrosine kinase that is heavily implicated in leukocyte regulation (Cornejo et al., 2009). In addition, JAK3 has a role in cell migration and actin cytoskeletal regulation, similar processes where PLD2 is also implicated (Henkels et al., 2011; Kumar et al., 2007; Mishra et al., 2012).
Fig. 5.
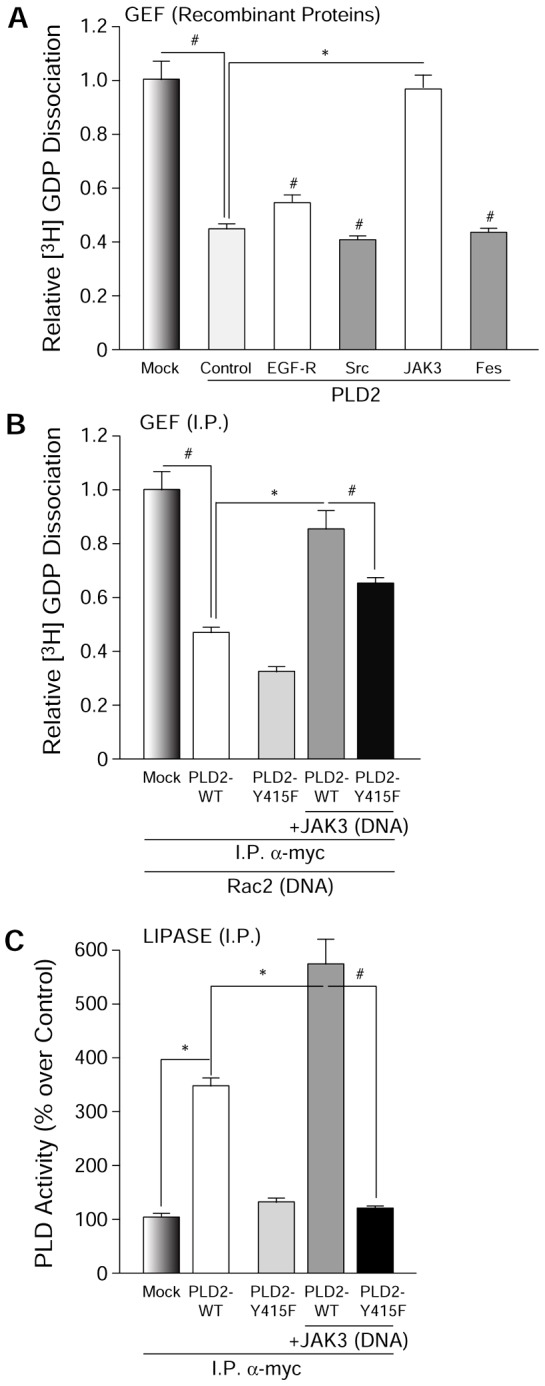
Only JAK3, out of a panel of four tyrosine kinases, inhibits PLD2 GEF. (A) Effect of tyrosine kinases Src, Fes, EGFR and JAK3 on PLD2 GEF activity in vitro (using recombinant proteins). (B) JAK3 has a negative effect on PLD2-GEF activity, whereas (C) it has a positive effect on lipase in vivo. COS-7 cells were overexpressed with PLD2-WT or PLD2-Y415F in the absence or presence of JAK3. Immunoprecipitated (I.P.) PLD2 from these cells was used for GEF assays (the smaller the bars, the higher the GEF activity). Triplicate results are means±s.e.m. and are expressed in terms of relative [3H]-GDP dissociation. *denotes significant differences (P<0.005) above controls; #denotes significant differences (P<0.005) below controls.
Second, to further establish the previous results, GEF and lipase assays were performed with immunoprecipitated (I.P.)-PLD2 from cells overexpressing PLD2-WT, PLD2-Y415F, both PLD2-WT and JAK3 or both PLD2-Y415F and JAK3. Fig. 5B,C indicate again that JAK3 has a negative effect on the GEF activity of I.P.-PLD2, whereas JAK3 has a positive effect on lipase activity, respectively. Contrarily, JAK3’s effects on PLD2 GEF and lipase activities are reversed to a large extent in the presence of PLD2-Y415F (PLD2 dephosphorylation mutant) (Fig. 5B,C), highlighting the key role of this residue to each of the two different PLD2 catalytic activities.
In conclusion, the data presented so far indicate the existence of an inter-regulation of the product of one catalytic activity on the other catalytic activity (PA on GEF and Rac2-GTP on lipase). PA has both a positive and a negative effect on the GEF activity of PLD2, depending on its concentration, while Rac2-GTP has a negative effect on PLD2-lipase activity. Apart from this, we also showed the phosphorylation-dependent mechanism of PLD2-GEF/lipase regulation where JAK3 has both positive and negative effects on lipase and GEF, respectively.
An oscillatory pattern of activation exists between GEF and lipase activities
Since PLD2 is an enzyme with both lipase and GEF activities, we next measured the two activities of PLD2 simultaneously as a function of time during cell stimulation with an extracellular agonist in two cell types, COS-7 and neutrophils. COS-7 cells overexpressing PLD2 were treated with epidermal growth factor (EGF), which is known to activate PLD2, and following increasing EGF treatment as a function of time, cellular PLD2 was immunoprecipitated and lipase and GEF activities were measured. Fig. 6A shows that both catalytic activities have an early peak (∼3 min) in COS-7 cells with GEF activity dominating over lipase activity. This is presumably due to the positive effect of PA on GEF, as seen in Fig. 1A.
Fig. 6.

PLD2 lipase and GEF cellular activities as a function of time. Effect of EGF (A) or IL-8 (B) stimulation on increasing time of the lipase and GEF activities of PLD2 overexpressed in COS-7 cells (A) and primary neutrophils (B). PLD2's lipase and GEF activities peak at early time following EGF stimulation, whereas at later times lipase activity continues to increase but GEF declines. (C) Chemotaxis of primary neutrophils. Peripheral blood neutrophils migrated towards IL-8 in a time-dependent manner. Cells were incubated in transwell permeable supports in multi-well plates over a period of 60 minutes. The dashed line represents accumulation of neutrophils to the bottom well where the chemoattractant was added. The solid line represents the ‘rate’ of migration (as number of cells migrated per unit of time). Values are means± s.e.m. of three independent experiments performed in duplicate.
As time progresses, more Rac2-GTP is produced, which has the effect of decreasing the lipase activity (as seen in Fig. 4A), consistent with a temporary loss of activity in Fig. 6A between 3 and 10 min. At later times of cellular stimulation (>10 min), this scenario is reversed, as the lipase activity supersedes the GEF activity. We believe that the reason for this is the involvement of JAK3, which has both positive and negative effects on GEF and lipase, respectively (as indicated in Fig. 5). Due to JAK3 action, the lipase activity is restored, which starts to peak again at 10 min and is still high at ∼30 min. This lipase activity leads to a steady PA accumulation. GEF activity is further impaired in the presence of high concentrations of PA (as presented earlier in Fig. 1B), and an effective GEF–lipase switch takes place. With this, the results observed in Figs 1–5 underlie the oscillatory pattern of GEF and lipase activities observed in Fig. 6A.
Physiological relevance: PLD2 GEF–lipase switch in primary human neutrophils
Next, to determine the physiological relevance of this model, we chose primary human neutrophils, as (a) Rac2 comprises >96% of Rac in neutrophils (Ambruso et al., 2000) and (b) PLD2's role in phagocyte chemotaxis is established (Lehman et al., 2006). Fig. 6B shows lipase and GEF activities of neutrophils over time in response to stimulation with interleukin-8 (IL-8), the main PMN chemoattractant. A similar oscillatory temporal pattern like that of COS-7 cells was observed where both the activities peak at earlier times of stimulation, and the GEF activity predominates over that of the lipase activity.
In order to confirm the physiological significance of the lipase/GEF switch, we performed chemotaxis in human neutrophils using 20 nM IL-8. Cells were incubated in transwell permeable supports in multi-well plates over a period of up to 60 minutes to migrate against the chemoattractant that was added to the bottom well. Fig. 6C (solid line) shows neutrophil migration from 5 to 60 min. Fig. 6C (dashed line) shows the rate of migration (as cells moved/unit of time as a proxy for migration velocity) consistent with a rapid, initial phase, and an increasingly stationary phase at later times. Observing the rate of migration it became evident that this parameter correlates with the first peak of GEF and Lipase activities in Fig. 6A,B. This means that in the initial stages of cell migration, the cells use both activities available to them to speed up the process. Chemotaxis rate diminishes after 15 minutes, but neutrophils still migrate (dashed line Fig. 6C).
The accumulation of the reaction products (PA and Rac2-GTP) over time reflects lipase–GEF switch pattern
The data presented above presumes the existence of “oscillatory” production of PA and/or Rac2-GTP. To clarify this, we used two approaches to address this issue. First, we documented the accumulation of PA at increasing times. For PA sensor experiments, COS-7 cells were seeded on coverslips, transfected with pEGFP-Spo20PABD-WT alone or along with PLD2. pEGFP-Spo20PABD-WT encodes for a fluorescent PA sensor consisting of EGFP fused to a 40 amino acid PA binding domain from the yeast Spo20 protein. Two days post-transfection, cells were treated with EGF for increasing time points, coverslips were fixed and the nuclei were stained with DAPI. Cells were visualized using fluorescence microscopy. As shown in Fig. 7A, PA accumulates at early times (1–3 min) and then increases at later time points (>15 min), albeit with different subcellular localizations. The appearance of PA in the cytosol implies its accumulation on internal membrane structures where it participates in a plethora of signaling pathways. Second, we documented the accumulation of Rac-GTP following increasing time of neutrophil stimulation. Fig. 7B shows Rac2-GTP levels were measured in neutrophils after cell stimulation with IL-8 as a function of time by PBD pulldown assay. Rac2 activation is highest at around 1 to 5 min (early peak) after which there is a gradual drop and by 10 min reaches basal levels (0 min), which is maintained until 15 min. Rac2 activation appears to decline between 15 and 30 min. Fig. 7C shows the densitometry line graph of the same with a pattern of products of the reaction that parallels the enzymatic activity behavior of PLD2's activities.
Fig. 7.

PA, Rac2-GTP and JAK3 as a function of time. (A) The panels show immunofluorescence of PA sensor (as reported in Frondorf et al., 2010) in COS-7 cells following EGF stimulation for increasing time. For PA sensor, cells were transfected with pEGFP-Spo20PABD-WT, which encodes for a fluorescent PA sensor consisting of EGFP fused to a 40 amino acid PA binding domain from the yeast Spo20 protein. Two days post-transfection, cells were treated with EGF for increasing time points. Cells were fixed to coverslips, the nuclei were stained with DAPI and cells were imaged using fluorescent microscope. PA accumulates at ∼3 min and then it increases with time, peaking at 15 min. Shown is a representative experiment among three performed with similar results (B) PBD-pulldown western blot showing Rac2-GTP levels upon IL-8 treatment of neutrophils for increasing time points. Result shown in B are representative of three experiments. (C) Densitometry line graph indicating Rac2-GTP levels measured by PBD pulldown (experiments performed in B). C is an average of three different experiments (D) JAK3 enzymatic activity was measured as indicated in the Materials and Methods section in COS-7 cells (closed circles) and neutrophils (open circles) that were stimulated with EGF or IL-8 respectively for the lengths of time indicated. Inset in D shows JAK3 localization in COS-7 cells upon EGF stimulation for the indicated time points. Results shown are representative of three different experiments. Fig. 7D was best fit by the Weibull (peak) distribution, 4-parameter equation. (E) Lipase and GEF activities of PLD2 are reduced and enhanced respectively (at 7 min after IL-8 stimulation) when JAK3 is silenced in dHL-60 cells. Cells were silenced with Negative-siRNA (si-Neg) or siJAK3. Immunoprecipitated PLD2 from these cells was used for GEF assays (the smaller the bars, the higher the GEF activity). (F) dHL-60s were silenced with Negative-siRNA or siJAK3, which was followed on the next day by overexpressing PLD2 (or they were mock-transfected). After a total of 4.5 days after the initial DMSO induction of differentiation, cells were taken for transwell assays for measurement of chemotaxis against IL-8. Triplicate results are means±s.e.m. and are expressed in terms of relative [3H]-GDP dissociation. *denotes significant differences (P<0.005) above controls; #denotes significant differences (P<0.005) below controls.
In addition to measuring PA and Rac2-GTP levels, we also measured JAK3 activity over time in both COS-7 cells and neutrophils (Fig. 7D). Fig. 7D confirms that JAK3 activity peaks at 5 to 10 min following cell stimulation, suggesting that JAK3 activity does indeed reflect the lipase/GEF switch pattern seen in COS-7 cells in Fig. 6A and in neutrophils in Fig. 6B. Additionally to confirm these results using an alternative approach that expands upon JAK3’s involvement in lipase/GEF switch, we performed PLD2 lipase and GEF activity assays using JAK3 silenced differentiated HL-60 cells (neutrophil-like). Fig. 7E shows that when JAK3 expression is silenced PLD2-lipase activity is significantly reduced, whereas the GEF activity is enhanced at 7 min after IL-8 stimulation. This is an indication of a switch: JAK3 specifically activates the lipase activity and inhibits GEF activity allowing GEF activity to return to basal levels while lipase activity is enhanced.
Further, we performed chemotaxis of dHL-60 cells overexpressing PLD2 with or without JAK3 silencing (Fig. 7F). dHL60s showed a marked difference when JAK3 is silenced when compared to that of negative siRNA-treated cells. In the presence of JAK3 silencing, basal or PLD2-mediated cell migration is significantly decreased. This suggests that JAK3-mediated regulation of PLD2 plays an important role in neutrophil cell motility.
From these results, it can be concluded that two distinct patterns of PLD2 enzymatic activities exist, ‘early’ and ‘late’ in an ‘oscillatory’ temporal pattern. During early activation, both of PLD2's activities are high, GEF being higher than the lipase, whereas at later time points lipase overtakes and supersedes GEF. JAK3 is one of the key players in determining this switch to lipase activity; the others are PA and Rac2-GTP.
A molecular model that explains the GEF–lipase cellular switch
Based on the data presented in this study, we propose the regulatory mechanism for dual (GEF/lipase) PLD2 activity. The inter-regulation occurs by the product of one reaction acting upon the other enzymatic activity (i.e. PA acting on GEF-PLD2 or Rac2-GTP acting on lipase-PLD2) and is time-dependent (Fig. 8). Fig. 8A shows re-plotting of Fig. 6A,B to depict the complex dynamics of lipase and GEF activities of PLD2 as a function of time. The numbers in the graph (Fig. 8A) represent the steps as outlined in the following panel (Fig. 8B). We explain this with four well-defined points for the new model of lipase–GEF inter-regulation (Fig. 8B): 1) at early times of cell stimulation, PA has a positive effect on PLD2-GEF activity, 2) as time progresses accumulation of Rac2-GTP due to GEF activity downregulates PLD2-lipase activity and 3) JAK3 tyrosine kinase then restores lipase activity by phosphorylating PLD2. At the same time JAK3 has an inhibitory effect on GEF activity. JAK3 phosphorylates Y415 on PLD2 (Henkels et al., 2010), which helps to enhance lipase activity. This is an indication of a switch: JAK3 specifically activates the lipase activity and inhibits GEF activity allowing GEF activity to return to basal levels, while lipase activity is enhanced. The final result on the physiology of neutrophil chemotaxis is that cell migration is maintained.
Fig. 8.

Model for the proposed regulatory mechanism for dual (GEF and lipase) PLD2 activity. The inter-regulation occurs as the product of one reaction acting upon the alternate enzymatic activity (i.e. PA acting on GEF-PLD2 or Rac2-GTP acting on lipase-PLD2). (A) Re-plotting of Fig. 6A,B showing the complex dynamics of lipase and GEF activities as a function of time. The numbers in the graph represent the steps as outlined in the following panel. The arrow indicates the time when a switch between GEF activity and lipase activity takes place, thanks to the intervention of JAK3. (B) The proposed model is well explained in four points for the lipase–GEF inter-regulation. (1) At early times (∼5 min) of cell stimulation both PA and Rac2-GTP are produced to begin chemotaxis. (2) As time progresses (∼10 min), accumulation of Rac2-GTP (due to GEF activity) negatively feeds back on PLD2-lipase activity. (3) JAK3 tyrosine kinase restores lipase activity while inhibiting GEF activity. (4) Upon cell stimulation at >30 min, lipase predominates and GEF is further inhibited. Eventually at 60 min chemotaxis plateaus and both of the activities slow down.
However, the mechanism behind the negative effect of JAK3 on PLD2 is not known at present. We speculate that it might be due to phosphorylation on a negative residue on PLD2 or it also can be due to a phosphorylation-independent event where a conformational change might be affecting the ability of the PX/PH (GEF domain) to bind to the substrate, 4) lastly GEF is further inhibited as the increase in PA interferes with GEF binding to Rac2. This is a novel regulatory mechanism of a dual activity enzyme, where the formation of product (PA) of one activity (lipase) is maximized by shutting down the other activity (GEF) that would synthesize the converse product (Fig. 8).
Discussion
We have described here the complex intra-molecular regulatory mechanisms in the dual enzyme, PLD2, which contains a C-terminal lipase domain and Rac GEF activity in its PX domain. Briefly, upon activation of PLD2, both enzymatic functions are co-activated and the moderate amounts of PA produced by the lipase act to further stimulate GEF activity. The Rac2-GTP thus produced is a negative regulator of the lipase activity and causes a dip in PA production. Lipase activity increases at later time points after PLD2 is phosphorylated on Y415 by activated JAK3 and this time around, the PA concentration is significantly higher and acts to inhibit the GEF activity of PLD2. This is an entirely novel mechanism of a GEF–lipase switch that explains neutrophil chemotaxis. A large rate of cell migration (cells migrated per unit of time) occurs in the first 10–15 minutes after the cells encounter a chemoattractant (Fig. 6C). This parameter correlates with the first peak of GEF and lipase activities in Fig. 6A,B. This means that in the initial stages of cell migration, the cells use both activities available to them to speed up the process. Chemotaxis rate diminishes after 15 minutes, but neutrophils still migrate (dashed line Fig. 6C). The maintenance of this late phase is possible as PLD lipase activity is kept high (in spite of low GEF activity) thanks to the intervention of the tyrosine kinase JAK3.
PLD via its dual activities (lipase and GEF) generates PA and Rac2-GTP, which are central in regulating actin cytoskeletal machinery and cell movement (Rudge and Wakelam, 2009). We present a novel reciprocal regulation of an enzyme with dual activity (GEF and lipase) in the same molecule. The regulation consists of a double allosteric mechanism on alternate activities by the products of the reaction of the opposite activity and also phosphorylation-dependent mechanisms. Specifically, Rac2-GTP, the product of GEF-PLD2 regulates the PLD2-lipase activity, whereas PA, the product of lipase-PLD2, regulates the PLD2-GEF activity.
The PLD2 dual activity regulatory mechanism operates in two phases. At early times (1–3 min) upon cell stimulation, PA is a positive effector and forms a reinforcing, positive feedback loop that ensures a quick and potent response of the cell. On the other hand, as Rac2-GTP accumulates in the cell, it has a negative effect on the PLD2-lipase activity. In addition, both PA and Rac2-GTP carry their messenger roles further downstream to key signal transduction pathways in the cell. A decline and slow rise in GEF and lipase activities, respectively, during the intermediate phase is due to JAK3 phosphorylation-dependent mechanisms. In the second phase of the regulatory mechanism, at sufficiently long periods of times (>15–30 min), PA accumulates in the cell and acts as negative regulator of GEF activity forming a negative feedback. As far as we know, this is the first description of such an alternate and temporally regulated dual mechanism in cell biology.
The mechanism of this temporal switch from a positive regulation to a negative regulation can be explained in molecular terms by experiments described in this report in conjunction with other known mechanisms documented by other researchers. Let us divide the GEF–lipase inter-regulation into two phases. Phase-I is the positive feedback of PA on GEF (alternate) activity at early times of cell stimulation followed by Rac2's negative effect on lipase activity. Phase-II is the negative feedback of PA on GEF activity at late times of cell stimulation.
Regarding the effect of lipase-generated PA on GEF in phase-I (positive feedback on alternate activities, i.e. PA on GEF), our data presented here shows that PA induces the formation of active Rac2 that in the presence of PA results in an increase in the Rac2-GTP species. The negative effect of GEF (Rac2-GTP) on the lipase activity can be explained by binding interference of Rac2-GTP, which accumulates in the cell. Rac2-GTP at high concentrations binds to PLD2 and impedes the access of lipase-PLD2 to its substrate PC in the cell membrane. Our laboratory has shown both molecular and cellular evidence for this (Peng et al., 2011b). Rac2 can be detected by immunofluorescence in an ‘arc’ preceding PLD2 in the cell membrane at the leading edge of chemotaxing leukocytes.
In phase-II of the GEF–lipase inter-regulation (PA's negative feedback on GEF activity at longer times of cell stimulation), the mechanism involves competitive binding, which leads to inhibition of activities. The negative effect of lipase on GEF during phase-II can be explained as follows: PA at high concentrations interferes with the binding of PLD2 to Rac2. As shown in the data of the present study, this is a sterical impediment created by PA due to its ability to bind to the PX domain where the GEF activity resides.
There are many examples of heterotropic allosteric regulation of enzymes of the carbohydrate metabolism, where sufficient accumulation of the product of a synthetic pathway inhibits the initial enzyme. However, as far as we know, this is the first description of the same enzyme regulated by the product of one activity on the other companion activity (PA on GEF and Rac2-GTP on lipase). It is also the first report of a temporal switch from positive feedback to a selective negative feedback to efficiently upregulate one activity and downregulate the other activity to maximize the production of the needed signaling product. This, we believe, has unexpected and profound consequences in cell motility.
Materials and Methods
Cell culture, neutrophil isolation and HL-60 cell differentiation
COS-7 cells were grown at 37°C in a 5% CO2 incubator in Dulbeccos's Modified Eagle's Media (DMEM)+10% (v/v) newborn calf serum (NCS). When cultured cells reached a confluence of ∼60% they were transfected with the plasmid of interest. Neutrophils were isolated from peripheral blood of volunteer donors, who signed an IRB-approved consent form. Promyelocytic leukemic HL-60 cells were grown at 37°C in a 5% CO2 incubator in Iscove's modified Eagle's medium+40% (v/v) fetal bovine serum. Cell density was maintained between 0.1 and 1.0×106/ml. HL-60 cells were induced to differentiate to the neutrophilic phenotype (dHL-60) by the addition of 1.25% (v/v) DMSO to the culture medium for 4 days.
Cell transfection
Cells were transiently transfected with PLD2 or Rac2 or JAK3 plasmid constructs. COS-7 cell transfections were performed using 5 μl Lipofectamine (Invitrogen, Carlsbad, CA) and 5 μl Plus reagent (Invitrogen) in Opti-MEM medium (Invitrogen). COS7 cells were transfected for 3 h, washed and were re-fed with prewarmed complete medium. After 36 h, cells were harvested for their respective experiment. When necessary, post-transfection before harvesting, cells were treated with 10 nM EGF for indicated time lengths. In case of dHL-60s, two days after induction of differentiation, cells were transfected with myc-PLD2 plasmid using nucleofection, as per the manufacturer's protocol (Amaxa, Gaithersburg, MD). Briefly, appropriate amounts of plasmid were added to 2×106 cells in 100 µl of nucleofection solution V in the electroporation cuvette. Cells were electroporated using program T-019 (as per the manufacturer's instructions). After nucleofection, cells were resuspended in fresh DMSO [1.25% (v/v)] containing media. Cells were immediately plated into six-well plates and incubated at 37°C for 48 hr to allow for maximum gene expression. Cells were harvested for the appropriate experiment, after 48 hrs of transfection.
Gene silencing
For JAK3, we used siRNA from Applied Biosystems that targeted exon 19 [locus s7653; sense, 5′-GUAUCGUGGUGUCAGCUAUd(TT)-3′]. As a negative control, cells were silenced using scrambled siRNA that lacks homology for any known gene sequences. For silencing experiments, HL-60 cells were differentiated for 2 days and on day 3 cells were transfected with siRNA using nucleofection, in a similar fashion as that of overexpression. Cells were harvested for the required experiment after 48 hrs of silencing.
GEF activity: GDP dissociation from Rac2
To examine the effect of kinases on PLD2-mediated [3H]-GDP dissociation from Rac2, 19 pmol of Rac2 was preloaded with 2 µM [3H]-GDP. Simultaneously, either recombinant or I.P.-PLD2 was incubated for 10 min in 20 mM Tris-HCl pH 7.5, 0.1 mM DTT, 80 mM NaCl, 0.5 mM MgCl2, 0.8 mM AMP-PNP and 1 mM GTP (80 µl volume). PLD2 alone (19 pmol) or Rac2 alone was used as positive and negative controls, respectively. Preloaded [3H]-GDP bound Rac2 samples were mixed with buffer containing PLD2 from the previous step (100 µl volume). Aliquots were taken at different times to measure the amount of radiolabeled [3H]-GDP bound to Rac2, spotted on Millipore BA85, air-dried, washed 3× for 5 min with ice-cold 20 mM Tris-HCl pH 7.5, 100 mM NaCl and 10 mM MgCl2. The amount of [3H]-GDP-bound to Rac2 was measured by scintillation spectrometry. The assay was performed similarly with immunoprecipitated PLD2 or full length PLD2.
GEF activity: GTP binding to Rac2
To examine the effect of PX or PXPH on GTP binding, [35S]-GTPγS bound to Rac2 in the presence of GST–PX or GST–PXPH was measured. Rac2 alone was used as the positive and negative control. Nineteen pmol of Rac2 was incubated with 8 µM GDP, 6 mM MgCl2 (20 µl volume) for 10 min at room temp. GDP-bound Rac2 was added to 25 µl 100 µM AMP-PNP, 1 mM MgCl2 and 1 µM [35S]-GTPγS in the absence or presence of GST–PX or GST–PXPH (75 µl volume). The relative amount of [35S]-GTPγS-bound to Rac2 was measured by scintillation spectrometry.
Immunoprecipitation, SDS-PAGE and western blot analyses
To confirm the presence of overexpressed protein, we performed SDS-PAGE and western blot analyses of myc- or HA-tagged mutant proteins that were overexpressed in dHL-6 neutrophilic cells. Two days post-transfection mutant proteins were immunoprecipitated using anti-myc or anti-HA antibodies. Samples were analyzed by SDS-PAGE and western blot analysis to confirm the presence of mutant PLD2 proteins in the cell lysates. Immunoprecipitated mutants were also used to measure relative GDP dissociation from Rac2.
Rac2 PBD pulldown assays
Rac2 PBD pulldown assays were also conducted with purified, recombinant (Rac2 and PLD2 fusion) proteins to measure the activation of Rac2. The assay uses an immobilized GTPase-binding domain that is recruited to GTP-bound, activated GTPase. Since the specificity of the interaction is dependent on the sequence of the binding domain, this assay is highly specific in detecting the activation of Rac. Five µl of PAK-1-PBD-agarose was added to each sample and incubated at 4°C for 30 min in the presence of magnesium lysis buffer (25 mM HEPES, 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA and 2% glycerol). Samples were loaded onto gels, transferred to blotting membranes and probed with α-HA antibodies to detect recombinant, GTP-bound Rac2. HRP-conjugated secondary antibodies were incubated with PVDFs and products visualized using ECL reagents.
Generation of GST-tagged fusion proteins using a bacterial expression system
The PX or PXPH domain of PLD2 was subcloned into the pGEX-4T-1 vector. Individual constructs were transformed into BL21 cells. BL21 culture was induced with 800 µM (GST–PX) or 100 µM IPTG (GST–PXPH) overnight at 16°C (GST–PX) or 25°C (GST–PXPH). Cells were harvested and cell lysates were prepared by resuspending the pellets in lysis buffer (5 mM HEPES, 100 µM sodium orthovanadate and 0.4% Triton X-100 supplemented with protease inhibitor cocktail just before use). Samples were sonicated to obtain crude lysates, which were then centrifuged at 5000 g for 5 min to obtain the clarified lysates. GST-tagged proteins in the clarified lysates were pulled down using glutathione-sepharose beads overnight at 4°C, after which samples were centrifuged to remove the unbound protein and the beads were washed three times with 1× PBS. GST proteins bound to glutathione–sepharose beads were analyzed via western blot analysis using anti-GST antibody. GST–PX or GST–PXPH bound to glutathione–sepharose beads were used to perform GTP binding assays. Rac2 alone was used as a negative control.
Liposome co-sedimentation assay
Liposome co-sedimentation assay was performed as explained in (Kurooka et al., 2011; Lee et al., 2002). Liposomes composed of phosphatidylcholine (PC), both PC and PA, both PC and PI(3)P or both PC and PI(4,5)P2 (weight ratio 6∶4) were resuspended in 100 μl buffer (50 mM Tris-HCl, pH 7.4 and 100 mM NaCl) to which 1 μg of myc-PLD2WT or GST-PX or GST-PH was added. Liposomes and proteins were incubated for 30 min at 30°C. After incubation, samples were centrifuged at 17,000 g for 15 min, supernatants were discarded and the pellets were analyzed by SDS-PAGE and western blot analyses with specific antibodies.
Lipase assay
COS-7 cells were overexpressed with PLD2-WT or mutants, which was immunoprecipitated with anti-myc antibody two days post-transfection. In case of neutrophils PLD2 was immunoprecipitated using anti-PLD2 antibody after cells were treated with IL-8 for indicated time lengths. Immunoprecipitated PLD2 was processed for PLD2 activity in PC8 liposomes and n- [3H] butanol beginning with the addition of the following reagents (final concentrations): 3.5 mM PC8 phospholipid, 1 µM PIP2, 45 mM HEPES (pH 7.8), and 1.0 µCi of n-[3H]butanol in a liposome form as indicated in (Liscovitch et al., 2000). Samples were incubated for 20 min at 30°C with continuous shaking. The addition of 0.3 ml of ice-cold chloroform/methanol (1∶2) stopped the reactions. Lipids were then isolated and resolved by thin layer chromatography. The amount of [3H] phosphatidylbutanol ([3H]PBut) that co-migrated with PBut standards was measured by scintillation spectrometry.
Immunofluorescence microscopy
COS-7 cells were seeded onto glass coverslips at a density of ∼10–20% confluency and then were either transfected with recombinant GFP-tagged PA sensor alone or co-transfected with both GFP-tagged PA sensor and myc-tagged PLD2. At 48 hours post-transfection, coverslips were fixed with 4% paraformaldehyde for 10 min at room temp, and then, nuclei were stained with 1∶2000 DAPI in PBS. Coverslips were washed with PBS and then distilled water, dried and mounted onto glass slides using Vectashield mounting solution. Cells were imaged using a Nikon 50i eclipse epifluorescence microscope and a 100× PlanFluor oil objective.
JAK3 kinase assay
Samples containing 2×106 cells were lysed and were incubated in the presence of the following final concentration of each: 4 mM MOPS, pH 7.0, 15 mM MgCl2, 1 mM EGTA, 0.2 mM Na Orthovanadate, 0.2 mM DTT, 1 µCi [32Pγ]-ATP, 100 µM cold ATP and 42 µM JAK3tide substrate to yield a 40 µl total kinase reaction volume. Reactions were incubated at 30°C for 20 min and stopped by spotting 20 µl reactions onto 2× 2.5 cm2 pieces of P81 Whatman filter paper for duplicate determinations. After filter papers were dry, each was washed in cold running water for 5 min total. Filters were dried and individual filters placed into scintillation vials containing Scintiverse II (Fisher) liquid scintillation cocktail. All samples were counted in a Beckman LS 6000TA liquid scintillation counter using the [32P] protocol for 1 min each. Results were quantified as DPMs and expressed in terms of percent over control.
Cell migration assay
Post-transfection, dHL-60 cells (or fresh neutrophils were appropriated) were harvested and resuspended at a density of 1×106 cells/ml in chemotaxis buffer (Rosewell Park Memorial Institute Medium (RPMI)+0.1% BSA). Cell migration was carried over in transwells (nucleopore polycarbonate membrane) as indicated in (Lehman et al., 2006). Two hundred µl of these cells were added to the 5 µm pore size, 6.5 mm diameter transwell inserts (that were prewetted with chemotaxis buffer). These transwell inserts were placed in the wells of a 24 well plate containing 14 nM IL-8 in 500 µl of chemotaxis buffer. Cells were incubated for the indicated periods of time at 37°C and 5% CO2, after which inserts were discarded. Cells that migrated to the bottom well were fixed with 4% paraformaldehyde and counted using phase contrast microscopy. The number of cells that migrated to the lower wells was calculated by placing 10 µl aliquots on a hemocytometer and counting four fields in duplicate. Cell rate (as a proxy for migration velocity) was calculated as number of cells migrated per unit of time (specifically 5 min), as reported earlier in (Gomez-Cambronero et al., 2003).
Determination of PA intracellular content with a GFP-based Spo20 PA sensor
The PA sensor plasmid we used (courtesy of Dr Michael Frohman) takes advantage of a 40-amino-acid sequence found in Spo20 that binds to cell membrane phospholipids, particularly PA. Spo20 (Sporulation-specific protein 20) is a yeast protein required for the fusion of exocytic vesicles with the plasma membrane during yeast sporulation through its interactions with the SNARE complex (Nakanishi et al., 2004; Neiman, 1998; Neiman et al., 2000). Spo has an inhibitory region that sequesters the protein in the nucleus (Nakanishi et al., 2004; Weimbs et al., 1997) and a positive regulatory region that binds to phospholipids (including PA) in the cell membrane. This region contains an amphipathic helix with hydrophobic and positive charged faces. The initial report of a plasmid construction of the PA sensor in yeast, with the coding sequence of the PA binding domain (PABD) from Spo20p amino acids 51–91 was in (Nakanishi et al., 2004). Cloning of the PABD in pEGFPC1 vector (Clontech) leading to pEGFP-Spo20PABD-wt for use in microscopy of mammalian cells and generation of the pEGFP-Spo20PABD-L67P mutant was performed in (Zeniou-Meyer et al., 2007), where the authors show that PA is not present in cell membrane in resting cells but it becomes visible upon cell stimulation (PA is present in the nucleus of resting cells but that could be non-specific, non-PA related). Another use in mammalian cells with PLD inhibitor was reported in (Su et al., 2009) and the first use in leukocytes and also restriction enzyme digestion analysis was documented in (Frondorf et al., 2010)(8) indicating that the construct is cut by ApaLRI at the pUC origin (in addition, AgeI and DraII are one cut, PstI is a two-cut, and NcoI is a four-cut restriction enzyme).
Statistical analysis
Data are presented as mean±s.e.m. The difference between means was assessed by the single factor analysis of variance (ANOVA) test. Probability of P<0.05 indicated a significant difference.
Acknowledgments
We thank Dr Michael Frohman (SUNY Stony Brook) for providing us with the GFP-based Spo20 PA sensor.
Footnotes
Author contributions
J.G.-C. designed research; M.M. and K.M.H. performed research; M.M., K.M.H. and J.G.-C. analyzed data; and J.G.-C. wrote the paper.
Funding
This work was supported by the National Institutes of Health [grant number HL056653 to J.G.C.]; the Boonshoft School of Medicine (BSOM) [grant number 229102 to J.G.C.]; and the State of Ohio Research Incentive [grant number 668372 to J.G.C.]. Deposited in PMC for release after 12 months.
References
- Ambruso D. R., Knall C., Abell A. N., Panepinto J., Kurkchubasche A., Thurman G., Gonzalez-Aller C., Hiester A., deBoer M., Harbeck R. J. et al. (2000). Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA 97, 4654–4659 10.1073/pnas.080074897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznar S., Lacal J. C. (2001). Rho signals to cell growth and apoptosis. Cancer Lett. 165, 1–10 10.1016/S0304-3835(01)00412-8 [DOI] [PubMed] [Google Scholar]
- Benitah S. A., Valerón P. F., Lacal J. C. (2003). ROCK and nuclear factor-kappaB-dependent activation of cyclooxygenase-2 by Rho GTPases: effects on tumor growth and therapeutic consequences. Mol. Biol. Cell 14, 3041–3054 10.1091/mbc.E03-01-0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld J., Nalbant P., Bohl B. P., Pertz O., Hahn K. M., Bokoch G. M. (2007). GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell 12, 699–712 10.1016/j.devcel.2007.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzolli R., Shemon A. N., Fang M. Q., Hughes W. E. (2006). Phospholipid signalling through phospholipase D and phosphatidic acid. IUBMB Life 58, 457–461 10.1080/15216540600871142 [DOI] [PubMed] [Google Scholar]
- Chae H. D., Lee K. E., Williams D. A., Gu Y. (2008a). Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood 111, 2597–2605 10.1182/blood-2007-06-093237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae Y. C., Kim J. H., Kim K. L., Kim H. W., Lee H. Y., Heo W. D., Meyer T., Suh P. G., Ryu S. H. (2008b). Phospholipase D activity regulates integrin-mediated cell spreading and migration by inducing GTP-Rac translocation to the plasma membrane. Mol. Biol. Cell 19, 3111–3123 10.1091/mbc.E07-04-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. Y., Rex C. S., Babayan A. H., Kramár E. A., Lynch G., Gall C. M., Lauterborn J. C. (2010). Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J. Neurosci. 30, 10977–10984 10.1523/JNEUROSCI.1077-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockcroft S. (2009). Phosphatidic acid regulation of phosphatidylinositol 4-phosphate 5-kinases. Biochim. Biophys. Acta 1791, 905–912 10.1016/j.bbalip.2009.03.007 [DOI] [PubMed] [Google Scholar]
- Cornejo M. G., Boggon T. J., Mercher T. (2009). JAK3: a two-faced player in hematological disorders. Int. J. Biochem. Cell Biol. 41, 2376–2379 10.1016/j.biocel.2009.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrotte M., Chasserot-Golaz S., Huang P., Du G., Ktistakis N. T., Frohman M. A., Vitale N., Bader M. F., Grant N. J. (2006). Dynamics and function of phospholipase D and phosphatidic acid during phagocytosis. Traffic 7, 365–377 10.1111/j.1600-0854.2006.00389.x [DOI] [PubMed] [Google Scholar]
- Cougoule C., Hoshino S., Dart A., Lim J., Caron E. (2006). Dissociation of recruitment and activation of the small G-protein Rac during Fcgamma receptor-mediated phagocytosis. J. Biol. Chem. 281, 8756–8764 10.1074/jbc.M513731200 [DOI] [PubMed] [Google Scholar]
- Delon C., Manifava M., Wood E., Thompson D., Krugmann S., Pyne S., Ktistakis N. T. (2004). Sphingosine kinase 1 is an intracellular effector of phosphatidic acid. J. Biol. Chem. 279, 44763–44774 10.1074/jbc.M405771200 [DOI] [PubMed] [Google Scholar]
- Foster D. A. (2009). Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim. Biophys. Acta 1791, 949–955 10.1016/j.bbalip.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frondorf K., Henkels K. M., Frohman M. A., Gomez-Cambronero J. (2010). Phosphatidic acid is a leukocyte chemoattractant that acts through S6 kinase signaling. J. Biol. Chem. 285, 15837–15847 10.1074/jbc.M109.070524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishiro S. H., Tanimura S., Mure S., Kashimoto Y., Watanabe K., Kohno M. (2008). ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem. Biophys. Res. Commun. 368, 162–167 10.1016/j.bbrc.2008.01.066 [DOI] [PubMed] [Google Scholar]
- Fumagalli L., Zhang H., Baruzzi A., Lowell C. A., Berton G. (2007). The Src family kinases Hck and Fgr regulate neutrophil responses to N-formyl-methionyl-leucyl-phenylalanine. J. Immunol. 178, 3874–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cambronero J. (2011). The exquisite regulation of PLD2 by a wealth of interacting proteins: S6K, Grb2, Sos, WASp and Rac2 (and a surprise discovery: PLD2 is a GEF). Cell. Signal. 23, 1885–1895 10.1016/j.cellsig.2011.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cambronero J., Horn J., Paul C. C., Baumann M. A. (2003). Granulocyte-macrophage colony-stimulating factor is a chemoattractant cytokine for human neutrophils: involvement of the ribosomal p70 S6 kinase signaling pathway. J. Immunol. 171, 6846–6855 [DOI] [PubMed] [Google Scholar]
- Gu Y., Filippi M. D., Cancelas J. A., Siefring J. E., Williams E. P., Jasti A. C., Harris C. E., Lee A. W., Prabhakar R., Atkinson S. J. et al. (2003). Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 302, 445–449 10.1126/science.1088485 [DOI] [PubMed] [Google Scholar]
- Guo F., Cancelas J. A., Hildeman D., Williams D. A., Zheng Y. (2008). Rac GTPase isoforms Rac1 and Rac2 play a redundant and crucial role in T-cell development. Blood 112, 1767–1775 10.1182/blood-2008-01-132068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler L. C., Hemsath L., Fiegen D., Blumenstein L., Herbrand U., Stege P., Dvorsky R., Ahmadian M. R. (2006). Purification and biochemical properties of Rac1, 2, 3 and the splice variant Rac1b. Methods Enzymol. 406, 1–11 10.1016/S0076-6879(06)06001-0 [DOI] [PubMed] [Google Scholar]
- Henkels K. M., Peng H. J., Frondorf K., Gomez-Cambronero J. (2010). A comprehensive model that explains the regulation of phospholipase D2 activity by phosphorylation-dephosphorylation. Mol. Cell. Biol. 30, 2251–2263 10.1128/MCB.01239-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels K. M., Frondorf K., Gonzalez-Mejia M. E., Doseff A. L., Gomez-Cambronero J. (2011). IL-8-induced neutrophil chemotaxis is mediated by Janus kinase 3 (JAK3). FEBS Lett. 585, 159–166 10.1016/j.febslet.2010.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels K. M., Mahankali M., Gomez-Cambronero J. (2013). Increased cell growth due to a new lipase-GEF (Phospholipase D2) fastly acting on Ras. Cell. Signal. 25, 198–205.3 10.1016/j.cellsig.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J. H., Lee C. S., Hwang D., Ryu S. H. (2012). Understanding of the roles of phospholipase D and phosphatidic acid through their binding partners. Prog. Lipid Res. 51, 71–81 10.1016/j.plipres.2011.12.003 [DOI] [PubMed] [Google Scholar]
- Jeon H., Kwak D., Noh J., Lee M. N., Lee C. S., Suh P. G., Ryu S. H. (2011). Phospholipase D2 induces stress fiber formation through mediating nucleotide exchange for RhoA. Cell. Signal. 23, 1320–1326 10.1016/j.cellsig.2011.03.014 [DOI] [PubMed] [Google Scholar]
- Jose Lopez-Andreo M., Gomez-Fernandez J. C., Corbalan-Garcia S. (2003). The simultaneous production of phosphatidic acid and diacylglycerol is essential for the translocation of protein kinase Cepsilon to the plasma membrane in RBL-2H3 cells. Mol. Biol. Cell 14, 4885–4895 10.1091/mbc.E03-05-0295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanasis E., Wilson T. E. (2002). Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics 161, 1015–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson R., Pedersen E. D., Wang Z., Brakebusch C. (2009). Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta 1796, 91–98 [DOI] [PubMed] [Google Scholar]
- Kumar N., Mishra J., Narang V. S., Waters C. M. (2007). Janus kinase 3 regulates interleukin 2-induced mucosal wound repair through tyrosine phosphorylation of villin. J. Biol. Chem. 282, 30341–30345 10.1074/jbc.C600319200 [DOI] [PubMed] [Google Scholar]
- Kurooka T., Yamamoto Y., Takai Y., Sakisaka T. (2011). Dual regulation of RA-RhoGAP activity by phosphatidic acid and Rap1 during neurite outgrowth. J. Biol. Chem. 286, 6832–6843 10.1074/jbc.M110.183772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E., Marcucci M., Daniell L., Pypaert M., Weisz O. A., Ochoa G. C., Farsad K., Wenk M. R., De Camilli P. (2002). Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196 10.1126/science.1071362 [DOI] [PubMed] [Google Scholar]
- Lehman N., Di Fulvio M., McCray N., Campos I., Tabatabaian F., Gomez-Cambronero J. (2006). Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 108, 3564–3572 10.1182/blood-2006-02-005959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman N., Ledford B., Di Fulvio M., Frondorf K., McPhail L. C., Gomez-Cambronero J. (2007). Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. FASEB J. 21, 1075–1087 10.1096/fj.06-6652com [DOI] [PubMed] [Google Scholar]
- Liscovitch M., Czarny M., Fiucci G., Tang X. (2000). Phospholipase D: molecular and cell biology of a novel gene family. Biochem. J. 345, 401–415 10.1042/0264-6021:3450401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahankali M., Peng H. J., Cox D., Gomez-Cambronero J. (2011a). The mechanism of cell membrane ruffling relies on a phospholipase D2 (PLD2), Grb2 and Rac2 association. Cell. Signal. 23, 1291–1298 10.1016/j.cellsig.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahankali M., Peng H. J., Henkels K. M., Dinauer M. C., Gomez-Cambronero J. (2011b). Phospholipase D2 (PLD2) is a guanine nucleotide exchange factor (GEF) for the GTPase Rac2. Proc. Natl. Acad. Sci. USA 108, 19617–19622 10.1073/pnas.1114692108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahankali M., Henkels K. M., Alter G., Gomez-Cambronero J. (2012). Identification of the catalytic site of phospholipase D2 (PLD2) newly described guanine nucleotide exchange factor activity. J. Biol. Chem. 287, 41417–41431 10.1074/jbc.M112.383596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S., Nakanishi H., Sasaki T., Takai Y. (1996). A membrane-associated GDP/GTP exchange protein specific for Rho small GTP-binding protein - partial purification and characterization from rat brain. Oncogene 12, 915–920 [PubMed] [Google Scholar]
- Mishra J., Karanki S. S., Kumar N. (2012). Identification of molecular switch regulating interactions of Janus kinase 3 with cytoskeletal proteins. J. Biol. Chem. 287, 41386–41391 10.1074/jbc.C112.363507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H., de los Santos P., Neiman A. M. (2004). Positive and negative regulation of a SNARE protein by control of intracellular localization. Mol. Biol. Cell 15, 1802–1815 10.1091/mbc.E03-11-0798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman A. M. (1998). Prospore membrane formation defines a developmentally regulated branch of the secretory pathway in yeast. J. Cell Biol. 140, 29–37 10.1083/jcb.140.1.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman A. M., Katz L., Brennwald P. J. (2000). Identification of domains required for developmentally regulated SNARE function in Saccharomyces cerevisiae. Genetics 155, 1643–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton L. J., Zhang Q., Saqib K. M., Schrewe H., Macura K., Anderson K. E., Lindsley C. W., Brown H. A., Rudge S. A., Wakelam M. J. (2011). PLD1 rather than PLD2 regulates phorbol-ester-, adhesion-dependent and Fcgamma-receptor-stimulated ROS production in neutrophils. J. Cell Sci. 124, 1973–1983 10.1242/jcs.082008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai S. Y., Kim C., Williams D. A. (2010). Rac GTPases in human diseases. Dis. Markers 29, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H. J., Henkels K. M., Mahankali M., Dinauer M. C., Gomez-Cambronero J. (2011a). Evidence for two CRIB domains in phospholipase D2 (PLD2) that the enzyme uses to specifically bind to the small GTPase Rac2. J. Biol. Chem. 286, 16308–16320 10.1074/jbc.M110.206672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H. J., Henkels K. M., Mahankali M., Marchal C., Bubulya P., Dinauer M. C., Gomez-Cambronero J. (2011b). The dual effect of Rac2 on phospholipase D2 regulation that explains both the onset and termination of chemotaxis. Mol. Cell. Biol. 31, 2227–2240 10.1128/MCB.01348-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K., Rudge S. A., Frohman M. A., Morris A. J., Engebrecht J. (1995). Phospholipase D signaling is essential for meiosis. Proc. Natl. Acad. Sci. USA 92, 12151–12155 10.1073/pnas.92.26.12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudge S. A., Wakelam M. J. (2009). Inter-regulatory dynamics of phospholipase D and the actin cytoskeleton. Biochim. Biophys. Acta 1791, 856–861 10.1016/j.bbalip.2009.04.008 [DOI] [PubMed] [Google Scholar]
- Song J. G., Pfeffer L. M., Foster D. A. (1991). v-Src increases diacylglycerol levels via a type D phospholipase-mediated hydrolysis of phosphatidylcholine. Mol. Cell. Biol. 11, 4903–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stace C. L., Ktistakis N. T. (2006). Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 1761, 913–926 10.1016/j.bbalip.2006.03.006 [DOI] [PubMed] [Google Scholar]
- Stace C., Manifava M., Delon C., Coadwell J., Cockcroft S., Ktistakis N. T. (2008). PA binding of phosphatidylinositol 4-phosphate 5-kinase. Adv. Enzyme Regul. 48, 55–72 10.1016/j.advenzreg.2007.11.008 [DOI] [PubMed] [Google Scholar]
- Su W., Yeku O., Olepu S., Genna A., Park J. S., Ren H., Du G., Gelb M. H., Morris A. J., Frohman M. A. (2009). 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75, 437–446 10.1124/mol.108.053298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Dong X., Li Z., Smith J. D., Wu D. (2008). Lack of a significant role of P-Rex1, a major regulator of macrophage Rac1 activation and chemotaxis, in atherogenesis. Prostaglandins Other Lipid Mediat. 87, 9–13 10.1016/j.prostaglandins.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimbs T., Low S. H., Chapin S. J., Mostov K. E., Bucher P., Hofmann K. (1997). A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc. Natl. Acad. Sci. USA 94, 3046–3051 10.1073/pnas.94.7.3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamahashi Y., Saito Y., Murata-Kamiya N., Hatakeyama M. (2011). Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization. J. Biol. Chem. 286, 44576–44584 10.1074/jbc.M111.267021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi A., Kim C., Li S., Marchal C. C., Towe J., Atkinson S. J., Dinauer M. C. (2004). Rac2-deficient murine macrophages have selective defects in superoxide production and phagocytosis of opsonized particles. J. Immunol. 173, 5971–5979 [DOI] [PubMed] [Google Scholar]
- Yotova I. Y., Quan P., Leditznig N., Beer U., Wenzl R., Tschugguel W. (2011). Abnormal activation of Ras/Raf/MAPK and RhoA/ROCKII signalling pathways in eutopic endometrial stromal cells of patients with endometriosis. Hum. Reprod. 26, 885–897 10.1093/humrep/der010 [DOI] [PubMed] [Google Scholar]
- Zeniou-Meyer M., Zabari N., Ashery U., Chasserot-Golaz S., Haeberlé A. M., Demais V., Bailly Y., Gottfried I., Nakanishi H., Neiman A. M. et al. (2007). Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J. Biol. Chem. 282, 21746–21757 10.1074/jbc.M702968200 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Du G. (2009). Phosphatidic acid signaling regulation of Ras superfamily of small guanosine triphosphatases. Biochim. Biophys. Acta 1791, 850–855 10.1016/j.bbalip.2009.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]