

Summary
During epithelial-to-mesenchymal transition (EMT), tightly associated, polarized epithelial cells become individual mesenchymal cells capable of migrating. Here, we investigate the role of the transmembrane protein tetraspanin18 (Tspan18) in chick cranial neural crest EMT. Tspan18 mRNA is expressed in premigratory cranial neural crest cells, but is absent from actively migrating neural crest cells. Tspan18 knockdown leads to a concomitant loss of cadherin-6B (Cad6B) protein, whereas Cad6B protein persists when Tspan18 expression is extended. The temporal profile of Cad6B mRNA downregulation is unaffected in these embryos, which indicates that Tspan18 maintains Cad6B protein levels and reveals that Cad6B is regulated by post-translational mechanisms. Although downregulation of Tspan18 is necessary, it is not sufficient for neural crest migration: the timing of neural crest emigration, basal lamina breakdown and Cad7 upregulation proceed normally in Tspan18-deficient cells. This emphasizes the need for coordinated transcriptional and post-translational regulation of Cad6B during EMT and illustrates that Tspan18-antagonized remodeling of cell–cell adhesions is only one step in preparation for cranial neural crest migration. Unlike Cad6B, which is transcriptionally repressed by Snail2, Tspan18 expression is downstream of the winged-helix transcription factor FoxD3, providing a new transcriptional input into cranial neural crest EMT. Together, our data reveal post-translational regulation of Cad6B protein levels by Tspan18 that must be relieved by a FoxD3-dependent mechanism in order for cranial neural crest cells to migrate. These results offer new insight into the molecular mechanisms of cranial neural crest EMT and expand our understanding of tetraspanin function relevant to metastasis.
Key words: Epithelial-to-mesenchymal transition, Neural crest, Tetraspanin, Cadherin
Introduction
Epithelial-to-mesenchymal transition (EMT) is a complex, multiple step process during which tightly joined epithelial cells undergo dramatic changes in cell polarity, cell–cell adhesion, and cytoskeletal arrangement to become motile, invasive mesenchymal cells (Hay, 2005; Thiery and Sleeman, 2006). Many of the events that create the intricate adult body plan during embryogenesis require EMT, including the formation of migratory neural crest cells (Nieto, 2011; Polyak and Weinberg, 2009; Thiery et al., 2009). Neural crest cells are a unique developmental cell population that arises in the developing CNS but disperses throughout the embryo to form diverse cell types that are crucial for vertebrate organisms (LeDouarin and Kalcheim, 1999). Despite the importance of migration for neural crest development, our understanding of neural crest EMT is incomplete. In fact, a recent study using time lapse imaging to track neural crest cells as they emigrate has revealed that our current model of neural crest EMT is oversimplified and emphasizes the need for closer examination (Ahlstrom and Erickson, 2009).
During EMT, cell–cell adhesion is disrupted by the altered expression of cadherins, Ca2+-dependent adhesion molecules that are the main structural component of epithelial cell membrane structures called adherens junctions (AJs) (Meng and Takeichi, 2009; Oda and Takeichi, 2011). In the chick embryo, cadherins relevant to neural crest EMT vary along the rostrocaudal axis. Cranial neural folds express cadherin-6B (Cad6B) and E-cadherin (E-cad), but not N-cadherin (N-cad). At cranial levels, Cad6B downregulation is necessary for neural crest EMT, while E-cad expression persists in migratory cells (Coles et al., 2007; Dady et al., 2012; Nakagawa and Takeichi, 1995; Nakagawa and Takeichi, 1998). On the other hand, trunk neural crest cells express Cad6B and N-cad but not E-cad, and N-cad is lost during EMT but Cad6B persists during migration (Dady et al., 2012; Nakagawa and Takeichi, 1995; Park and Gumbiner, 2010; Shoval et al., 2007). Subsequently, the less adhesive, type II cadherin-7 (Cad7) is upregulated in migratory neural crest cells all along the axis (Chu et al., 2006; Nakagawa and Takeichi, 1995; Nakagawa and Takeichi, 1998). As a result of this AJ remodeling, neural crest cells transition from tightly adherent epithelial cells to mesenchymal cells capable of distinct adhesive interactions including chain formations and collective cell migration (Alfandari et al., 2003; Carmona-Fontaine et al., 2011; Friedl and Wolf, 2003; Kulesa and Fraser, 2000; Nishimura and Takeichi, 2009; Theveneau and Mayor, 2012).
While AJ remodeling is the first step in EMT (Thiery et al., 2009), the mechanisms that regulate cadherin levels in neural crest cells during this transition remain incomplete. Transcriptional downregulation of cadherin expression is crucial to EMT (Nieto, 2011), and in the cranial neural crest, the well-documented EMT transcription factor Snail2 directly binds and represses the Cad6B promoter (Taneyhill et al., 2007). Meanwhile, in the trunk neural tube, ectopic expression of the neural crest transcription factor FoxD3 leads to N-cad downregulation and elicits features of EMT (Cheung et al., 2005; Dottori et al., 2001; Kos et al., 2001). However, FoxD3 is not a classical EMT transcription factor (Yang and Weinberg, 2008) and a role for FoxD3 in cranial neural crest EMT has not been evaluated. Moreover, cadherins typically undergo post-translational regulation through processing, trafficking, or stabilization (Nishimura and Takeichi, 2009; Thiery et al., 2009). For example, N-cad levels in chick trunk neural crest cells are regulated by processing prior to EMT (Shoval et al., 2007), and cadherin-11 cleavage is required for Xenopus cranial neural crest migration (McCusker et al., 2009). However, post-translational regulation of cadherins during cranial neural crest EMT has not been determined.
Tetraspanins are transmembrane scaffolding proteins that have been implicated in the control of cell–cell adhesion and motility (Hemler, 2005). Tetraspanins organize membrane microdomains through intracellular interactions with other membrane proteins, including cadherins, integrins, membrane-bound proteases, and cell surface receptors (Levy and Shoham, 2005). By clustering proteins and facilitating their interactions, tetraspanins affect protein function (Yáñez-Mó et al., 2009). Despite evidence that tetraspanins promote cadherin-dependent cell–cell adhesion and act as metastasis suppressors (Abe et al., 2008; Chattopadhyay et al., 2003; Greco et al., 2010; Johnson et al., 2009; Tsai and Weissman, 2011; Zöller, 2009), the role of tetraspanins in preventing EMT, and in regulating cadherins during neural crest development, has not been investigated.
We identified Tetraspanin18 (Tspan18) in a screen for genes upregulated as a consequence of neural crest induction (Adams et al., 2008; Gammill and Bronner-Fraser, 2002). Tspan18 was originally cloned from chick spinal cord, however, its function was unknown (Perron and Bixby, 1999). Here we report that Tspan18 is expressed in chick cranial premigratory neural crest cells in a pattern similar to that of Cad6B. Our analysis of Tspan18 knockdown and overexpression reveals a novel role for Tspan18 in stabilizing Cad6B protein levels to antagonize EMT and subsequent neural crest migration. Strikingly, Tspan18 must be downregulated for cranial neural crest cells to migrate, but FoxD3, rather than Snail2, is required for this repression. Collectively, our data reveal post-translational regulation of Cad6B protein levels by Tspan18, and identify FoxD3 as a novel transcriptional regulator of cranial neural crest EMT, providing new insight into the complex regulation of neural crest EMT.
Results
Tspan18, like Cad6B, is expressed by premigratory neural crest cells, but is downregulated prior to migration
To determine whether Tspan18 is expressed at the correct time and place to regulate neural crest cadherins, we visualized Tspan18 mRNA localization in chick embryos by whole mount in situ hybridization. Between 5 and 8 somites, Tspan18 transcripts were apparent in the neural tube, head mesenchyme, epithelial somites, and developing vasculature (Fig. 1A–D). Transverse sections revealed that Tspan18 was abundantly expressed in cranial premigratory neural crest cells in the neural folds at 3 somites (supplementary material Fig. S1B) and the dorsal neural tube at 5, 6 and 7 somites (Fig. 1A′–C′, arrowheads). However Tspan18 was absent in the dorsal neural tube at 8 somites (Fig. 1D′, arrow), after cranial neural crest cells have emigrated. This expression pattern resembled that of the epithelial cell adhesion molecule Cad6B that must be downregulated in order for cranial neural crest cells to migrate (Fig. 1E–H) (Coles et al., 2007; Taneyhill et al., 2007). Interestingly, both Tspan18 and Cad6B persisted in the forebrain, which does not produce neural crest cells (Fig. 1D,H). Tspan18 expression was never apparent in neural crest cells migrating away from the neural tube, and was absent from HNK-1-positive neural crest cells in the head mesenchyme (supplementary material Fig. S1D,E) and branchial arches, a cranial target (supplementary material Fig. S1G,H). Tspan18 transcripts were not detectable by in situ hybridization in premigratory trunk neural crest cells at any stage examined (supplementary material Fig. S1F) (C.L.F., unpublished). Tspan18 mRNA expression persisted in the head mesenchyme, epithelial somites, and developing vasculature at both 10 and 16 somites (supplementary material Fig. S1). However, Tspan18 was downregulated in rostral somites that had dissociated into sclerotome and dermomyotome (supplementary material Fig. S1D,G). Thus, Tspan18 is expressed by premigratory cranial neural crest cells and generally correlates with epithelial and not mesenchymal cell types.
Fig. 1.
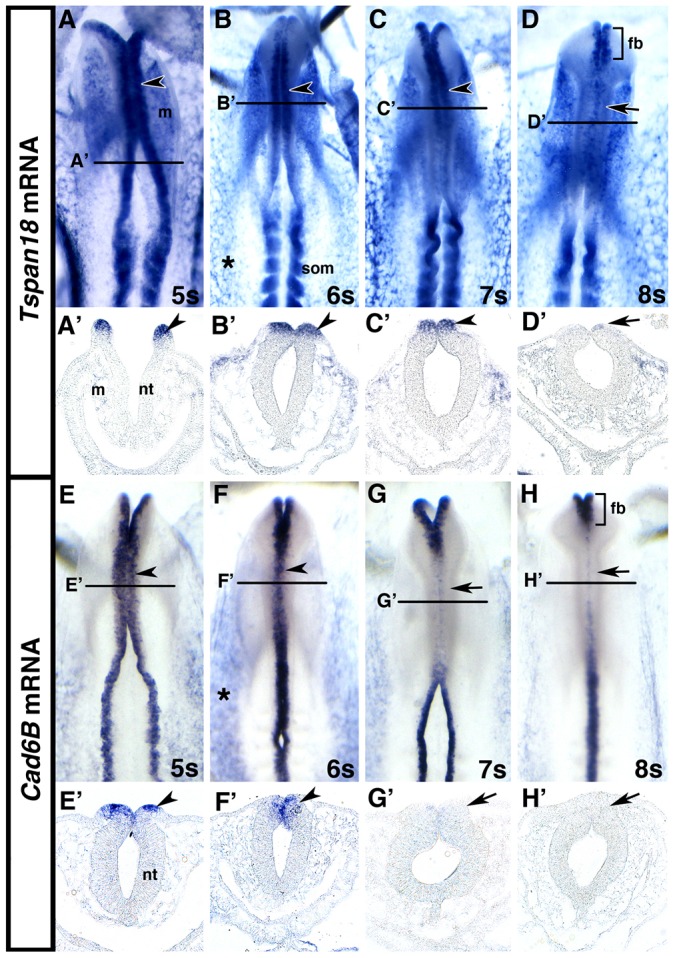
Tspan18 and Cad6B expression in cranial premigratory neural crest cells is downregulated prior to migration. (A–H) Whole mount in situ hybridization for Tspan18 (A–D) or Cad6B (E–H) in embryos at 5 somite (s; A,E), 6 somites (B,F), 7 somites (C,G) and 8 somites (D,H) (dorsal view, anterior embryo half). Tspan18 and Cad6B expression in the cranial dorsal neural tube (A–C,E,F; arrowheads) is downregulated (D,G,H; arrows) prior to neural crest migration. Tspan18 and Cad6B expression in the forebrain (brackets in D,H) persists during midbrain neural crest migration. Tspan18 and Cad6B are also expressed in the developing vasculature (asterisks in B,F) and Tspan18 is expressed in the head mesenchyme and epithelial somites. (A′–H′). Transverse sections at the levels indicated in A–H reveal Tspan18 (A′–D′) and Cad6B (E′–H′) expression in premigratory neural crest cells in the dorsal neural tube (arrowheads in A′–C′ and E′,F′) that is downregulated upon neural crest migration (D′,G′,H′; arrows). Tspan18 expression in the cranial mesenchyme is also apparent. fb, forebrain; m, mesenchyme; nt, neutral tube; som, somite.
Tspan18 knockdown leads to premature loss of Cad6B protein
Given that Tspan18 and Cad6B were similarly expressed (Fig. 1), and that tetraspanins are known to influence cadherin localization and function (Abe et al., 2008; Greco et al., 2010; Hemler, 2005), we postulated that Tspan18 would affect Cad6B. To investigate this possibility, we designed a FITC-tagged, antisense morpholino oligonucleotide (MO) to block Tspan18 translation (TS18MO). FITC-tagged standard control MO (ContMO) or TS18MO were electroporated into presumptive neural crest cells and Cad6B protein was visualized by immunofluorescence at 5–7 somites in whole mount and cryosections. Electroporation of ContMO had no effect on Cad6B protein levels (Fig. 2A–C; equivalent average fluorescence intensity on targeted and untargeted sides, O). However, when Tspan18 was knocked down, Cad6B protein was diminished in the forebrain (Fig. 2D; 81.3% of embryos; P = 2.2×10−4, n = 16), a region that continuously expressed Tspan18 and Cad6B (Fig. 1) and does not produce neural crest cells. Meanwhile, following TS18MO electroporation into the premigratory neural crest-containing midbrain, Cad6B protein was largely depleted (Fig. 2E; 68.7% of embryos; P = 6.5×10−4, n = 16) and the average intensity of Cad6B immunofluorescence on the targeted side was significantly reduced (Fig. 2O; n = 5; P = 8.0×10−3). Whole mount images of TS18MO-electroporated embryos further confirmed the reduction of Cad6B protein levels throughout the cranial premigratory neural crest (Fig. 2F). Embryos electroporated with a TS18MO containing a 5 base pair mismatch (mmTS18MO) did not exhibit this severe phenotype (7/9 unaffected; 2/9 mildly affected; C.L.F., unpublished), indicating the TS18MO concentration was in the effective and specific dose range (Moulton and Yan, 2008). Furthermore, these effects were specific to Cad6B as electroporation of TS18MO did not affect the level or localization of N-cad (supplementary material Fig. S2; n = 4) or E-cad (supplementary material Fig. S3; n = 16). These results indicate that Tspan18 is required to maintain premigratory Cad6B protein levels.
Fig. 2.

Tspan18 knockdown leads to a reduction in Cad6B protein levels with variable effects on Cad6B mRNA expression. (A–F) Transverse sections (A,B,D,E) and whole mount images (C,F; midbrain dorsal view) reveal that Cad6B immunofluorescence (A–F, red; A′–F′) is reduced on the targeted side of the neural tube (green) in the forebrain (arrowhead in D′) and midbrain (arrowheads in E′,F′) of embryos at 6–7 somites unilaterally electroporated with TS18MO (D–F) but not with ContMO (A–C). (G–M) Whole mount in situ hybridization (G,J; dorsal view, anterior embryo half) and transverse sections (H,I,K,L,M) reveal that Cad6B mRNA expression (purple) is reduced on the targeted side (green) compared with the untargeted side in the forebrain (arrowhead in K) but is unaffected in the midbrain of the majority of TS18MO-electroporated embryos (L). In 27.8% of embryos, Cad6B mRNA was reduced in neural folds electroporated with TS18MO (arrowhead in M). (N) Bar graph representing the frequency of electroporated embryos exhibiting affected versus unaffected Cad6B protein levels or mRNA expression. (O) Quantification of Cad6B immunofluorescence intensity comparing the MO-targeted (targ) and untargeted (untarg) sides of the midbrain neural tube in individual sections (n = 5). nt, neural tube; mb, midbrain; ns, not significant. Dotted lines in C′,F′ indicate embryo midline. Scale bars: 50 µm (A,C,D,F).
To verify that the loss of Cad6B protein in TS18MO-electroporated embryos was efficient and not due to off-target effects of TS18MO, we assessed efficacy and specificity of the knockdown phenotype, and also visualized cell proliferation and survival. Because commercially available human Tspan18 antibodies do not cross-react with chick Tspan18 and the chick Tspan18 antibody we raised was ineffective, we were unable to visualize endogenous Tspan18 knockdown. However, as TS18MO anneals to nucleotides +1 to +25 of the Tspan18 open reading frame, we could assess knockdown of C-terminal myc-tagged Tspan18 expressed from a DNA construct (pCIG-Tspan18MT). Co-electroporation of 1 µg/µl pCIG-Tspan18MT with TS18MO dramatically reduced translation of the tagged protein (supplementary material Fig. S4). To assess the specificity of TS18MO, we co-electroporated 1 µg/µl untagged pCIG-Tspan18 with TS18MO, and evaluated Cad6B protein. Driving additional Tspan18 expression in TS18MO electroporated cells rescued Cad6B protein levels on the targeted side of the neural tube (supplementary material Fig. S5; Cad6B protein levels reduced in 13.6% of TS18MO + Tspan18 coelectroporated embryos and 88.9% of TS18MO electroporated embryos; P = 4.9×10−4, n = 22). Moreover, embryos electroporated with either ContMO or TS18MO exhibited no statistically significant difference in the number of phospho-histone H3 (pH3) positive proliferating cells between the targeted and untargeted sides of the neural tube (supplementary material Fig. S6A–C; P = 0.27; n = 3). Likewise, no change in cell death, visualized by TUNEL staining, was apparent between the targeted and untargeted sides of the neural tube (supplementary material Fig. S6D–F; P = 0.42; n = 4). These data indicate that Tspan18 knockdown is efficient, and TS18MO phenotypes are not due to off-target effects or changes in cell number through proliferation or death.
Tspan18 knockdown has variable effects on Cad6B mRNA expression that may be a consequence of increased nuclear β-catenin
Tetraspanins can modulate membrane proteins that elicit intracellular signaling cascades and lead to indirect transcriptional changes in the nucleus (Berditchevski, 2001; Chairoungdua et al., 2010; Hemler, 2005; Huang et al., 2004). To determine whether loss of Cad6B protein following Tspan18 knockdown (Fig. 2E,F) was due to reduced Cad6B mRNA expression, we electroporated embryos with either ContMO or TS18MO and assessed Cad6B mRNA levels by whole mount in situ hybridization. Although Cad6B expression was unaffected in ContMO-electroporated embryos at 5s (Fig. 2G–I), Cad6B mRNA levels were reduced on the targeted side of the neural tube in the forebrain of TS18MO-electroporated embryos (Fig. 2N; P = 3.4×10−4, n = 16). Interestingly, midbrain Cad6B mRNA expression was unaffected in the majority of TS18MO-electroporated embryos, and the infrequent inhibition was not statistically significant (Fig. 2J–N; P = 0.14, n = 18). Thus, although Tspan18 knockdown can impact Cad6B gene expression, it does not account for the significant decrease in midbrain Cad6B protein levels in the majority of embryos (Fig. 2E′,F′,N). Furthermore, the differential effect in the forebrain and midbrain suggest that Cad6B mRNA expression is differentially regulated along the rostrocaudal axis.
To understand how a membrane protein like Tspan18 with no known signaling domains might affect Cad6B transcription in the nucleus (Fig. 2), we investigated two potential scenarios. We first assessed whether Tspan18 knockdown increased levels of the Cad6B transcriptional repressor Snail2 (Taneyhill et al., 2007). On the contrary, Snail2 protein levels were reduced in TS18MO-electroporated cells (supplementary material Fig. S7A–E), inconsistent with a reduction in Cad6B mRNA. Next we determined whether Tspan18 knockdown indirectly increased nuclear β-catenin levels. β-catenin interacts with cadherins in AJs (Meng and Takeichi, 2009) and additionally regulates gene expression, including repression of cadherin transcription (Huber et al., 1996; Jamora et al., 2003), as a downstream effector of the Wnt signaling pathway (Heuberger and Birchmeier, 2010). Because AJ disassembly can indirectly affect nuclear β-catenin levels (Heuberger and Birchmeier, 2010; Kam and Quaranta, 2009; Kuphal and Behrens, 2006; Onder et al., 2008; Orsulic et al., 1999; Shtutman et al., 2006), we reasoned that loss of Cad6B protein could elevate nuclear β-catenin. Interestingly, line scans revealed increased nuclear-localized β-catenin in TS18MO-targeted compared to untargeted neural fold cells (supplementary material Fig. S7F–H), providing a possible mechanism by which Tspan18 knockdown sometimes leads to reduced Cad6B transcription.
Tspan18 knockdown does not trigger neural crest migration
A previous report showed that Cad6B knockdown enhances neural crest migration (Coles et al., 2007). In turn, we reasoned that Tspan18 knockdown, which leads to premature loss of Cad6B protein (Fig. 2), would also promote neural crest migration. Specifically, we postulated that Tspan18-deficient embryos would exhibit precocious cranial neural crest migration. To investigate this possibility, neural crest cells electroporated with either ContMO or TS18MO were visualized by Sox10 in situ hybridization at 8–10 somites. Neural crest migration was unaffected in ContMO-electroporated embryos (Fig. 3A,B; n = 22). In 28.1% of TS18MO-electroporated embryos, Sox10-positive neural crest cells migrated noticeably farther on the targeted side compared to the untargeted side of the neural tube (Fig. 3D,F; n = 32). Unexpectedly, neural crest migration distance in the remaining 71.9% of TS18MO-electroporated embryos was similar to controls (Fig. 3C,E,G) and the enhanced migration phenotype we occasionally observed was not statistically significant (P = 0.11). Therefore, while Tspan18 knockdown leads to premature loss of Cad6B protein, in most embryos this is insufficient to stimulate cranial neural crest migration.
Fig. 3.

Tspan18 knockdown inconsistently enhances neural crest migration. (A–F) Embryos unilaterally electroporated with ContMO (A,B) or TS18MO (C–F) were processed by whole mount in situ hybridization for Sox10 at 8 or 9 somites (A,C,D, dorsal view, anterior embryo half; A′,C′,D′, construct targeting; B,E,F, midbrain transverse sections at the level indicated). Neural crest migration is unaffected by ContMO (A,B, arrows). TS18MO-electroporated neural crest cells migrated normally in most embryos (C,E, arrows), but was enhanced in 28.1% of embryos (D,F, arrowheads). (G) Bar graph represents the frequency of embryos with enhanced or normal migration.
Loss of Tspan18 does not interfere with the timing of subsequent steps in EMT
Although downregulation of Cad6B is required for cranial neural crest migration (Coles et al., 2007), AJ remodeling is not the only step in EMT; cells must also break down their restrictive basal lamina and express genes that establish their motility and mesenchymal characteristics (Hay, 2005). Thus, we reasoned that most Tspan18-deficient embryos did not migrate precociously because other steps in EMT were unaffected. Normally, cranial premigratory neural crest cells were enclosed within a basal lamina extending from the basal surface of the neural tube to the non-neural ectoderm (supplementary material Fig. S8A); at 8–10 somites, this basal lamina broke down, leaving a laminin-deficient void where HNK-1-positive migratory neural crest cells escaped the neural tube (supplementary material Fig. S8B,C) (Tosney, 1982). The same spatiotemporal pattern of laminin immunostaining was observed in embryos electroporated with either ContMO (Fig. 4A,B; n = 13) or TS18MO (Fig. 4C,D; n = 12). This indicates Tspan18 knockdown does not alter basal lamina integrity during cranial neural crest delamination.
Fig. 4.

Loss of Tspan18 does not alter basal lamina breakdown or the acquisition of mesenchymal character. Transverse midbrain sections of ContMO (A,B,E–G) or TS18MO (C,D,H–J) electroporated embryos immunostained for laminin (A–D, red; A′–D′) or Cad7 (E–J, red; E′–J′). (A–D) In all conditions, the intact basal lamina connecting the basal surface of the neural tube with the non-neural ectoderm at 7 somites (arrowheads in A′,C′) becomes discontinuous at 8 somites (arrows in B′,D′, insets in B′ and D′) in the region that cranial neural crest cells will exit the neural tube. (E–J) Cad7 protein is undetectable in early migrating neural crest cells at 8 somites (arrows in E′ and H′) but accumulates by 10 somites in both ContMO and TS18MO targeted and untargeted migratory cranial neural crest cells (arrowheads in F′,G′,I′,J′). G and J are the unelectroporated halves of embryos shown in F and G. nt, neural tube. Scale bars: 50 µm.
Once neural crest cells downregulate Cad6B, they activate expression of the mesenchymal cell adhesion molecule, Cad7 (Nakagawa and Takeichi, 1998). Although Cad7 protein was undetectable in early migrating cranial neural crest cells at 8 somites (supplementary material Fig. S8D), Cad7 protein was apparent in HNK-1-positive migratory neural crest cells by 10 somites (supplementary material Fig. S8E; staining was variable at 9 somites). This was consistent with the pattern of Cad7 expression in trunk migratory neural crest cells, although levels are higher in the trunk (supplementary material Fig. S8F) (Nakagawa and Takeichi, 1998). The timing of Cad7 protein accumulation was unaffected by Tspan18 knockdown: Cad7 was undetectable at 8 somites in migratory cranial neural crest cells electroporated with ContMO (Fig. 4E; n = 9) or TS18MO (Fig. 4H; n = 9), but present at 10 somites in cranial neural crest cells electroporated with either ContMO (Fig. 4F,G; n = 4) or TS18MO (Fig. 4I,J; n = 4). Altogether, these results suggest that Tspan18 regulates Cad6B protein levels specifically, and is not required for other steps in EMT like delamination and acquisition of mesenchymal fate.
Sustained expression of Tspan18 prevents neural crest migration
Tspan18 mRNA was absent in migratory neural crest cells (Fig. 1; supplementary material Fig. S1). To determine whether downregulation of Tspan18 was a prerequisite for cranial neural crest migration, we forced Tspan18 expression in neural crest cells past the stage it is normally lost. Electroporation of the chick expression construct pCIG (Megason and McMahon, 2002) at low (3 µg/µl) or high (5 µg/µl) concentrations did not affect migration of Sox10- or HNK-1-positive neural crest cells (Fig. 5A,B,E,G,K). In contrast, neural crest migration was inhibited on the targeted side of the neural tube in over 90% of embryos electroporated with pCIG-TS18 at either concentration [Fig. 5C,D,F,H,K, arrowheads; P = 5.3×10−6 (low); P = 1.9×10−6 (high)]. When analyzed by HNK-1 immunofluorescence, it was apparent that sometimes all neural crest migration was blocked (Fig. 5F, arrowhead) compared to the unelectroporated side (arrow). Meanwhile in other embryos, GFP-positive Tspan18-expressing cells were stopped near the dorsal neural tube (Fig. 5H, arrowhead), while GFP-negative neural crest cells migrated normally (Fig. 5H, arrow). This suggests that scoring migration without distinguishing between electroporated and unelectroporated cells (as in Fig. 5C,D,K) under-represents the severity of the phenotype. These effects were not due to changes in cell proliferation or death, as no differences were observed between targeted and untargeted sides of the neural tube in pCIG or pCIG-TS18 electroporated embryos (supplementary material Fig. S9). Together, these data indicate that Tspan18 expression is incompatible with migration.
Fig. 5.

Tspan18 prevents neural crest migration. (A–D) Whole mount in situ hybridization for Sox10 in embryos at 8–9 somites unilaterally electroporated with low (A,C; 3 µg/µl) or high (B,D; 5 µg/µl) concentrations of empty pCIG (A,B) or pCIG-TS18 (C,D; dorsal view, anterior embryo half; A′–D′, construct targeting). pCIG-TS18 impedes neural crest migration (arrowheads in C,D). Brightfield images (B″,D″) reveal an additional neural tube defect (D″, asterisk) in embryos electroporated with pCIG-TS18. (E–J) Transverse sections of embryos at 9–10 somites electroporated with 3 µg/µl pCIG (E,G,I) or pCIG-TS18 (F,H,J). pCIG-TS18-electroporated cells (F,H; green, arrowhead) clump near the neural fold, whereas pCIG-electroporated (E,G, green, arrow) and unelectroporated (F,H, arrows) HNK-1-positive (red) neural crest cells migrate normally. In contrast to pCIG-electroporated embryos (I), pCIG-TS18-electroporated embryos sometimes exhibit flat, open neural tubes (J, asterisk). (K) Bar graph represents the frequency of neural crest migration defects. (L) Bar graph represents the frequency of neural tube defects. nt, neural tube. Scale bars: 50 µm.
While sustained Tspan18 expression dorsally in the neural folds prevented neural crest migration, ectopic expression of Tspan18 in the ventral neural tube altered neural tube morphogenesis. In about 55% of 3 µg/µl pCIG-TS18-electroporated embryos (P = 5.9×10−7, n = 37) and 95% of, 5 µg/µl pCIG-TS18-electroporated embryos (P = 2.2×10−16, n = 29), the neuroepithelium on the targeted side remained flat and exhibited a severe neural tube defect (Fig. 5D″,J; asterisks). This phenotype was never observed in control embryos (Fig. 5B″,I,L). Even when a neural tube formed, more subtle effects on neural fold shape were sometimes apparent (see for example Fig. 5F,H). However, morphogenetic abnormalities did not account for neural crest migration defects since pCIG-TS18-electroporated neural folds produced migratory neural crest cells (Fig. 5F,H, arrow).
Cad6B protein persists following Tspan18 overexpression, whereas Cad6B mRNA is downregulated on time
Tspan18 knockdown reduced Cad6B protein levels (Fig. 2) while Tspan18 overexpression impeded neural crest migration (Fig. 5). Because Cad6B overexpression inhibits neural crest migration (Coles et al., 2007), we postulated that sustained Tspan18 expression prevented neural crest migration through effects on Cad6B. Cad6B protein was absent in the midbrain neural folds of 7-somite embryos electroporated with 3 µg/µl pCIG (Fig. 6A,D; n = 10). However, we detected Cad6B protein in 92.5% of embryos at 7–10 somites electroporated with 3 µg/µl pCIG-TS18 (Fig. 6B–D; P = 4.4×10−5, n = 14 embryos). Importantly, Cad6B protein was still restricted to the dorsal neural tube in pCIG-TS18 electroporated embryos, suggesting that Tspan18 overexpression maintains existing protein and does not induce de novo Cad6B translation in the neurectoderm (Fig. 6C).
Fig. 6.

Tspan18 overexpression maintains Cad6B protein, while Cad6B mRNA downregulates on schedule. Embryos unilaterally electroporated with empty pCIG (A,E,G) or pCIG-TS18 (B,C,F,H) were immunostained for Cad6B protein at 7 somites (A,B, wholemount midbrain dorsal view; C, transverse midbrain section) or processed by whole mount in situ hybridization for Cad6B mRNA (E–H, dorsal view, anterior embryo half; green in A–C,E′–H′, construct targeting). (A–C) Cad6B protein (A–C, red; A′–C′) is downregulated normally in pCIG electroporated embryos (A′) but maintained in the midbrain dorsal neural tube of pCIG-TS18 electroporated embryos (white arrowheads in B′,C′). Ectopic Cad6B protein was not observed in the ventral neural tube or unelectroporated side of the embryo (white arrows in C′). Dotted lines in A′,B′ indicate embryo midline. (D) Bar graph represents the number of embryos at 7–10 somites exhibiting normal downregulation versus maintenance of Cad6B protein. (E–H) Although pCIG-TS18-electroporated embryos at 5 somites exhibit Cad6B mRNA expression in the neural folds that is dispersed (F) and sometimes ectopic (arrowhead in F″) compared with pCIG electroporated embryos (E), Cad6B mRNA is downregulated at 7 somites with the same temporal profile in pCIG (G) and TS18MO-electroporated embryos (H). (I) Bar graph represents the number of embryos exhibiting ectopic Cad6B mRNA expression. Scale bars: 50 µm (A,C).
To determine whether maintenance of Cad6B protein in the neural folds reflected persistent Cad6B transcription, we visualized Cad6B mRNA by in situ hybridization after electroporation with either pCIG or pCIG-TS18. In unmanipulated embryos (Fig. 1E–G) or embryos electroporated with 3 µg/µl pCIG (Fig. 6E,G; n = 16), Cad6B mRNA was expressed at 5 somites, but downregulated by 7 somites. Likewise, Cad6B expression was present, albeit dispersed, on the targeted side of the neural tube in 5-somite embryos electroporated with pCIG-TS18 (Fig. 6F; n = 22). In turn, Cad6B mRNA expression in the midbrain was downregulated on time at 7 somites on both the pCIG-TS18 targeted and untargeted sides of the neural tube (Fig. 6H; n = 11). Because Cad6B mRNA downregulated normally following Tspan18 overexpression (Fig. 6H), but Cad6B protein persisted (Fig. 6B–D), these results indicate that Tspan18 maintains Cad6B protein levels directly, without indirectly modulating Cad6B mRNA expression. Moreover, expression of neural crest markers Cad6B (Fig. 6B–D,F,H), Sox10 (Fig. 5C,D,F), and HNK1 (Fig. 5F,H) indicate that neural crest cells form following Tspan18 overexpression, but simply fail to migrate.
Interestingly, 31.6% of pCIG-TS18 targeted embryos exhibited ectopic Cad6B mRNA expression in the adjacent non-neural ectoderm (arrowhead in Fig. 6F″,I; P = 4.3×10−2, n = 19). Since pCIG-TS18 embryos are electroporated at HH4+, it is possible that the ectopic Cad6B expressing cells reflect incomplete morphogenetic movements and displacement of neural crest precursors that are specified during gastrulation (Basch et al., 2006). Alternatively, if Cad6B mRNA expression is induced, only the non-neural ectoderm is competent to respond, as ectopic Cad6B mRNA was never observed in the neural tube. In either case, we cannot detect ectopic Cad6B protein, indicating it is below the limits of detection, some factor is missing, and/or Cad6B translation is repressed outside the neural folds.
FoxD3 negatively regulates Tspan18 expression
Downregulation of Tspan18 was required for neural crest cells to migrate (Fig. 5). Thus, characterizing the transcriptional regulation of Tspan18 is essential to understanding how neural crest cells prepare for migration. During cranial neural crest EMT, Snail2 represses transcription of Cad6B (Taneyhill et al., 2007). Given the role of Tspan18 in stabilizing Cad6B protein levels (Fig. 6), we investigated whether Tspan18 expression, like Cad6B, was repressed by Snail2. However, real time qRT-PCR analysis of Tspan18 mRNA expression after knockdown of Snail2 suggests that Tspan18 is not a Snail2 target (L.A. Taneyhill, personal communication), leading us to investigate other possible regulators of Tspan18 expression.
Previous studies have shown that ectopic expression of the winged helix transcription factor FoxD3 in trunk neural tube leads to changes in cell–cell adhesion and promotes neural crest delamination (Cheung et al., 2005; Dottori et al., 2001; Kos et al., 2001); thus FoxD3 was also a candidate to regulate Tspan18. To investigate this possibility, we examined the effect of FoxD3 overexpression on Tspan18 mRNA expression at 5–8 somites. As expected, Tspan18 mRNA levels were unaffected by electroporation with 5 µg/µl of the chick expression construct pMES (Swartz et al., 2001) (Fig. 7A; n = 16). However, Tspan18 mRNA expression was dramatically reduced on the targeted side of the dorsal neural tube in 76.5% of embryos electroporated with 5 µg/µl pMES-FoxD3 (Fig. 7C; P = 5.1×10−6, n = 17). Transverse sections of the midbrain confirmed this observation (compare arrowhead in Fig. 7D and arrow in Fig. 7B) and furthermore revealed a neural tube defect similar to that of pCIG-TS18 electroporated embryos (Fig. 5D″,J). These results suggest that Tspan18 lies downstream of FoxD3.
Fig. 7.

FoxD3 negatively regulates Tspan18. Embryos unilaterally electroporated with 5 µg/µl of empty pMES (A,B) or pMES-FoxD3 (C,D), or with ContMO (F,G) or FoxD3MO (H,I) were processed by whole mount in situ hybridization for Tspan18 at 7 somites (A–D) or 9 somites (F–I). A,C,F,H shows dorsal view, anterior embryo half; A′,C′,F′,H′, construct targeting; B,D,G,I, midbrain transverse sections. (A–D) Tspan18 expression in the neural folds (A,B, arrow) is drastically reduced on the targeted side of the neural tube in pMES-FoxD3 electroporated embryos (C,D, arrowheads). (E) Bar graph represents the frequency of pMES-FoxD3-electroporated embryos exhibiting reduced Tspan18 mRNA expression. (F–I) Tspan18 expression is normally downregulated at 9 somites (F,G, arrow), but is maintained on the targeted side of FoxD3MO-electroporated embryos (H,I, arrowheads). Neural crest migration was inhibited in FoxD3MO-electroporated embryos (I′, arrowhead). (J) Bar graph represents the frequency of FoxD3MO-electroporated embryos with persistent Tspan18 expression.
To confirm that FoxD3 negatively impacts Tspan18 expression, we also determined whether FoxD3 was necessary for Tspan18 mRNA downregulation. To this end, we electroporated neural crest precursors with the previously characterized FoxD3 MO (Kos et al., 2001) and analyzed expression of Tspan18 mRNA at 9–10 somites. While Tspan18 mRNA was absent from the midbrain of ContMO electroporated embryos (Fig. 7F,G), Tspan18 transcripts persisted following FoxD3 knockdown (Fig. 7H,I; P = 9.8×10−4, n = 14). Moreover, these FoxD3-deficient, Tspan18-expressing cells failed to migrate (Fig. 7I′) (Kos et al., 2001). This indicates that FoxD3 is required for Tspan18 transcriptional downregulation, and moreover, suggests that persistent endogenous Tspan18 expression, like Tspan18 overexpression (Fig. 5), prevents neural crest migration.
Discussion
Although neural crest cells must undergo EMT to become motile and AJ remodeling is a crucial step in this process, a complete understanding of cadherin regulation in neural crest cells is lacking. In this study, we have revealed Tspan18 as a novel post-translational regulator of Cad6B in cranial neural crest cells. We show that Tspan18 is abundantly expressed in premigratory but not in migratory cranial neural crest cells, and that Tspan18 downregulation is required for cranial neural crest cells to migrate. Because Tspan18 overexpression maintains, and Tspan18 knockdown leads to premature loss of Cad6B protein without affecting Cad6B mRNA expression, we conclude that Tspan18 post-translationally regulates Cad6B-dependent cell adhesion to antagonize cranial neural crest EMT. Snail2 does not regulate Tspan18, as it does Cad6B; rather Tspan18 is downstream of FoxD3. Taken together, our data suggest that Tspan18 post-translationally regulates Cad6B protein levels and must be downregulated by FoxD3 during neural crest EMT in a pathway parallel to Snail2-dependent Cad6B transcriptional regulation.
Tspan18 prevents neural crest migration
Tspan18 overexpression blocks neural crest migration (Fig. 5C,D,F,H) and affects neural tube morphogenesis (Fig. 5D″,J), however, these effects are unlikely to be interdependent. First of all, migration defects are more common than neural tube defects and there is not a one-to-one correlation between these phenotypes (Fig. 5K,L). Second, abnormal neural folds produce migratory neural crest cells (Fig. 5H, arrow); it is the expression of Tspan18 that prevents migration (Fig. 5H, arrowhead). Third, generally speaking, neural crest migration does not require neural tube closure either developmentally (e.g. mouse cranial neural tube) or experimentally (C.L.F. and L.S.G., unpublished observations). Thus, persistent Tspan18 expression prevents neural crest migration. Since Tspan18 is not normally expressed in the ventral neural tube or neural folds past 7 somites (Fig. 1), Tspan18 does not normally regulate neural tube closure; however, when misexpressed, Tspan18 must affect proteins involved in neurepithelial morphogenesis. These likely include cadherins, as N-cadherin gain- or loss-of-function causes similar defects (Detrick et al., 1990; Fujimori et al., 1990; Nandadasa et al., 2009) and the dynamic regulation of AJ positioning is required for epithelial folding during Drosophila gastrulation (Wang et al., 2012).
Tspan18 and Cad6B expression persist in the forebrain even after they are downregulated in the midbrain (Fig. 1D,H). As the rostral-most neural tube does not produce neural crest cells (Creuzet et al., 2005), it is possible that the expression of these epithelial markers is one factor that prevents neural crest migration from this region. Interestingly, Tspan18 knockdown led to a statistically significant reduction of Cad6B mRNA expression in forebrain neural folds, despite Cad6B mRNA being unaffected in the midbrain of the majority of embryos (Fig. 2N). Meanwhile, Cad6B protein is more effectively diminished by Tspan18 knockdown in the midbrain (Fig. 2E) than the forebrain (Fig. 2D′). This suggests regional complexity in Cad6B transcriptional and post-translational regulation, and the regulatory relationship between Tspan18 and Cad6B.
Tspan18 post-translationally maintains Cad6B protein
Tspan18 loss-of-function impacts both Cad6B mRNA and protein levels, however our results suggest these effects are separable. First, Tspan18 knockdown has a significant effect on Cad6B protein and minimal effects on Cad6B mRNA levels in the midbrain (Fig. 2). Second, Cad6B protein persists when Tspan18 is overexpressed despite temporally normal downregulation of Cad6B mRNA (Fig. 6H). Third, when Tspan18 is overexpressed, Cad6B protein persists only in the dorsal neural tube where it is normally expressed (Fig. 6C) indicating Tspan18 affects existing Cad6B protein rather than eliciting de novo expression. Altogether, these results suggest that Tspan18 maintains Cad6B protein levels post-translationally. Transcriptionally, we propose that Tspan18 knockdown leads to Cad6B mRNA downregulation as a secondary consequence of AJ remodeling that results in increased nuclear β-catenin (supplementary material Fig. S7F–H).
The ability of Tspan18 to affect Cad6B post-translationally is consistent with existing knowledge of tetraspanins and cadherins. For example, in human cancer cells, the tetraspanin CD82 promotes E-cad-dependent cell–cell adhesion by stabilizing E-cad protein–protein interactions without markedly altering E-cad protein levels (Abe et al., 2008). However, in this study, CD82 was concluded to promote epithelial barrier formation to contain metastatic cells, rather than to antagonize EMT (Abe et al., 2008). Nevertheless, post-translational regulation of cadherins during EMT is not unprecedented. N-cad protein is cleared by processing during trunk neural crest EMT (Shoval et al., 2007). Moreover, during mouse gastrulation, p38 destabilizes and EPB41L5 alters the localization of E-cad, acting in conjunction with E-cad transcriptional repression to enable EMT (Hirano et al., 2008; Zohn et al., 2006). Thus, coupled transcriptional and post-translational regulation of cadherin levels appears to be a common mechanism to tightly regulate AJ remodeling and cell adhesion during dynamic, rapid events like neural crest migration and gastrulation (Thiery et al., 2009).
The means by which Tspan18 maintains Cad6B protein levels is unclear. One possibility is that Cad6B is processed, and Tpsan18 protects Cad6B from processing enzymes to stabilize it at the membrane. Tetraspanins can associate with membrane proteases such as ADAM metalloproteases (Yáñez-Mó et al., 2011), and Tspan18 could alter ADAM-dependent Cad6B processing. N-cad is processed in trunk neural crest cells by ADAM10 (Shoval et al., 2007), and cadherin-11 cleavage regulates Xenopus cranial neural crest migration (McCusker et al., 2009). However, Cad6B processing has not been defined, precluding evaluation of this scenario. Another possibility is that Tspan18 protects Cad6B from degradation. In either case, Tspan18 could interact with Cad6B directly, or it could promote the formation of a complex that stabilizes Cad6B. Evaluating these mechanisms are important future experiments.
Tspan18 knockdown does not ensure premature neural crest migration
Loss of Tspan18 does not promote early neural crest migration (Fig. 3) despite a consistent reduction in Cad6B protein levels (Fig. 2), an event that was previously shown to augment neural crest migration (Coles et al., 2007). There are several likely explanations for this. First and foremost, Tspan18 knockdown may deplete Cad6B protein, but in a majority of embryos, Cad6B mRNA persists in the midbrain (Fig. 2N). Thus, in contrast to Cad6B knockdown (Coles et al., 2007), new Cad6B protein will continue to be translated, barely detectable by immunofluorescence (see Fig. 2O) but presumably sufficient to maintain adhesion. Only in the minority of cases where Cad6B mRNA is also lost in the midbrain would an effect on migration be anticipated. Incidentally, the frequency of embryos in which Cad6B mRNA is downregulated in the midbrain (Fig. 2N) is roughly equivalent to the frequency of embryos with precocious migration (Fig. 3G). Unfortunately we cannot visualize Cad6B mRNA levels in premigratory neural crest cells and subsequently assay those same cells for precocious migration in order to test this correlation directly.
It is unclear why only some Tspan18 morphant embryos show the more dramatic phenotype. One possibility is that severely affected embryos are those with neural folds uniformly targeted with high levels of MO. Tspan18 knockdown is efficient (supplementary material Fig. S4), however, MO electroporation is by nature mosaic. Given that cadherins interact homophilically, when MO targeting is variable in the neural fold, cells containing low levels of TS18MO and residual Cad6B could stabilize AJs in adjacent, well-targeted cells, prevent β-catenin nuclear translocation (supplementary material Fig. S7F–H), and thus reduce the penetrance of the enhanced migration phenotype.
Another reason Tspan18 knockdown may not reliably elicit precocious migration is that loss of an epithelial cadherin is not the sole feature of EMT; to emigrate from the neural tube, the basal lamina must also break down, and neural crest cells must remodel their AJs to include cadherins that allow mesenchymal cell–cell adhesion during collective cell migration (Friedl and Wolf, 2003; Park and Gumbiner, 2010; Theveneau and Mayor, 2012). Although Cad6B protein is lost prematurely following Tspan18 knockdown, basal lamina break down and upregulation of the mesenchymal cadherin Cad7 still occur on the proper developmental timeline (Fig. 4). As neural crest cells will not invade an intact basal lamina (Erickson, 1987), this likely prevents precocious neural crest migration. In this respect, Tspan18 and Cad6B knockdown are similar: although Cad6B knockdown leads to increased numbers of migratory neural crest cells, it has minimal effects on the extent of migration away from the neural tube (Coles et al., 2007), supporting this interpretation. The diversity and non-linearity of cellular behaviors during trunk neural crest emigration also suggest that it is not possible to change the time course of EMT by disrupting any one individual feature (Ahlstrom and Erickson, 2009).
Finally, it is also possible that Tspan18 knockdown does not elicit premature cranial neural crest migration because of genetic redundancy. The tetraspanin family is large, including 33 members with broad, overlapping expression domains, and mutants often exhibit only minor defects (Rubinstein, 2011). In the chick embryo, Tspan18 is coexpressed with Tspan4 and CD9/Tspan29 during later development of the spinal cord, thus it is possible other tetraspanins compensate for Tspan18 loss of function during cranial neural crest development (Perron and Bixby, 1999). However, this would not explain why neural crest cells occasionally do migrate prematurely (Fig. 3), thus, it would seem that other mechanistic explanations are more likely.
Tspan18 is downstream of FoxD3: a novel transcriptional input into cranial neural crest EMT
While they have similar temporal expression patterns (Fig. 1), and although Tspan18 maintains Cad6B protein levels (Fig. 6), transcriptional regulation of Tspan18 and Cad6B is distinct: Cad6B is directly repressed by Snail2 in cranial neural crest cells (Taneyhill et al., 2007), and Tspan18 is downstream of FoxD3 (Fig. 7). Although its role in regulating neural crest multipotency and cell fate is well known (Kos et al., 2001; Lister et al., 2006; Mundell and Labosky, 2011; Sasai et al., 2001), a role for FoxD3 in EMT is not unexpected. When overexpressed in trunk neural tube cells, FoxD3 promotes cell adhesive changes associated with EMT, leading to loss of N-cad expression and upregulation of Cad7 and β1-integrin in a Snail2-independent fashion (Cheung et al., 2005; Dottori et al., 2001). As downregulation of Tspan18 is necessary for cranial neural crest cells to migrate (Figs 5,7), our results show that FoxD3 is an indirect transcriptional regulator of cranial crest EMT that negatively affects Tspan18 expression. Thus the action of Snail2 and FoxD3 converge to impact Cad6B transcriptionally and post-translationally (through downregulation of Tspan18), resulting in AJ remodeling during cranial neural crest migration. As only cranial neural crest cells express detectable Tspan18 (Fig. 1), and as Snail2 transcriptionally downregulates Cad6B prior to cranial but not trunk neural crest migration (Park and Gumbiner, 2010; Taneyhill et al., 2007), it appears that this coordinated transcriptional and postranslational regulation of Cad6B is a cranial-specific mechanism. Additional experiments are currently underway to further characterize the transcriptional regulation of Tspan18 by FoxD3.
A new model for cranial neural crest EMT
Our study of Tspan18 provides new insight into the unique molecular mechanisms of cranial neural crest EMT (Fig. 8). Premigratory cranial neural crest cells express Tspan18 and Cad6B, two markers that distinguish their epithelial character. Our data indicate that, in premigratory cranial neural crest cells, Tspan18 maintains Cad6B-dependent AJs to antagonize EMT. During EMT, Snail2 downregulates Cad6B transcription (Taneyhill et al., 2007). In parallel, we demonstrate that FoxD3 downregulates Tspan18 expression, alleviating Tspan18-dependent Cad6B stabilization. Together, Cad6B transcriptional repression and post-translational destabilization result in AJ remodeling that changes cell adhesion and allows for mesenchymal transition and subsequent neural crest migration (Fig. 8A). Tspan18 downregulation is necessary for EMT, as prolonging endogenous Tspan18 expression (Fig. 7) or driving exogenous Tspan18 expression in the neural folds (Fig. 5) maintains Cad6B protein levels and/or prevents migration (Fig. 8B). However, Tspan18 downregulation is not sufficient for migration: loss of Tspan18 (and destabilization of Cad6B protein) does not alter Cad6B transcription and fails to trigger neural crest migration in the majority of cases (Fig. 8C). This underscores the importance of coordinated transcriptional and post-translational regulation of cadherin expression during EMT, and emphasizes that while cadherin switching and AJ remodeling is a critical step in EMT, it is not the only step in the process of producing migratory neural crest cells (Ahlstrom and Erickson, 2009; Lim and Thiery, 2012). In summary, post-translational stabilization of Cad6B by Tspan18 that must be downregulated for cells to undergo EMT is a novel mechanism that provides new insight into the developmental function of tetraspanins and has important implications for cancer metastasis.
Fig. 8.
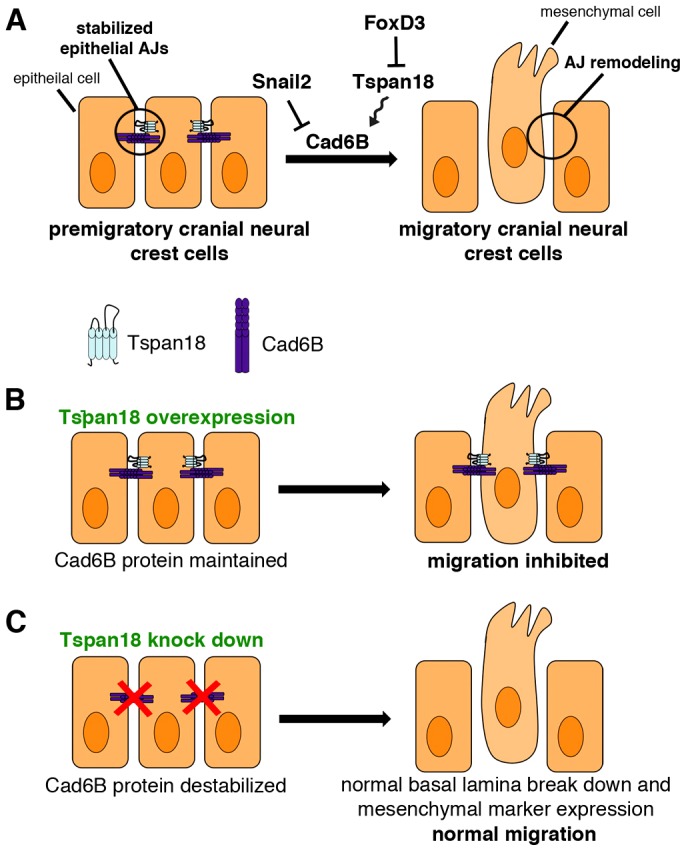
Role of Tspan18 in cranial neural crest EMT. (A) Epithelial, premigratory cranial neural crest cells are tightly joined by Tspan18-stabilized, Cad6B-containing adherens junctions (AJs). During EMT, neural crest cells transcriptionally downregulate Cad6B via Snail2 and post-translationally destabilize Cad6B by FoxD3-dependent downregulation of Tspan18 to remodel their AJs in preparation for migration. (B) Evidence for the role of Tspan18. Sustained expression of Tspan18 maintains Cad6B protein and promotes Cad6B-dependent AJs, inhibiting neural crest migration. (C) By contrast, loss of Tspan18 results in premature downregulation of Cad6B protein but normal cranial neural crest migration, presumably reflecting a need for coordinated transcriptional and post-translational regulation of Cad6B and temporally normal delamination and expression of mesenchymal markers. Thus, loss of Tspan18 is necessary, but not sufficient, for cranial neural crest migration.
Materials and Methods
Embryos
Fertile chicken embryos were incubated in a humidified incubator (G. Q. F. Manufacturing; Savannah, GA) at 37–38°C. Embryos were staged according to Hamburger and Hamilton (Hamburger and Hamilton, 1951) or by counting somite pairs.
Morpholino design, DNA constructs and electroporation
FITC-tagged, antisense morpholinos (MO) synthesized by GeneTools, LLC (Philomath, OR) included a Tspan18 translation blocking MO (TS18MO: 5′-TGCAGCTCAGACAGTCTCCCTCCAT-3′), a 5 base pair mismatch TS18MO (mmTS18MO: 5′-TGgAcCTCAcACAcTCTgCCTCCAT-3′), a FoxD3 translation blocking MO (FoxD3 MO: 5′-CGCTGCCGCCGCCCGATAGAGTCAT-3′; (Kos et al., 2001)) and a standard control MO (ContMO: 5′-CCTCTTACCTCAGTTACAATTTATA-3′). To produce pCIG-TS18 and pCIG-TS18MT, full length Tspan18 without or with 6×myc tags was cloned by PCR into the GFP bicistronic expression plasmid pCIG (Megason and McMahon, 2002) using 5–10-somite chick cDNA (RNA prepared by TRIZOL extraction, cDNA synthesized with Superscript III; Life Technologies, Carlsbad, CA) as a template. To produce pMES-FoxD3, full length FoxD3 (Kos et al., 2001) was cloned into the EcoRI site of the GFP bicistronic expression plasmid pMES (Swartz et al., 2001). FITC-tagged morpholinos at 750 µM (with 0.2 µg/µl pCS2+MycTag DNA as carrier) or DNA at the indicated concentrations were unilaterally electroporated into the presumptive neural crest at Hamburger and Hamilton (HH) stage 4+ as previously described (Gammill and Krull, 2011). After electroporation, embryos were incubated until the desired stages and fixed in 4% paraformaldehyde at room temperature for 15 minutes (Snail2 experiments) or 1 hour (all other experiments) before washing with PBS + 0.1% Tween. FITC or GFP targeting was verified by fluorescent microscopy before embryos were either immediately used for whole mount immunofluorescence, dehydrated into methanol and stored at −20°C for in situ hybridization, or embedded and prepared for sectioning.
In situ hybridization
Whole-mount in situ hybridization was performed as previously described (Wilkinson, 1992). Digoxigenin-labeled RNA probes were transcribed from the following templates: cTspan18 (Adams et al., 2008), cCad6B (Gammill and Bronner-Fraser, 2002), and cSox10 (Cheng et al., 2000). After processing, embryos were imaged in whole mount using a Zeiss Discovery V8 stereoscope, then embedded in gelatin, sectioned using a Leica CM1900 cryostat at 12–18 µm, and imaged on a Zeiss AxioImager A1 with a Zeiss AxioCam MRc5 digital camera and Axiovision software.
Immunohistochemistry
Immunofluorescence was performed as previously described (Roffers-Agarwal et al., 2012) with the following antibodies: anti-HNK-1 (ATTC, Manassas, VA; 1∶25), anti-Cad6B (DSHB, Iowa City, IA; clone CCD6B-1; 1∶100 (Nakagawa and Takeichi, 1998)), anti-laminin (DSHB clone 31 or 31-2; 1∶50), anti-Cad7 (DSHB clone CCD7-1; 1∶50 (Nakagawa and Takeichi, 1998)), anti-N-cad (DSHB clone 6B3; 1∶100), anti-E-cad (BD Transduction Laboratories; 1∶250 (Dady et al., 2012)), anti-Snail2 (DSHB clone 62.1E6; 1∶100), anti-β-catenin (Ctnnb1, BD Transduction Laboratories; 1∶200 (Matson et al., 2011)) and anti-phosphohistone H3 (pH 3; Millipore; Billerica, MA; 1∶250). Primary antibody was detected using donkey anti-mouse or donkey anti-rat secondary antibodies at 1∶250 (Jackson Labs; West Grove, PA). Slides were mounted in PermaFluor (Thermo Fisher Scientific; Waltham, MA) containing 1 µg/ml DAPI and imaged on a Zeiss LSM 710 laser scanning confocal microscope. Images were processed in Photoshop (Adobe).
Analysis
The statistical significance of the observed phenotypes was calculated by Fisher's exact test in R (R Development Core Team, 2012). For all expression analyses, staining on the targeted and untargeted sides of the neural tube were compared in individual images to rule out any difference in exposure time or staining efficiency between images/sections. To quantify Cad6B fluorescence intensity, the mean gray value was calculated in ImageJ for the Cad6B expression domain of three sections per embryo (n = 5 embryos). Statistical evaluation of intensity measurements was performed using SPSS 16.0 for Windows (Chicago, IL) using paired comparisons in a MANOVA. P<0.05 was considered statistically significant. Error bars indicate the standard error of the mean.
Supplementary Material
Acknowledgments
We are grateful to Yi-Chuan Cheng, Sean Megason, Cathy Krull and Carol Erickson for the kind gift of plasmids. We thank Lisa Taneyhill for technical advice and collaboration. Many thanks to the members of the Gammill lab for their input and support and Wuming Gong and Rebecca Pulver for their help with statistical analysis. The monoclonal antibodies used in this study, developed by M. Takeichi and S. Nakagawa (Cad6B, N-Cad, Cad7) and D. Fambrough (laminin), were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA.
Footnotes
Author contributions
L.S.G. and C.L.F. conceived and designed the experiments. C.L.F. performed the experiments. L.S.G. and C.L.F. analyzed the data and wrote the paper.
Funding
This work was supported by the National Institutes of Health [grant numbers F31 NRSA GM087951 to C.L.F., K22 DE015309 to L.S.G.]; and by a University of Minnesota Grant-in-Aid. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.120915/-/DC1
References
- Abe M., Sugiura T., Takahashi M., Ishii K., Shimoda M., Shirasuna K. (2008). A novel function of CD82/KAI-1 on E-cadherin-mediated homophilic cellular adhesion of cancer cells. Cancer Lett. 266, 163–170 10.1016/j.canlet.2008.02.058 [DOI] [PubMed] [Google Scholar]
- Adams M. S., Gammill L. S., Bronner-Fraser M. (2008). Discovery of transcription factors and other candidate regulators of neural crest development. Dev. Dyn. 237, 1021–1033 10.1002/dvdy.21513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlstrom J. D., Erickson C. A. (2009). The neural crest epithelial-mesenchymal transition in 4D: a ‘tail’ of multiple non-obligatory cellular mechanisms. Development 136, 1801–1812 10.1242/dev.034785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfandari D., Cousin H., Gaultier A., Hoffstrom B. G., DeSimone D. W. (2003). Integrin alpha5beta1 supports the migration of Xenopus cranial neural crest on fibronectin. Dev. Biol. 260, 449–464 10.1016/S0012-1606(03)00277-X [DOI] [PubMed] [Google Scholar]
- Basch M. L., Bronner-Fraser M., García-Castro M. I. (2006). Specification of the neural crest occurs during gastrulation and requires Pax7. Nature 441, 218–222 10.1038/nature04684 [DOI] [PubMed] [Google Scholar]
- Berditchevski F. (2001). Complexes of tetraspanins with integrins: more than meets the eye. J. Cell Sci. 114, 4143–4151 [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C., Theveneau E., Tzekou A., Tada M., Woods M., Page K. M., Parsons M., Lambris J. D., Mayor R. (2011). Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev. Cell 21, 1026–1037 10.1016/j.devcel.2011.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chairoungdua A., Smith D. L., Pochard P., Hull M., Caplan M. J. (2010). Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 190, 1079–1091 10.1083/jcb.201002049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay N., Wang Z., Ashman L. K., Brady-Kalnay S. M., Kreidberg J. A. (2003). alpha3beta1 integrin-CD151, a component of the cadherin-catenin complex, regulates PTPmu expression and cell-cell adhesion. J. Cell Biol. 163, 1351–1362 10.1083/jcb.200306067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Cheung M., Abu-Elmagd M. M., Orme A., Scotting P. J. (2000). Chick sox10, a transcription factor expressed in both early neural crest cells and central nervous system. Brain Res. Dev. Brain Res. 121, 233–241 10.1016/S0165-3806(00)00049-3 [DOI] [PubMed] [Google Scholar]
- Cheung M., Chaboissier M. C., Mynett A., Hirst E., Schedl A., Briscoe J. (2005). The transcriptional control of trunk neural crest induction, survival, and delamination. Dev. Cell 8, 179–192 10.1016/j.devcel.2004.12.010 [DOI] [PubMed] [Google Scholar]
- Chu Y. S., Eder O., Thomas W. A., Simcha I., Pincet F., Ben-Ze'ev A., Perez E., Thiery J. P., Dufour S. (2006). Prototypical type I E-cadherin and type II cadherin-7 mediate very distinct adhesiveness through their extracellular domains. J. Biol. Chem. 281, 2901–2910 10.1074/jbc.M506185200 [DOI] [PubMed] [Google Scholar]
- Coles E. G., Taneyhill L. A., Bronner-Fraser M. (2007). A critical role for Cadherin6B in regulating avian neural crest emigration. Dev. Biol. 312, 533–544 10.1016/j.ydbio.2007.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creuzet S., Couly G., Le Douarin N. M. (2005). Patterning the neural crest derivatives during development of the vertebrate head: insights from avian studies. J. Anat. 207, 447–459 10.1111/j.1469-7580.2005.00485.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dady A., Blavet C., Duband J. L. (2012). Timing and kinetics of E- to N-cadherin switch during neurulation in the avian embryo. Dev. Dyn. 241, 1333–1349 10.1002/dvdy.23813 [DOI] [PubMed] [Google Scholar]
- Detrick R. J., Dickey D., Kintner C. R. (1990). The effects of N-cadherin misexpression on morphogenesis in Xenopus embryos. Neuron 4, 493–506 10.1016/0896-6273(90)90108-R [DOI] [PubMed] [Google Scholar]
- Dottori M., Gross M. K., Labosky P., Goulding M. (2001). The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development 128, 4127–4138 [DOI] [PubMed] [Google Scholar]
- Erickson C. A. (1987). Behavior of neural crest cells on embryonic basal laminae. Dev. Biol. 120, 38–49 10.1016/0012-1606(87)90101-1 [DOI] [PubMed] [Google Scholar]
- Friedl P., Wolf K. (2003). Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374 10.1038/nrc1075 [DOI] [PubMed] [Google Scholar]
- Fujimori T., Miyatani S., Takeichi M. (1990). Ectopic expression of N-cadherin perturbs histogenesis in Xenopus embryos. Development 110, 97–104 [DOI] [PubMed] [Google Scholar]
- Gammill L. S., Bronner-Fraser M. (2002). Genomic analysis of neural crest induction. Development 129, 5731–5741 10.1242/dev.00175 [DOI] [PubMed] [Google Scholar]
- Gammill L. S., Krull C. E. (2011). Embryological and genetic manipulation of chick development. Methods Mol. Biol. 770, 119–137 10.1007/978-1-61779-210-6_5 [DOI] [PubMed] [Google Scholar]
- Greco C., Bralet M. P., Ailane N., Dubart-Kupperschmitt A., Rubinstein E., Le Naour F., Boucheix C. (2010). E-cadherin/p120-catenin and tetraspanin Co-029 cooperate for cell motility control in human colon carcinoma. Cancer Res. 70, 7674–7683 10.1158/0008-5472.CAN-09-4482 [DOI] [PubMed] [Google Scholar]
- Hamburger V., Hamilton H. L. (1951). A series of normal stages in the development of the chick embryo. J. Morphology 88, 231–272 [DOI] [PubMed] [Google Scholar]
- Hay E. D. (2005). The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 233, 706–720 10.1002/dvdy.20345 [DOI] [PubMed] [Google Scholar]
- Hemler M. E. (2005). Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811 10.1038/nrm1736 [DOI] [PubMed] [Google Scholar]
- Heuberger J., Birchmeier W. (2010). Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2, a002915 10.1101/cshperspect.a002915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M., Hashimoto S., Yonemura S., Sabe H., Aizawa S. (2008). EPB41L5 functions to post-transcriptionally regulate cadherin and integrin during epithelial-mesenchymal transition. J. Cell Biol. 182, 1217–1230 10.1083/jcb.200712086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. L., Liu D., Masuya D., Kameyama K., Nakashima T., Yokomise H., Ueno M., Miyake M. (2004). MRP-1/CD9 gene transduction downregulates Wnt signal pathways. Oncogene 23, 7475–7483 10.1038/sj.onc.1208063 [DOI] [PubMed] [Google Scholar]
- Huber O., Korn R., McLaughlin J., Ohsugi M., Herrmann B. G., Kemler R. (1996). Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 59, 3–10 10.1016/0925-4773(96)00597-7 [DOI] [PubMed] [Google Scholar]
- Jamora C., DasGupta R., Kocieniewski P., Fuchs E. (2003). Links between signal transduction, transcription and adhesion in epithelial bud development. Nature 422, 317–322 10.1038/nature01458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. L., Winterwood N., DeMali K. A., Stipp C. S. (2009). Tetraspanin CD151 regulates RhoA activation and the dynamic stability of carcinoma cell-cell contacts. J. Cell Sci. 122, 2263–2273 10.1242/jcs.045997 [DOI] [PubMed] [Google Scholar]
- Kam Y., Quaranta V. (2009). Cadherin-bound beta-catenin feeds into the Wnt pathway upon adherens junctions dissociation: evidence for an intersection between beta-catenin pools. PLoS ONE 4, e4580 10.1371/journal.pone.0004580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos R., Reedy M. V., Johnson R. L., Erickson C. A. (2001). The winged-helix transcription factor FoxD3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development 128, 1467–1479 [DOI] [PubMed] [Google Scholar]
- Kulesa P. M., Fraser S. E. (2000). In ovo time-lapse analysis of chick hindbrain neural crest cell migration shows cell interactions during migration to the branchial arches. Development 127, 1161–1172 [DOI] [PubMed] [Google Scholar]
- Kuphal F., Behrens J. (2006). E-cadherin modulates Wnt-dependent transcription in colorectal cancer cells but does not alter Wnt-independent gene expression in fibroblasts. Exp. Cell Res. 312, 457–467 10.1016/j.yexcr.2005.11.007 [DOI] [PubMed] [Google Scholar]
- LeDouarin N., Kalcheim C. (1999). The Neural Crest Cambridge: Cambridge University Press [Google Scholar]
- Levy S., Shoham T. (2005). Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 20, 218–224 10.1152/physiol.00015.2005 [DOI] [PubMed] [Google Scholar]
- Lim J., Thiery J. P. (2012). Epithelial-mesenchymal transitions: insights from development. Development 139, 3471–3486 10.1242/dev.071209 [DOI] [PubMed] [Google Scholar]
- Lister J. A., Cooper C., Nguyen K., Modrell M., Grant K., Raible D. W. (2006). Zebrafish Foxd3 is required for development of a subset of neural crest derivatives. Dev. Biol. 290, 92–104 10.1016/j.ydbio.2005.11.014 [DOI] [PubMed] [Google Scholar]
- Matson C. K., Murphy M. W., Sarver A. L., Griswold M. D., Bardwell V. J., Zarkower D. (2011). DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476, 101–104 10.1038/nature10239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker C., Cousin H., Neuner R., Alfandari D. (2009). Extracellular cleavage of cadherin-11 by ADAM metalloproteases is essential for Xenopus cranial neural crest cell migration. Mol. Biol. Cell 20, 78–89 10.1091/mbc.E08-05-0535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason S. G., McMahon A. P. (2002). A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development 129, 2087–2098 [DOI] [PubMed] [Google Scholar]
- Meng W., Takeichi M. (2009). Adherens junction: molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 1, a002899 10.1101/cshperspect.a002899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton J. D., Yan Y. L. (2008). Using Morpholinos to control gene expression. Curr. Protoc. Mol. Biol. Chapter 26, Unit 26 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundell N. A., Labosky P. A. (2011). Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development 138, 641–652 10.1242/dev.054718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S., Takeichi M. (1995). Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development 121, 1321–1332 [DOI] [PubMed] [Google Scholar]
- Nakagawa S., Takeichi M. (1998). Neural crest emigration from the neural tube depends on regulated cadherin expression. Development 125, 2963–2971 [DOI] [PubMed] [Google Scholar]
- Nandadasa S., Tao Q., Menon N. R., Heasman J., Wylie C. (2009). N- and E-cadherins in Xenopus are specifically required in the neural and non-neural ectoderm, respectively, for F-actin assembly and morphogenetic movements. Development 136, 1327–1338 10.1242/dev.031203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M. A. (2011). The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 27, 347–376 10.1146/annurev-cellbio-092910-154036 [DOI] [PubMed] [Google Scholar]
- Nishimura T., Takeichi M. (2009). Remodeling of the adherens junctions during morphogenesis. Curr. Top. Dev. Biol. 89, 33–54 10.1016/S0070-2153(09)89002-9 [DOI] [PubMed] [Google Scholar]
- Oda H., Takeichi M. (2011). Evolution: structural and functional diversity of cadherin at the adherens junction. J. Cell Biol. 193, 1137–1146 10.1083/jcb.201008173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder T. T., Gupta P. B., Mani S. A., Yang J., Lander E. S., Weinberg R. A. (2008). Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 68, 3645–3654 10.1158/0008-5472.CAN-07-2938 [DOI] [PubMed] [Google Scholar]
- Orsulic S., Huber O., Aberle H., Arnold S., Kemler R. (1999). E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 112, 1237–1245 [DOI] [PubMed] [Google Scholar]
- Park K. S., Gumbiner B. M. (2010). Cadherin 6B induces BMP signaling and de-epithelialization during the epithelial mesenchymal transition of the neural crest. Development 137, 2691–2701 10.1242/dev.050096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron J. C., Bixby J. L. (1999). Tetraspanins expressed in the embryonic chick nervous system. FEBS Lett. 461, 86–90 10.1016/S0014-5793(99)01429-5 [DOI] [PubMed] [Google Scholar]
- Polyak K., Weinberg R. A. (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9, 265–273 10.1038/nrc2620 [DOI] [PubMed] [Google Scholar]
- R Development Core Team(2012). R: A Language and Environment for Statistical Computing R Foundation for Statistical Computing, Vienna, Austria. Available at: http://www.R-project.org/ [Google Scholar]
- Roffers-Agarwal J., Hutt K. J., Gammill L. S. (2012). Paladin is an antiphosphatase that regulates neural crest cell formation and migration. Dev. Biol. 371, 180–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein E. (2011). The complexity of tetraspanins. Biochem. Soc. Trans. 39, 501–505 10.1042/BST0390501 [DOI] [PubMed] [Google Scholar]
- Sasai N., Mizuseki K., Sasai Y. (2001). Requirement of FoxD3-class signaling for neural crest determination in Xenopus. Development 128, 2525–2536 [DOI] [PubMed] [Google Scholar]
- Shoval I., Ludwig A., Kalcheim C. (2007). Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development 134, 491–501 10.1242/dev.02742 [DOI] [PubMed] [Google Scholar]
- Shtutman M., Levina E., Ohouo P., Baig M., Roninson I. B. (2006). Cell adhesion molecule L1 disrupts E-cadherin-containing adherens junctions and increases scattering and motility of MCF7 breast carcinoma cells. Cancer Res. 66, 11370–11380 10.1158/0008-5472.CAN-06-2106 [DOI] [PubMed] [Google Scholar]
- Swartz M. E., Eberhart J., Pasquale E. B., Krull C. E. (2001). EphA4/ephrin-A5 interactions in muscle precursor cell migration in the avian forelimb. Development 128, 4669–4680 [DOI] [PubMed] [Google Scholar]
- Taneyhill L. A., Coles E. G., Bronner-Fraser M. (2007). Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development 134, 1481–1490 10.1242/dev.02834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E., Mayor R. (2012). Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 366, 34–54 10.1016/j.ydbio.2011.12.041 [DOI] [PubMed] [Google Scholar]
- Thiery J. P., Sleeman J. P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 10.1038/nrm1835 [DOI] [PubMed] [Google Scholar]
- Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- Tosney K. W. (1982). The segregation and early migration of cranial neural crest cells in the avian embryo. Dev. Biol. 89, 13–24 10.1016/0012-1606(82)90289-5 [DOI] [PubMed] [Google Scholar]
- Tsai Y. C., Weissman A. M. (2011). Dissecting the diverse functions of the metastasis suppressor CD82/KAI1. FEBS Lett. 585, 3166–3173 10.1016/j.febslet.2011.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. C., Khan Z., Kaschube M., Wieschaus E. F. (2012). Differential positioning of adherens junctions is associated with initiation of epithelial folding. Nature 484, 390–393 10.1038/nature10938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D. (1992). Whole mount in situ hybridization of vertebrate embryos. In Situ Hybridization: A Practical Approach 75–83Oxford: Oxford University Press [Google Scholar]
- Yáñez-Mó M., Barreiro O., Gordon-Alonso M., Sala-Valdés M., Sánchez-Madrid F. (2009). Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 19, 434–446 10.1016/j.tcb.2009.06.004 [DOI] [PubMed] [Google Scholar]
- Yáñez-Mó M., Gutiérrez-López M. D., Cabañas C. (2011). Functional interplay between tetraspanins and proteases. Cell. Mol. Life Sci. 68, 3323–3335 10.1007/s00018-011-0746-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Weinberg R. A. (2008). Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829 10.1016/j.devcel.2008.05.009 [DOI] [PubMed] [Google Scholar]
- Zohn I. E., Li Y., Skolnik E. Y., Anderson K. V., Han J., Niswander L. (2006). p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell 125, 957–969 10.1016/j.cell.2006.03.048 [DOI] [PubMed] [Google Scholar]
- Zöller M. (2009). Tetraspanins: push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 9, 40–55 10.1038/nrc2543 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.