

The death ligand TRAIL plays a role in physiologic resolution of inflammation and exogenous TRAIL has potential therapeutic benefits in neutrophil-dominant inflammation.
Keywords: death receptor, lung injury, innate immunity, LPS affiliation/site
Abstract
Novel therapeutics targeting neutrophilic inflammation are a major unmet clinical need in acute and chronic inflammation. The timely induction of neutrophil apoptosis is critical for inflammation resolution, and it is thought that acceleration of apoptosis may facilitate resolution at inflammatory sites. We previously demonstrated that a death receptor ligand, TRAIL, accelerates neutrophil apoptosis in vitro. We examined the role of TRAIL in neutrophil-dominant inflammation in WT and TRAIL-deficient mice. TRAIL deficiency did not alter constitutive neutrophil apoptosis, whereas exogenous TRAIL accelerated apoptosis of murine peripheral blood neutrophils. We compared TRAIL-deficient and WT mice in two independent models of neutrophilic inflammation: bacterial LPS-induced acute lung injury and zymosan-induced peritonitis. In both models, TRAIL-deficient mice had an enhanced inflammatory response with increased neutrophil numbers and reduced neutrophil apoptosis. Correction of TRAIL deficiency and supraphysiological TRAIL signaling using exogenous protein enhanced neutrophil apoptosis and reduced neutrophil numbers in both inflammatory models with no evidence of effects on other cell types. These data indicate the potential therapeutic benefit of TRAIL in neutrophilic inflammation.
Introduction
The resolution of acute inflammation requires recruited neutrophils to undergo programmed cell death by apoptosis in a timely manner after completion of effective host defense functions. This response limits the potential for bystander tissue damage. There is evidence that the onset of apoptosis is tightly coupled to inflammation resolution [1, 2]. Conversely, neutrophil lifespan is prolonged in tissues [3], and neutrophil apoptosis may be delayed at sites of inflammation by a wide range of proinflammatory stimuli [4]. Recent data emphasize the potential of pharmacological strategies to “drive” neutrophil apoptosis in vivo and thus, to aid clearance of the inflammatory infiltrate [2].
The molecular mechanisms governing the onset of neutrophil apoptosis are surprisingly ill-defined. Few stimuli inducing physiological neutrophil apoptosis have been described. Moreover, many factors modulate other neutrophil functions in addition to apoptosis, emphasizing the importance of elucidating mechanisms that are selective for the control of apoptosis. The death receptor FasL induces neutrophil apoptosis [5, 6] with evidence for a role in acute lung injury [7] but is also a stimulus to neutrophil recruitment [6]. Another death receptor ligand, TNF-α, has a modest, proapoptotic effect, which is later opposed by its downstream, prosurvival effects, mediated via NF-κB activation [8], resulting in a net delay of neutrophil apoptosis [9]. We have previously shown that the death receptor ligand TRAIL induces neutrophil apoptosis without NF-κB activation in vitro and without the chemoattractant effects of FasL [10]. More recently, TRAIL has been implicated in eliminating senescent, circulating neutrophils [11].
TRAIL is a type II membrane protein principally expressed by leukocyte populations [12]. In humans, TRAIL interacts with four membrane receptors belonging to the TNFR family. TRAIL-R1 and TRAIL-R2 have cytoplasmic death domains and can activate caspases and NF-κB [13]. The decoy receptors TRAIL-R3 and TRAIL-R4 have truncated death domains and do not activate caspases but may activate NF-κB [13]. A soluble decoy receptor for TRAIL, osteoprotegerin, is also described. Mice have a single TRAIL death receptor, which shares 79% sequence homology with human TRAIL-R2 [14], together with two decoy receptors.
TRAIL-deficient mice have demonstrated an increased adaptive immune response in mouse models of autoimmune arthritis [15] and encephalomyelitis [16]. TRAIL deficiency has been associated with increased neutrophil survival in a murine model of listeriosis [17] and in a pneumococcal meningitis model [18], in the latter case, with reduced cerebral inflammation and improved outcomes following rTRAIL treatment. These studies suggest TRAIL-induced apoptosis may be a key mechanism regulating the neutrophilic response to bacterial infection.
To determine the potential of TRAIL signaling to modulate neutrophil apoptosis during neutrophil-dominant inflammation, we have examined its role in two well-characterized models: LPS-mediated acute lung injury and zymosan-induced peritonitis. We show mice deficient in TRAIL have no defect of neutrophil recruitment but have delayed neutrophil apoptosis and delayed inflammation resolution. Importantly, administration of exogenous TRAIL not only restores a WT phenotype to TRAIL-deficient mice but also accelerates inflammation resolution in WT mice.
MATERIALS AND METHODS
Animals
TRAIL-deficient mice on a C57/BL6 background were a kind gift from Amgen (Seattle, WA, USA) via Dr. Mark Smyth (University of Melbourne, Australia) and have been described previously [19]. TRAIL-deficient mice and WT controls (8–12 weeks) were anesthetized with an i.p injection of ketamine (Ketaset, 100 mg/kg; Willows Francis Veterinary, Crawley, UK), xylazine hydrochloride (10 mg/kg; Bayer, Suffolk, UK), and atropine (0.02 mg/kg; Phoenix Pharmaceuticals, Burlingame, CA, USA). All procedures were approved by the University of Sheffield Ethics Committee (UK) and were performed in accordance with the Home Office Animal (Scientific Procedures) Act 1986.
Materials
Goat anti-rat IgG microbeads were obtained from Miltenyl Biotec (Bisley, UK). Rat anti-mouse antibodies to CD2 (Rm2-5), CD5 (53-7.3), and CD45R (RA3-6B2) were obtained from BD PharMingen (Oxford, UK). Rat anti-mouse F4/80 antigen (Clone A3-1) and CD-115 were purchased from Serotec (Kidlington, UK). LPS from Escherichia coli serotype 10 and zymosan were obtained from Sigma-Aldrich (Poole, UK). Murine rTRAIL was purchased from Biomol International (UK).
Preparation of peripheral blood neutrophils
This method has been described previously [20]. Briefly, 1 ml blood was collected via cardiac puncture from anesthetized mice using a heparinized syringe and was transferred into dextran T500 (Amersham Pharmacia Biotech, Buckinghamshire, UK), 1.25% w/v in saline, to a final volume of 10 ml. Following erythrocyte sedimentation, leukocyte-containing supernatants from three mice were pooled and washed in PBS buffer with 0.5% BSA, pH 7.4. After cytocentrifugation of an aliquot to obtain differential cell counts, leukocytes were incubated with anti-CD2 (1.5 μg/106 lymphocytes), -CD5 (2 μg/106 lymphocytes), -CD45R (10 μg/106 lymphocytes), -F4/80 (2 μg/106 monocytes), and -CD115 (15 μg/106 lymphocytes) prior to negative selection of neutrophils using a cooled LD column attached to a MACS magnet (Miltenyl Biotec). The final yield was ∼1 × 106 neutrophils for each group of mice. Neutrophil purity was assessed by differential counts of cytocentrifuge preparations, and samples of >90% purity were obtained for subsequent experiments. Neutrophil viability was assessed by trypan blue staining and was always >98.5%.
Neutrophil culture
Neutrophils were cultured at 1.0 × 106/ml in RPMI 1640 (Sigma-Aldrich) with 10% FCS with added glutamine, penicillin, and streptomycin (100 U/L), all from Life Technologies (Paisley, UK). Aliquots (100 μl) of cells were cultured with and without 100 ng/ml rTRAIL in nontissue culture-treated Falcon “Flexiwell” plates (BD PharMingen) at 37°C in a 5% CO2 atmosphere. Cells were harvested from culture at 6, 12, and 18 h.
Assessment of neutrophil viability and apoptosis
At the time-points described, cytocentrifuge preparations were made and the proportion of apoptotic neutrophils determined by counting duplicate cytospins (>300 cells/slide), stained by Diff-Quick (Merck, Dorset, UK). In keeping with previous work [1], we found that the morphological features of apoptotic and nonapoptotic murine neutrophils could be clearly distinguished by light microscopy (see Fig. 1A). In addition, membrane integrity was assessed at all time-points by exclusion of the vital dye trypan blue, and necrosis, defined as trypan blue-positive cells without morphological features of apoptosis, was <5% unless otherwise stated. Apoptosis was also assessed by flow cytometry, detecting externalization of phosphatidylserine using Annexin V (BD PharMingen) and costaining with To-Pro 3 (Molecular Probes, Leiden, The Netherlands) to distinguish late-apoptotic or necrotic cells by failure of the latter to exclude this vital dye [21]. Both fluorescent dyes were used according to the manufacturer's instructions. Neutrophils were identified by staining with FITC-1A8 (BD PharMingen) [21]. Cells were analyzed on a dual-laser FACSCalibur flow cytometer (BD PharMingen) and a minimum of 10,000 events recorded and analyzed using CellQuest software (BD PharMingen).
Figure 1. Effects of TRAIL on apoptosis of murine peripheral blood neutrophils.

(A) Cytocentrifuge preparation of purified peripheral blood neutrophils from TRAIL-deficient mice shows apoptotic neutrophils (arrows) readily distinguishable from neutrophils of nonapoptotic morphology. (B) Neutrophils were purified from peripheral blood and sampled after 6, 12, and 18 h in culture. Apoptosis was assessed by cytospin morphology of neutrophils from control (open bars) and TRAIL-deficient (filled bars) and plotted against time (h); n=4. At t = 0 h, apoptosis was <1.0%. There were no statistically significant differences between WT and TRAIL-deficient in constitutive neutrophil apoptosis. (C) Addition of rTRAIL accelerated apoptosis of WT neutrophils. Freshly isolated peripheral blood neutrophils were cultured with or without TRAIL (100 ng/ml). Mean ± sem of the percentage of apoptotic cells is shown for control (open bars) and TRAIL-treated cells (filled bars); n = 4. In both populations at both time-points, TRAIL treatment caused a significant increase in levels of neutrophil apoptosis.
Model of LPS-mediated acute lung injury
The model of i.t. instillation of LPS has been described in detail previously [22]. A 24-gauge catheter (Jelco; Johnson and Johnson Medical, Ascot, UK) was inserted into the trachea of anesthetized mice and LPS (0.3 μg), or PBS as a control was instilled into the lungs using a pipette gel-loading tip and then flushed through the catheter with air. At the relevant time-points, experiments were terminated by giving the mice an overdose of sodium pentabarbitone.
For experiments where rTRAIL was administered i.t., the protocol was modified to avoid repeat cannulation of the trachea. LPS (3 mg) was delivered by nebulization at t = 0 and then at t = 24 h rTRAIL (1 mg/kg), was given by i.t. cannulation as above.
Model of zymosan-mediated acute peritonitis
Peritoneal inflammation was induced as described previously [23]. Zymosan A was prepared (2 mg/ml) in sterile, 0.9% normal saline, and 0.5 ml was injected i.p. (t=0). A subgroup of mice was then injected with 100 μg rTRAIL at 6 h. At 24 h and 48 h, the animals were killed and the peritoneal cavity lavaged with 2 ml saline. Hemocytometer counts were performed and cytospin preparations made for differential cell counts and assessment of apoptosis. The supernatant was removed and stored at –70°C.
BAL
BAL was performed using five 0.8 ml aliquots of 10 U/ml ice-cold heparinized saline (Leo Laboratories, Ireland), which were then pooled [21]. A total of 10 μl BAL was diluted in 90 μl 3% acetic acid for a hemocytometer total cell count. Cytospin preparations were made from each BAL sample (100 μl), stained with Diff-Quick, and assessed by blinded reviewers. Differential cell counts, including the proportion of neutrophils that was apoptotic, were then calculated using the fractions for each leukocyte population estimated by analysis of cytospin preparations. The remainder of the BALF was centrifuged (300 g for 6 min), supernatant was stored at –70°C, and the cell pellet was resuspended for flow cytometry.
Cytokine and IgM measurements in BAL
Cytokine/chemokine concentrations were measured by CBA flex sets and a FACSArray bioanalyzer (BD Biosciences, San Jose, CA, USA), in accordance with the manufacturer′s protocols. As a marker of pulmonary vascular leak and thus, of lung injury [24], IgM concentration in BAL was determined by ELISA (Bethyl Laboratories, Montgomery, TX, USA), in accordance with the manufacturer′s protocol.
Immunohistochemistry and TUNEL staining
Unlavaged lungs were fixed via the trachea with 10% buffered formalin at 20 cm H2O, processed, and embedded in paraffin wax blocks. Lung samples were serially sectioned at 5 μm and processed using 0.6 mol/L sodium citrate buffer (pH 6) antigen retrieval where required, as described previously [25]. Serial sections were stained with mouse mAb to TRAIL (Vector Laboratories, Peterborough, UK), as described previously [26], incubated for 1 h, labeled using a streptavidin avidin biotin complex peroxidase technique (Vector Laboratories), and visualized with 3,3′-diaminobenzidine (DakoCytomation, Denmark). Negative controls involved incubation with isotype antibody/secondary antibody alone. TUNEL was performed using an ApopTag peroxidase in situ apoptosis detection kit (Chemicon International, Temecula, CA, USA), as per the manufacturer's instructions.
Statistical analysis
Results are expressed as mean ± se and were analyzed for statistical variance using a one- or two-way ANOVA as appropriate, followed by a Bonferroni's post-test for multiple comparisons. Results were considered significant if P < 0.05. In all figures, *P < 0.05; **P < 0.01; and ***P < 0.001.
RESULTS
Constitutive apoptosis of peripheral blood neutrophils from TRAIL-deficient mice
We confirmed that there were no differences in total leukocyte numbers and numbers of circulating neutrophils between TRAIL-deficient and WT mice (data not shown), as described previously [19]. In freshly isolated peripheral blood neutrophils, there was no difference in numbers of apoptotic neutrophils: percentage of WT neutrophils showing morphological features of apoptosis was 0.53 ± 0.29% (mean±sem) versus 0.40 ± 0.20% for TRAIL-deficient mice; n = 3. When cultured in vitro, neutrophils underwent constitutive apoptosis (Fig. 1A), with no significant difference in the percentage of apoptotic cells between WT and TRAIL-deficient neutrophils at 6, 12, or 18 h (Fig. 1B).
TRAIL induces neutrophil apoptosis in vitro
Having previously shown that TRAIL induces apoptosis of human neutrophils [10], we evaluated the effect of exogenous TRAIL on murine neutrophils. WT neutrophils were isolated and cultured with rTRAIL (100 ng/ml) for 6 h and 18 h. rTRAIL treatment resulted in an increase in neutrophil apoptosis at 6 h and 18 h in WT and TRAIL-deficient neutrophils (Fig. 1C).
LPS-mediated neutrophilic inflammation is enhanced and neutrophil apoptosis decreased in TRAIL-deficient mice
As described previously [1], i.t. LPS induced rapid neutrophil migration into the lungs of challenged mice, as assessed by BAL at time-points up to 144 h following instillation. Total cell counts and percent neutrophil counts were increased in WT and TRAIL-deficient mice by 24 h after LPS instillation when compared with mice instilled with PBS alone. The numbers of neutrophils in BALF from control mice were <0.1 × 105 neutrophils at 24 h and at all subsequent time-points in WT and TRAIL-deficient strains (data not shown). There was an enhanced inflammatory response in TRAIL-deficient compared with WT mice, apparent from 48 h after LPS instillation, with significantly increased total cell counts and total and percent neutrophil counts (Fig. 2, A–C). There were small reciprocal changes in BAL percent macrophages (Fig. 2D), but total numbers of macrophages were unchanged (data not shown). Very few lymphocytes were present, and no significant difference in total lymphocyte counts was observed between WT and TRAIL-deficient mice (data not shown). Apoptosis of BAL neutrophils was assessed using morphologic criteria as described previously [1]. From the 48-h time-point onward, there was a significantly lower proportion of apoptotic neutrophils in the TRAIL-deficient compared with WT mice (Fig. 2E). These findings were confirmed at the 48-h time-point by identification of Annexin-V-positive cells within the IA8-positive neutrophil population by flow cytometry (Fig. 2F), but this method was not reliable at later time-points as a result of reduced cell numbers. Apoptosis of tissue cells was quantified using TUNEL staining (Fig. 2, G–I) and appeared largely confined to the influxing inflammatory cells, which were predominantly neutrophils.
Figure 2. Inflammatory cell numbers and apoptosis following i.t. administration of LPS in WT and TRAIL-deficient mice.

Cell counts were obtained from cytocentrifuge preparations and hemocytometer counts of BALF lavaged from the lungs at time-points up to 144 h; n = 10 animals in each group. In all panels, WT mice are depicted as a solid line and TRAIL-deficient mice as a dashed line. (A) Total leukocyte counts. At 48 h, there were significantly more leukocytes in the TRAIL-deficient when compared with WT BAL. (B) Total neutrophil counts. At 48 h, there were significantly more neutrophils in the TRAIL-deficient when compared with WT BAL. (C) The percentage of neutrophils in TRAIL-deficient mice was significantly increased after LPS instillation when compared with WT mice. (D) The percentage of macrophages rose in WT and TRAIL-deficient mice over time as inflammation resolved, and TRAIL-deficient mice had a significantly lower percentage. (E) The percentage of BALF neutrophils with morphological appearances of apoptosis was assessed. The percentage of apoptotic neutrophils increased in both populations, but at 48, 72, and 96 h, there was a significantly lower proportion of apoptotic neutrophils in the TRAIL-deficient mice. Neutrophil apoptosis was also assessed by flow cytometry. (F) The percentage of apoptotic neutrophils (IA8+/Annexin V+) was significantly reduced in TRAIL-deficient mice at 48 h compared with WT mice (P<0.05). Histologic sections of mice 24 h post-LPS treatment were TUNEL-stained for detection of apoptotic events. (G) Numbers of TUNEL-positive (+ve) cells were significantly higher in WT (H) than in TRAIL-deficient mice (I). HPF, per high power field.
Cytokine and chemokine levels in BAL
To investigate whether TRAIL might have an effect on pulmonary neutrophilia other than through reduced neutrophil apoptosis, chemokines and cytokines involved in neutrophil recruitment were measured in BAL (Table 1). There were no significant differences in levels of KC at any of the time-points studied. MIP-2 was markedly raised at 24 h, in keeping with previous studies [27], but levels did not differ between WT and TRAIL-deficient mice. IL-17 was not detected at either time-point in WT or TRAIL-deficient mice (data not shown). These data, combined with the observation that there was no difference in neutrophil numbers until 48 h after LPS instillation, suggested that neutrophil persistence in the TRAIL-deficient animals was unlikely to be a result of excessive, ongoing recruitment. No significant differences in TNF-α, IL-1β, or MIP-1α levels were observed at any time-point. MIP-1β and IL-1α levels were significantly higher in TRAIL-deficient mice at 24 h, but although statistically significant, the differences were not large in comparison with previous studies [1, 28].
Table 1. Levels of Chemokines and Cytokines in BALF.
| 24 h |
48 h |
|||||
|---|---|---|---|---|---|---|
| C57BL6 | TRAIL–/– | P value | C57BL6 | TRAIL–/– | P value | |
| TNF-α pg/ml | 262 (104–751) | 244 (175–398) | NS | 80.7 (7–126) | 77.8 (9.2–154) | NS |
| KC (CXCL1) pg/ml | 8.2 (7.9–9.7) | 7.57 (4.46–11.1) | NS | 3.44 (1.7–4.26) | 4.0 (3.7–4.96) | NS |
| IL-1α pg/ml | 21.8 (12.3–24) | 31.2 (24.1–40.6) | <0.05 | 17.9 (9.1–31.7) | 28.8 (12.1–63.5) | NS |
| IL-1β pg/ml | 10.3 (9.9–11.9) | 11.4 (10.2–14.3) | NS | 9.97 (9.97–13.6) | 9.97 (9.97–19.8) | NS |
| MIP-1α (CCL3) pg/ml | 34.6 (19.7–45) | 47.5 (42.1–49.5) | NS | 27.2 (21.2–40.3) | 27.9 (10.4–70.3) | NS |
| MIP-1β (CCL4L1) pg/ml | 57.5 (32–61.5) | 97.5 (89.3–115) | <0.05 | 63.60 (10.3–72.3) | 63.6 (10.6–172) | NS |
| MIP-2 ηg/ml | 44.2 (15.4–87.3) | 48.5 (32.2–75.9) | NS | 15 (3.8–36.2) | 10.6 (4.1–21) | NS |
A range of cytokines and chemokines was measured at 24 h and 48 h following LPS instillation. All values are shown as median(range) and represent the values obtained from five independent experiments (n = 5). TRAIL–/–, TRAIL-deficient.
IgM levels in BAL were increased in TRAIL-deficient mice following LPS instillation
An increase in neutrophil numbers can be associated with increased vascular permeability and lung injury in murine models of pulmonary inflammation [24, 28]. As described previously [24], we measured IgM levels in BAL between the two murine strains and found significantly increased IgM in BAL of TRAIL-deficient mice at 48 h and 72 h following LPS instillation, the time-points at which we also saw significant differences in neutrophil numbers in BAL (Fig. 3).
Figure 3. IgM levels in BALF following i.t. administration of LPS in WT and TRAIL-deficient mice.

Total IgM levels measured by ELISA in BAL at 48 h and 72 h were significantly higher in TRAIL-deficient (filled bars) compared with WT mice (open bars; P<0.05; n=5).
TRAIL immunolocalization after LPS-induced lung injury/TRAIL expression
Immunohistochemical examination of TRAIL expression in the lung was performed at key time-points in WT mice. In WT animals receiving i.t. saline, TRAIL staining was observed only in small pulmonary arteries (Fig. 4A). After LPS administration to WT mice, immunostaining was seen predominantly in bronchial epithelial cells but with occasional staining of alveolar macrophages (Fig. 4B). Staining of sections from TRAIL-deficient mice was negative (Fig. 4C). Importantly, in TUNEL staining of lung sections, there was no evidence of excess epithelial cell death, either alveolar or in the airways (Fig. 2, G–H), following LPS treatment in WT compared with TRAIL-deficient mice, despite enhanced TRAIL immunostaining in these cells.
Figure 4. TRAIL expression following i.t. LPS.
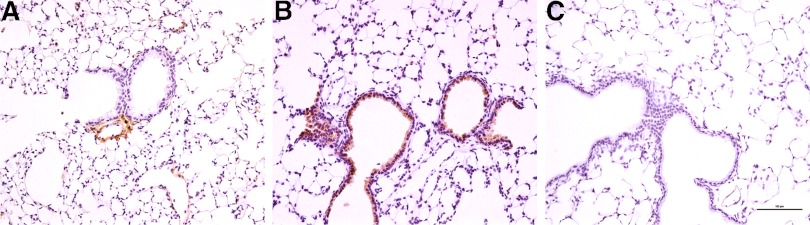
Lung sections 48 h after i.t. instillation of saline (control) or LPS were immunostained for TRAIL. (A) Lung section demonstrating lack of positive TRAIL staining in the airways of a WT mouse following saline administration. TRAIL staining is identified only where associated with small pulmonary arteries. (B) Lung section demonstrating TRAIL expression principally in the bronchial epithelium following LPS challenge of WT mice, with occasional staining of alveolar macrophages. (C) Lung section demonstrating absence of specific TRAIL staining in a TRAIL-deficient lung (negative control). These sections are representative of multiple sections cut in a minimum of three experiments. All images, 200× original magnification; scale bar represents 100 μm.
Zymosan-mediated peritoneal inflammation is enhanced in TRAIL-deficient mice
As described previously [23], i.p. zymosan induced neutrophil migration into the peritoneum of challenged mice, assessed by peritoneal lavage at time-points up to 48 h following instillation. Total neutrophil counts were increased in WT and TRAIL-deficient mice when compared with mice instilled with PBS alone (Fig. 5A). No significant differences between the two strains in macrophage or lymphocyte numbers were observed (data not shown). The proportion of the neutrophil lavage population that had the microscopic appearances of apoptosis at 24 h and 48 h was significantly lower in the TRAIL-deficient mice (Fig. 5B).
Figure 5. Neutrophil counts and apoptosis in peritoneal lavage fluid following i.p. administration of zymosan in WT and TRAIL-deficient mice.

(A) Total neutrophil counts were obtained from cytocentrifuge preparations of lavage fluid from the peritoneum at 24 h and 48 h (n=5 experiments), multiplying the differential count by the total leukocyte number obtained from hemocytometer counts. (B) The percentage of lavage neutrophils with morphological appearances of apoptosis was calculated. At 24 h and 48 h, the percentage of apoptotic neutrophils was increased in both populations, but there was a significantly lower proportion of apoptotic neutrophils in the TRAIL-deficient (filled bars) compared with WT (open bars) mice.
Administration of rTRAIL induces neutrophil apoptosis and reduces neutrophil numbers in pulmonary and peritoneal inflammatory models in vivo
To explore the effects of exogenous TRAIL on neutrophilic inflammation and apoptosis in vivo, we first established a model of acute pulmonary inflammation induced by nebulized LPS. This required a larger dose of LPS (3 mg), compared with 0.3 μg by i.t. administration, and slightly higher levels of neutrophilia were induced, together with the expected difference in neutrophil counts at 48 h between WT and TRAIL-deficient mice (Fig. 6, A and B). Neutrophilic inflammation was established over 24 h and then rTRAIL delivered i.t., seeking evidence of accelerated inflammation resolution at 48 h after the initial challenge. TRAIL treatment significantly reduced neutrophil numbers (total cell count and percent) in TRAIL-deficient and WT mice (Fig. 6, A and B). There was no significant effect on macrophage numbers (data not shown). There was an increase in the proportion of apoptotic neutrophils detected in BAL following TRAIL treatment (Fig. 6C). Moreover, TUNEL staining of WT lung sections revealed an increase in the numbers of apoptotic cells detected following TRAIL treatment (Fig. 6, D–F). Again, these apoptotic events appeared largely confined to the inflammatory infiltrate, with no evidence that TRAIL treatment resulted in significant epithelial cell death. We also determined whether TRAIL treatment induced a neutrophil chemokine response. KC and IL-17 were undetectable at this time-point in the presence or absence of TRAIL treatment, whereas MIP-2 levels were raised as before but not significantly different (data not shown).
Figure 6. i.t. administration of rTRAIL induces neutrophil apoptosis and reduces neutrophil numbers in vivo.

TRAIL (5 μg) was administered i.t. 24 h after nebulized administration of 3 mg LPS; n = 6 animals in each group. Total neutrophil counts at 48 h from WT mice are shown as open bars and from TRAIL-deficient mice as filled bars. There were significantly more (A) cells and (B) neutrophils in the TRAIL-deficient mice lavages when compared with WT, which significantly reduced in response to TRAIL therapy. (C) At 48 h, the percentage of apoptotic neutrophils was reduced in TRAIL-deficient mice compared with WT mice but significantly increased in response to TRAIL treatment. Histologic sections of WT mice 24 h post-TRAIL treatment were TUNEL-stained for detection of apoptotic events. (D) Numbers of TUNEL-positive cells were significantly higher in WT (E and F) mice following TRAIL treatment (tx). Neb, Nebulized.
In the peritonitis model, rTRAIL was injected 6 h following i.p. administration of zymosan and neutrophil counts and levels of apoptosis assessed at 24 h. TRAIL treatment again resulted in significantly reduced neutrophil numbers in TRAIL-deficient and WT mice (Fig. 7, A and B), with increased neutrophil apoptosis, as assessed by morphologic criteria (Fig. 7C), indicating efficacy of TRAIL treatment in pulmonary and systemic inflammation.
Figure 7. i.p. administration of rTRAIL induces neutrophil apoptosis and restores neutrophil numbers in vivo.

TRAIL (5 μg) was injected into the peritoneal cavity 6 h after zymosan administration; n = 6 animals in each group. Total neutrophil counts from WT mice are shown as open bars and from TRAIL-deficient mice, as filled bars. There were significantly more (A) cells and (B) neutrophils in the TRAIL-deficient mice lavage when compared with WT, which significantly reduced in response to TRAIL therapy. (C) At 24 h, the percentage of apoptotic neutrophils was reduced in TRAIL-deficient mice compared with WT mice but significantly increased in response to TRAIL treatment.
DISCUSSION
In these studies, we show that neutrophils deficient in the death receptor ligand TRAIL have normal constitutive apoptosis. However, in two independent inflammatory models, animals deficient in TRAIL show delayed neutrophil apoptosis and increased neutrophilic inflammation. In both models, administration of exogenous TRAIL restored the WT phenotype in TRAIL-deficient mice and importantly, accelerated neutrophil apoptosis and reduced neutrophil numbers in WT mice (Fig. 8). In contrast to the effects of other death receptor ligands that cause death or inflammatory responses in multiple cell populations, the effect of TRAIL appeared to be restricted to the selective induction of neutrophil apoptosis.
Figure 8. TRAIL regulates inflammatory neutrophil apoptosis.

A proposed model for TRAIL regulating inflammation resolution by modulation of neutrophil lifespan. In WT mice, TRAIL expression by tissue cells, e.g., the respiratory epithelium, can induce apoptosis of recruited neutrophils. In TRAIL-deficient mice, inflammatory cell survival is prolonged until engagement of a constitutive program of neutrophil apoptosis. Exogenous TRAIL accelerates the apoptotic death of neutrophils in TRAIL-deficient and WT mice.
Although the role of death receptor ligands such as FasL and TNF-α in accelerating neutrophil apoptosis is well recognized, each also has significant proinflammatory effects, with FasL inducing neutrophil chemotaxis [29] and TNF-α having an antiapoptotic effect mediated via NF-κB [8, 9]. We previously showed that TRAIL had neither of these effects in human neutrophils in vitro and postulated that TRAIL treatment might regulate neutrophil apoptosis in vivo [10]. The TRAIL system has since been implicated in the elimination of senescent circulating neutrophils [11], suggesting a role in physiological regulation of neutrophil lifespan. Further interest in TRAIL arose, as the majority of primary cell types, although not all, is resistant to TRAIL, in part, as a result of expression of cell surface decoy receptors TRAIL-R3 and -R4, reducing the likelihood of unwanted cytotoxicity in other cell types with TRAIL treatment in vivo [12]. In contrast, FasL induces apoptosis of bronchial epithelial cells [30], and pulmonary overexpression of TNF-α induces chronic pulmonary inflammation and fibrosis [31].
In agreement with earlier studies [18], we found no difference in neutrophil numbers in the circulation of WT and TRAIL-deficient mice. When cultured in vitro, there was no difference in constitutive neutrophil apoptosis between WT and TRAIL-deficient cells, and both cell types showed an equivalent apoptotic response to exogenous TRAIL. Examination of an experimental model of LPS-induced lung inflammation revealed an increase in the number of neutrophils present in the lung in response to LPS, which peaked at 48 h and resolved thereafter. Multiple studies of LPS-induced acute lung inflammation have confirmed the transient nature of the inflammatory response and that modification of individual factors provides a transient delay rather than complete cessation of resolution [32, 33]. The total number of neutrophils present in the lavage in the WT mice was in keeping with previous data published by our group and others [1, 34]. There were clear differences in neutrophil numbers at 24 and 48 h post-LPS instillation between WT and TRAIL-deficient mice, but by 72 h, total neutrophil numbers were not significantly different, although in TRAIL-deficient mice, neutrophils remained a higher proportion of the inflammatory exudate (Fig. 2C), and fewer of them were apoptotic, as assessed by annexin V/To-Pro 3 staining, than in the WT (Fig. 2E). Murine models involving manipulation of neutrophil apoptosis, as we have described previously [1, 35], show prolonged inflammation that then resolves, rather than an indefinite failure of resolution, consistent with what we now demonstrate in the TRAIL-deficient mice. This convergence between WT and TRAIL-deficient mice at later time-points could also reflect reduced neutrophil recruitment in the TRAIL-deficient mice. The recruitment of neutrophils to the lung cannot be measured directly in murine models. There were no significant differences, however, between the murine strains in levels of cytokines typically associated with neutrophil recruitment (KC, MIP-2, IL-17) or with NF-κB activation (KC, MIP-2) [36]. In other organs, such as the inflamed joint, reverse endothelial migration of neutrophils has been described [37], but to our knowledge, retrograde movement of neutrophils back across the pulmonary epithelium has not been described. Other proresolution molecules, particularly endogenous lipid mediators, such as resolvins and protectins [38], were not measured in our model and could have contributed to the resolution of the neutrophilic response, including effects on neutrophil lifespan. Ingestion of apoptotic cells can also significantly reprogram the cytokine network in infection-related pulmonary models of inflammation [39], reducing key cytokines involved in driving the polarization of classically activated macrophages [40].
Importantly, we showed a significant reduction in neutrophil apoptosis in the TRAIL-deficient mice at time-points coincident with inflammation resolution, demonstrating that reduced neutrophil apoptosis contributes to the increased neutrophil numbers detected in these mice. The rates of apoptosis that we observed, although low, are comparable with those that we have observed previously following i.t. LPS in caspase-1-deficient mice, which have an intrinsic delay of neutrophil apoptosis [1], in a murine model of acute Pseudomonas aeruginosa pneumonia [35], and also are comparable with levels of apoptosis in zebrafish models of neutrophilic inflammation [41]. Other authors have also shown that small increases in rates of neutrophil apoptosis, e.g., induced by the cyclin-dependent kinase inhibitor Roscovitine [2], can substantially accelerate inflammation resolution. Conversely, Bax inhibition delays resolution, with similar relative changes in apoptosis to those observed in this manuscript [42]. Very small, absolute changes in apoptosis can have major differences for cell populations over time. For example, a 0.5% difference from 3.8% to 3.3% of apoptotic CD4+ T cells has been shown over time to be associated with a very profound effect in immune reconstitution in HIV infection, resulting in alterations in mean CD4+ T cell counts from 249 to 632 cells/mL [43]. Early modeling studies of CD4+ T cell apoptosis within LNs showed that a very small increase in percentage of apoptotic cells following antibody treatment, from 0.5% to 1%, was sufficient to substantially reduce cell numbers [44]. Modeling data suggested that each apoptotic cell was visible only for ∼50 min, presumably prior to efferocytosis and degradation, so the transient nature of the “free” apoptotic population likely explains why small numbers of apoptotic cells reflect substantial shifts in population numbers [45].
We measured IgM, a recognized marker of a pulmonary vascular leak in murine models of lung injury [24, 46] and also, importantly, of ARDS in humans [47]. Changes in BAL levels of IgM correlate well with changes in albumin and total protein in BAL [24, 46]. We observed a doubling of IgM levels in TRAIL-deficient compared with WT mice at 48 h and 72 h postinjury, a difference comparable with other studies [24, 48] and demonstrating increased vascular permeability in TRAIL-deficient compared with WT mice.
Histological analysis of murine lungs instilled with LPS revealed up-regulation of TRAIL on the airway epithelium when compared with mice instilled with saline. This epithelial expression of TRAIL could affect cell recruitment to the lung, as work by Li et al. [49] showed that TRAIL induces the expression of E-selectin, ICAM-1, and IL-8, promoting adhesion of leukocytes to the vascular endothelium. However, our data do not suggest that this is a major contributor to the neutrophilic inflammation and instead, raise the possibility that epithelial TRAIL could down-regulate neutrophilic inflammation by induction of apoptosis. Importantly, TUNEL staining of lung sections did not suggest any excess of epithelial cell death as a result of these cells expressing TRAIL. Macrophage expression of death receptor ligands also plays a role in apoptosis induction. A role for macrophage FasL induction of target cell killing has been described for lymphocytes [50] and monocytes [51]. TRAIL-mediated killing of lymphocytes is also described [52]. However, we observed only infrequent TRAIL expression by alveolar macrophages in this model over a range of time-points, and again, this was not associated with any loss of macrophage numbers. Recent data show that levels of soluble TRAIL in BAL samples from ARDS patients are significantly elevated and correlate with neutrophil numbers in the BAL [53]. Neutrophils express TRAIL [54] and up-regulate it in response to certain proinflammatory cytokines [55]. However, histological data suggested that epithelial cells and to a lesser extent, macrophages were the major source of TRAIL in our model, and we were unable to detect BAL neutrophil expression of TRAIL by flow cytometry (data not shown).
We also investigated whether the persistent neutrophilic inflammation observed in TRAIL-deficient mice is restricted to the lung or is a more widespread phenomenon. Zymosan-induced peritonitis caused increased total neutrophil numbers and reduced neutrophil apoptosis in TRAIL-deficient compared with WT mice to a similar extent to the acute lung injury model. Taken together, these data suggest that TRAIL may play a physiological role in regulating the neutrophil lifespan at sites of inflammation. Nonetheless, it should be noted that neutrophilic inflammation still resolves in the absence of TRAIL, in keeping with a role for other endogenous mediators, such as resolvins and related lipids [56] in physiologic resolution, mediators that may potentially also compensate for the absence of TRAIL in limiting the lifespan of inflammatory neutrophils.
Treatment with exogenous TRAIL was evaluated in both models of inflammation and resulted in increased levels of neutrophil apoptosis and reduced neutrophil numbers, particularly in TRAIL-deficient but also significantly in WT mice. These findings fit with previous studies using TRAIL-deficient mice in models of listeriosis [17] and meningitis [18], which provided evidence that TRAIL-deficient mice show prolonged inflammation [18] and delayed neutrophil apoptosis [17], which could be abrogated by TRAIL treatment. Yao et al. [57] have also demonstrated that intra-articular injection of TRAIL reduces leukocyte infiltration in a rabbit knee model of arthritis. More recently, Weckmann et al. [58], in a murine model of asthma, showed increased pulmonary epithelial expression of TRAIL, as in our studies, but also, that inhibition of TRAIL function attenuated allergic inflammation. In contrast, we have shown that the absence of TRAIL is associated with enhanced inflammation in the lung, as in other models of nonallergic inflammation [18]. One theoretical advantage of TRAIL treatment for neutrophilic inflammation is that it induces death in a limited range of cell types [12]. We found no evidence of increased loss of alveolar macrophages from BAL and no evidence of excess epithelial cell death on TUNEL staining. Moreover, although we did not directly assess NF-κB activation of BAL cells, there was no increase in BAL levels of NF-κB-dependent neutrophil chemokines following TRAIL treatment.
Our data therefore suggest TRAIL treatment may have beneficial effects in acute lung injury by reducing the acute inflammatory response. In contrast, the FasL-Fas system appears to have a predominantly proinflammatory effect, up-regulating neutrophil chemokines [7] and inducing epithelial cell apoptosis [59]. Indeed, intrapulmonary instillation of FasL is sufficient to induce neutrophilic inflammation in mice [60].
In summary, we show that the absence of TRAIL results in enhancement of neutrophilic inflammation, suggesting that this ligand may have a role in regulating physiologic inflammation. In two robust and well-characterized models of inflammation, we show that TRAIL treatment restores the phenotype in TRAIL-deficient mice but importantly, also reduced neutrophilic inflammation in WT mice. Recent data from Rossi et al. [2] have shown that acceleration of neutrophil apoptosis can ameliorate inflammation in multiple murine models and are supported by data from other strategies for modulation of neutrophil apoptosis in inflammation in vivo, e.g., via hypoxia signaling pathways [61]. The data presented similarly suggest a possible therapeutic role for TRAIL that can be explored further in more injurious models of persistent neutrophilic inflammation.
ACKNOWLEDGMENTS
This work was funded by a Wellcome Trust Clinical Research Training Fellowship to E.E.M. (075776) and by the National Institute of Health Research Cardiovascular Biomedical Research Unit Sheffield (I.S. and M.K.B.W.). A.L. holds a MRC Career Development Award Fellowship (G0800318), S.A.R. is a MRC Senior Clinical Fellow (G0701932), and D.H.D. is a Wellcome Trust Senior Clinical Fellow (076945).
SEE CORRESPONDING EDITORIAL ON PAGE 841
- ARDS
- acute respiratory distress syndrome
- BALF
- BAL fluid
- FasL
- Fas ligand
- i.t.
- intratracheal
- KC
- keratinocyte-derived chemokine
- MRC
- Medical Research Council
- t
- time
AUTHORSHIP
E.E.M. performed the bulk of experimental work, with H.M.M., D.H.D., and S.E.F. involved in establishing and conducting in vivo experiments. A.L. performed and interpreted immunohistochemistry. M.K.B.W., I.S., S.A.R., and D.H.D. were involved in all aspects of experimental design and together with E.E.M., wrote the manuscript. All authors reviewed and commented on the manuscript.
REFERENCES
- 1. Rowe S. J., Allen L., Ridger V. C., Hellewell P. G., Whyte M. K. (2002) Caspase-1-deficient mice have delayed neutrophil apoptosis and a prolonged inflammatory response to lipopolysaccharide-induced acute lung injury. J. Immunol. 169, 6401–6407 [DOI] [PubMed] [Google Scholar]
- 2. Rossi A. G., Sawatzky D. A., Walker A., Ward C., Sheldrake T. A., Riley N. A., Caldicott A., Martinez-Losa M., Walker T. R., Duffin R., Gray M., Crescenzi E., Martin M. C., Brady H. J., Savill J. S., Dransfield I., Haslett C. (2006) Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064 [DOI] [PubMed] [Google Scholar]
- 3. Jones H. A., Clark R. J., Rhodes C. G., Schofield J. B., Krausz T., Haslett C. (1994) In vivo measurement of neutrophil activity in experimental lung inflammation. Am. J. Respir. Crit. Care Med. 149, 1635–1639 [DOI] [PubMed] [Google Scholar]
- 4. Bianchi S. M., Dockrell D. H., Renshaw S. A., Sabroe I., Whyte M. K. (2006) Granulocyte apoptosis in the pathogenesis and resolution of lung disease. Clin. Sci. (Lond.) 110, 293–304 [DOI] [PubMed] [Google Scholar]
- 5. Liles W. C., Kiener P. A., Ledbetter J. A., Aruffo A., Klebanoff S. J. (1996) Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J. Exp. Med. 184, 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Renshaw S. A., Timmons S. J., Eaton V., Usher L. R., Akil M., Bingle C. D., Whyte M. K. B. (2000) Inflammatory neutrophils retain susceptibility to apoptosis mediated via the Fas death receptor. J. Leukoc. Biol. 67, 662–668 [DOI] [PubMed] [Google Scholar]
- 7. Neff T. A., Guo R. F., Neff S. B., Sarma J. V., Speyer C. L., Gao H., Bernacki K. D., Huber-Lang M., McGuire S., Hoesel L. M., Riedemann N. C., Beck-Schimmer B., Zetoune F. S., Ward P. A. (2005) Relationship of acute lung inflammatory injury to Fas/FasL system. Am. J. Pathol. 166, 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ward C., Chilvers E. R., Lawson M. F., Pryde J. G., Fujihara S., Farrow S. N., Haslett C., Rossi A. G. (1999) NF-κB is a critical regulator of granulocyte apoptosis in vitro. J. Biol. Chem. 274, 4309–4318 [DOI] [PubMed] [Google Scholar]
- 9. Murray J., Barbara J. A., Dunkley S. A., Lopez A. F., Van Ostade X., Condliffe A. M., Dransfield I., Haslett C., Chilvers E. R. (1997) Regulation of neutrophil apoptosis by tumor necrosis factor-α: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood 90, 2772–2783 [PubMed] [Google Scholar]
- 10. Renshaw S. A., Parmar J. S., Singleton V., Rowe S. J., Dockrell D. H., Dower S. K., Bingle C. D., Chilvers E. R., Whyte M. K. (2003) Acceleration of human neutrophil apoptosis by TRAIL. J. Immunol. 170, 1027–1033 [DOI] [PubMed] [Google Scholar]
- 11. Lum J. J., Bren G., McClure R., Badley A. D. (2005) Elimination of senescent neutrophils by TNF-related apoptosis-inducing [corrected] ligand. J. Immunol. 175, 1232–1238 [DOI] [PubMed] [Google Scholar]
- 12. Sheridan J. P., Marsters S. A., Pitti R. M., Gurney A., Skubatch M., Baldwin D., Ramakrishnan L., Gray C. L., Baker K., Wood W. I., Goddard A. D., Godowski P., Ashkenazi A. (1997) Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277, 818–821 [DOI] [PubMed] [Google Scholar]
- 13. Johnstone R. W., Frew A. J., Smyth M. J. (2008) The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 8, 782–798 [DOI] [PubMed] [Google Scholar]
- 14. Wu G. S., Burns T. F., Zhan Y., Alnemri E. S., El-Deiry W. S. (1999) Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 59, 2770–2775 [PubMed] [Google Scholar]
- 15. Lamhamedi-Cherradi S. E., Zheng S. J., Maguschak K. A., Peschon J., Chen Y. H. (2003) Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL−/− mice. Nat. Immunol. 4, 255–260 [DOI] [PubMed] [Google Scholar]
- 16. Cretney E., McQualter J. L., Kayagaki N., Yagita H., Bernard C. C., Grewal I. S., Ashkenazi A., Smyth M. J. (2005) TNF-related apoptosis-inducing ligand (TRAIL)/Apo2L suppresses experimental autoimmune encephalomyelitis in mice. Immunol. Cell Biol. 83, 511–519 [DOI] [PubMed] [Google Scholar]
- 17. Zheng S. J., Jiang J., Shen H., Chen Y. H. (2004) Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J. Immunol. 173, 5652–5658 [DOI] [PubMed] [Google Scholar]
- 18. Hoffmann O., Priller J., Prozorovski T., Schulze-Topphoff U., Baeva N., Lunemann J. D., Aktas O., Mahrhofer C., Stricker S., Zipp F., Weber J. R. (2007) TRAIL limits excessive host immune responses in bacterial meningitis. J. Clin. Invest. 117, 2004–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cretney E., Takeda K., Yagita H., Glaccum M., Peschon J. J., Smyth M. J. (2002) Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 168, 1356–1361 [DOI] [PubMed] [Google Scholar]
- 20. Cotter M. J., Norman K. E., Hellewell P. G., Ridger V. C. (2001) A novel method for isolation of neutrophils from murine blood using negative immunomagnetic separation. Am. J. Pathol. 159, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dockrell D. H., Marriott H. M., Prince L. R., Ridger V. C., Ince P. G., Hellewell P. G., Whyte M. K. (2003) Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J. Immunol. 171, 5380–5388 [DOI] [PubMed] [Google Scholar]
- 22. Ridger V. C., Wagner B. E., Wallace W. A., Hellewell P. G. (2001) Differential effects of CD18, CD29, and CD49 integrin subunit inhibition on neutrophil migration in pulmonary inflammation. J. Immunol. 166, 3484–3490 [DOI] [PubMed] [Google Scholar]
- 23. Doherty N. S., Poubelle P., Borgeat P., Beaver T. H., Westrich G. L., Schrader N. L. (1985) Intraperitoneal injection of zymosan in mice induces pain, inflammation and the synthesis of peptidoleukotrienes and prostaglandin E2. Prostaglandins 30, 769–789 [DOI] [PubMed] [Google Scholar]
- 24. Mei S. H., McCarter S. D., Deng Y., Parker C. H., Liles W. C., Stewart D. J. (2007) Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 4, e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang X., Long L., Southwood M., Rudarakanchana N., Upton P. D., Jeffery T. K., Atkinson C., Chen H., Trembath R. C., Morrell N. W. (2005) Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 96, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 26. Lawrie A., Waterman E., Southwood M., Evans D., Suntharalingam J., Francis S., Crossman D., Croucher P., Morrell N., Newman C. (2008) Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am. J. Pathol. 172, 256–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abraham E., Carmody A., Shenkar R., Arcaroli J. (2000) Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L1137–L1145 [DOI] [PubMed] [Google Scholar]
- 28. Marriott H. M., Jackson L. E., Wilkinson T. S., Simpson A. J., Mitchell T. J., Buttle D. J., Cross S. S., Ince P. G., Hellewell P. G., Whyte M. K. B., Dockrell D. H. (2008) Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 177, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seino K., Iwabuchi K., Kayagaki N., Miyata R., Nagaoka I., Matsuzawa A., Fukao K., Yagita H., Okumura K. (1998) Chemotactic activity of soluble Fas ligand against phagocytes. J. Immunol. 161, 4484–4488 [PubMed] [Google Scholar]
- 30. Nakamura M., Matute-Bello G., Liles W. C., Hayashi S., Kajikawa O., Lin S. M., Frevert C. W., Martin T. R. (2004) Differential response of human lung epithelial cells to Fas-induced apoptosis. Am. J. Pathol. 164, 1949–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miyazaki Y., Araki K., Vesin C., Garcia I., Kapanci Y., Whitsett J. A., Vassalli P. (1995) Expression of a tumor necrosis factor-α transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J. Clin. Invest. 96, 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hilberath J. N., Carlo T., Pfeffer M. A., Croze R. H., Hastrup F., Levy B. D. (2011) Resolution of Toll-like receptor 4-mediated acute lung injury is linked to eicosanoids and suppressor of cytokine signaling 3. FASEB J., Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. D′Alessio F. R., Tsushima K., Aggarwal N. R., West E. E., Willett M. H., Britos M. F., Pipeling M. R., Brower R. G., Tuder R. M., McDyer J. F., King. L. S. (2009) CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Invest. 119, 2898–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee J. S., Wurfel M. M., Matute-Bello G., Frevert C. W., Rosengart M. R., Ranganathan M., Wong V. W., Holden T., Sutlief S., Richmond A., Peiper S., Martin T. R. (2006) The Duffy antigen modifies systemic and local tissue chemokine responses following lipopolysaccharide stimulation. J. Immunol. 177, 8086–8094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Allen L., Dockrell D. H., Pattery T., Lee D. G., Cornelis P., Hellewell P. G., Whyte M. K. (2005) Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J. Immunol. 174, 3643–3649 [DOI] [PubMed] [Google Scholar]
- 36. Everhart M. B., Han W., Sherrill T. P., Arutiunov M., Polushiokin V. V., Burke J. R., Sadikot R. T., Christman J. W., Yull F. E., Blackwell T. S. (2006) Duration and intensity of NF-κB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 176, 4995–5005 [DOI] [PubMed] [Google Scholar]
- 37. Buckley C. D., Ross E. A., McGettrick H. M., Osborne C. E., Haworth O., Schmutz C., Stone P. C., Salmon M., Matharu N. M., Vohra R. K., Nash G. B., Rainger G. E. (2006) Identification of a phenotypically and functionally distinct population of longlived neutrophils in a model of reverse endothelial migration. J. Leukoc. Biol. 79, 303–311 [DOI] [PubMed] [Google Scholar]
- 38. Serhan C. N. (2010) Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am. J. Pathol. 177, 1576–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marriott H. M., Hellewell P. G., Cross S. S., Ince P. G., Whyte M. K., Dockrell D. H. (2006) Decreased alveolar macrophage apoptosis is associated with increased pulmonary inflammation in a murine model of pneumococcal pneumonia. J. Immunol. 177, 6480–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mosser D. M., Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Loynes C. A., Martin J. S., Robertson A., Trushell D. M., Ingham P. W., Whyte M. K., Renshaw S. A. (2010) Pivotal advance: pharmacological manipulation of inflammation resolution during spontaneously resolving tissue neutrophilia in the zebrafish. J. Leukoc. Biol. 87, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sawatzky D. A., Willoughby D. A., Colville-Nash P. R., Rossi A. G. (2006) The involvement of the apoptosis-modulating proteins ERK 1/2, Bcl-xL and Bax in the resolution of acute inflammation in vivo. Am. J. Pathol. 168, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Negredo E., Massanella M., Puig J., Pérez-Alvarez N., Gallego-Escuredo J. M., Villarroya J., Villarroya F., Moltó J., Santos J. R., Clotet B., Blanco J. (2010) Nadir CD4 T cell count as predictor and high CD4 T cell intrinsic apoptosis as final mechanism of poor CD4 T cell recovery in virologically suppressed HIV-infected patients: clinical implications. Clin. Infect. Dis. 50, 1300–1308 [DOI] [PubMed] [Google Scholar]
- 44. Howie S. E., Sommerfield A. J., Gray E., Harrison D. J. (1994) Peripheral T lymphocyte depletion by apoptosis after CD4 ligation in vivo: selective loss of DC44- and ″activating″ memory T cells. Clin. Exp. Immunol. 95, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bellamy C. O., Malcolmson R. D., Harrison D. J., Wyllie A. H. (1995) Cell death in health and disease: the biology and regulation of apoptosis. Semin. Cancer Biol. 6, 3–16 [DOI] [PubMed] [Google Scholar]
- 46. Smith L. S., Gharib S. A., Frevert C. W., Martin T. R. (2010) Effects of age on the synergistic interactions between lipopolysaccharide and mechanical ventilation in mice. Am. J. Respir. Cell Mol. Biol. 43, 475–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holter J. F., Weiland J. E., Pacht E. R., Gadek J. E., David W. B. (1986) Protein permeability in the adult respiratory distress syndrome. Loss of size selectivity of the alveolar epithelium. J. Clin. Invest. 78, 1513–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lipke A. B., Matute-Bello G., Herrero R., Kurahashi K., Wong V. A., Mongovin S. M., Martin T. R. (2010) Febrile-range hyperthermia augments lipopolysaccharide-induced lung injury by a mechanism of enhanced alveolar epithelial apoptosis. J. Immunol. 184, 3801–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li J. H., Kirkiles-Smith N. C., McNiff J. M., Pober J. S. (2003) TRAIL induces apoptosis and inflammatory gene expression in human endothelial cells. J. Immunol. 171, 1526–1533 [DOI] [PubMed] [Google Scholar]
- 50. Badley A. D., Dockrell D., Simpson M., Schut R., Lynch D. H., Leibson P., Paya C. V. (1997) Macrophage-dependent apoptosis of CD4+ T lymphocytes from HIV-infected individuals is mediated by FasL and tumor necrosis factor. J. Exp. Med. 185, 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dockrell D. H., Lee M., Lynch D. H., Read R. C. (2001) Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J. Infect. Dis. 184, 713–722 [DOI] [PubMed] [Google Scholar]
- 52. Lum J. J., Pilon A. A., Sanchez-Dardon J., Phenix B. N., Kim J. E., Mihowich J., Jamison K., Hawley-Foss N., Lynch D. H., Badley A. D. (2001) Induction of cell death in human immunodeficiency virus-infected macrophages and resting memory CD4 T cells by TRAIL/Apo2l. J. Virol. 75, 11128–11136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee K. S., Choi Y. H., Kim Y. S., Baik S. H., Oh Y. J., Sheen S. S., Park J. H., Hwang S. C., Park K. J. (2008) Evaluation of bronchoalveolar lavage fluid from ARDS patients with regard to apoptosis. Respir. Med. 102, 464–469 [DOI] [PubMed] [Google Scholar]
- 54. Simons M. P., Leidal K. G., Nauseef W. M., Griffith T. S. (2008) TNF-related apoptosis-inducing ligand (TRAIL) is expressed throughout myeloid development, resulting in a broad distribution among neutrophil granules. J. Leukoc. Biol. 83, 621–629 [DOI] [PubMed] [Google Scholar]
- 55. Cassatella M. A., Huber V., Calzetti F., Margotto D., Tamassia N., Peri G., Mantovani A., Rivoltini L., Tecchio C. (2006) Interferon-activated neutrophils store a TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) intracellular pool that is readily mobilizable following exposure to proinflammatory mediators. J. Leukoc. Biol. 79, 123–132 [DOI] [PubMed] [Google Scholar]
- 56. Uddin M., Levy B. D. (2011) Resolvins: natural agonists for resolution of pulmonary inflammation. Prog. Lipid Res. 50, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yao Q., Seol D. W., Mi Z., Robbins P. D. (2006) Intra-articular injection of recombinant TRAIL induces synovial apoptosis and reduces inflammation in a rabbit knee model of arthritis. Arthritis Res. Ther. 8, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weckmann M., Collison A., Simpson J. L., Kopp M. V., Wark P. A., Smyth M. J., Yagita H., Matthaei K. I., Hansbro N., Whitehead B., Gibson P. G., Foster P. S., Mattes J. (2007) Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nat. Med. 13, 1308–1315 [DOI] [PubMed] [Google Scholar]
- 59. Matute-Bello G., Winn R. K., Jonas M., Chi E. Y., Martin T. R., Liles W. C. (2001) Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implications for acute pulmonary inflammation. Am. J. Pathol. 158, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wortinger M. A., Foley J. W., Larocque P., Witcher D. R., Lahn M., Jakubowski J. A., Glasebrook A., Song H. Y. (2003) Fas ligand-induced murine pulmonary inflammation is reduced by a stable decoy receptor 3 analogue. Immunology 110, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Walmsley S. R., Chilvers E. R., Thompson A. A., Vaughan K., Marriott H. M., Parker L. C., Shaw G., Parmar S., Schneider M., Sabroe I., Dockrell D. H., Milo M., Taylor C. T., Johnson R. S., Pugh C. W., Ratcliffe P. J., Maxwell P. H., Carmeliet P., Whyte M. K. (2011) Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J. Clin. Invest. 121, 1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]