

Abstract
Despite advances in cardiopulmonary resuscitation (CPR) methods including therapeutic hypothermia (TH), long-term neurological outcomes and survival after sudden cardiac arrest (CA) remains to be dismal. While nitric oxide (NO) prevents organ injury induced by ischemia and reperfusion (I/R), systemic vasodilation induced by intravenous NO-donor compounds typically precludes its use in post-CA patients in whom blood pressure is often low and unstable. Although developed as a selective pulmonary vasodilator, inhaled NO has systemic benefits in a variety of pre-clinical and clinical studies without causing potentially harmful systemic vasodilation. Breathing NO after CPR may prevent post-CA brain injury and improve long-term outcomes after CA and CPR.
Out-of-hospital cardiac arrest (OHCA) claims the lives of an estimated 310,000 Americans each year (Roger et al., 2012). Despite advances in cardiopulmonary resuscitation (CPR) methods, including the introduction of the automatic electrical defibrillator (AED) and therapeutic hypothermia (TH) (Bernard et al., 2002; The Hypothermia after Cardiac Arrest Study Group, 2002), only about 10% of adults treated for OHCA survive to hospital discharge, and up to 60% of survivors have moderate to severe cognitive deficits 3 months after resuscitation (Roine et al., 1993). Although the greatest proportion of in-hospital post-CA mortality and morbidity is caused by global ischemic brain injury, the severity of both myocardial dysfunction and systemic inflammation correlates with poor neurological outcome (Laver et al., 2004). The mechanisms responsible for post-CA brain injury include excitotoxicity, free radical formation, pathological activation of proteases, and cell death signalling (Neumar, 2000; Neumar et al., 2008). Many of the injurious pathways are executed over hours to days following return of spontaneous circulation (ROSC) causing disruption of blood–brain barrier (BBB), neuroinflammation, and delayed neurodegeneration (Fujioka et al., 2003; Sharma et al., 2011). While the protracted time-course of brain injury suggests a broad therapeutic window for neuroprotective strategies following CA (Neumar et al., 2008), no pharmacological agents have been proven effective in improving neurological outcomes in post-CA patients. Although TH confers significant protective effects when applied for 12–24 h after ventricular fibrillation (VF)-induced CA in adults, TH has been shown to benefit (improvement of neurological outcome), at most, 20% of victims in whom ROSC is achieved (Bernard et al., 2002; The Hypothermia after Cardiac Arrest Study Group, 2002). Therefore, additional therapies are urgently needed (Peberdy et al., 2010).
Nitric oxide (NO) is produced from NO synthases (NOS1, NOS2, and NOS3). One of the primary targets of NO is soluble guanylate cyclase (sGC) that generates the second messenger cGMP upon activation. sGC is a heme-containing heterodimeric enzyme composed of one α and one β subunit. In most tissues, including heart, lung, and vascular smooth muscle cells, the sGCα1β1 heterodimer is the predominant isoform. NO binds to the heme moiety of sGC and stimulates the synthesis of cGMP (Friebe and Koesling, 2003). cGMP exerts its effects by interacting with cGMP-dependent protein kinases (PKG), cGMP-regulated phosphodiesterases (PDE), and cGMP-regulated ion channels (Fig. 1). cGMP is metabolized to the relatively inactive GMP by PDEs. Increasing evidence has demonstrated the importance of cGMP-independent signaling in the biological effects of NO. NO can elicit effects by reacting with a variety of molecules, typically via thiol groups (–SH) or transition metal centers (Stamler, 1994). NO modulates functions of a number of proteins by S-nitrosylation (Jaffrey et al., 2001).
Fig. 1.
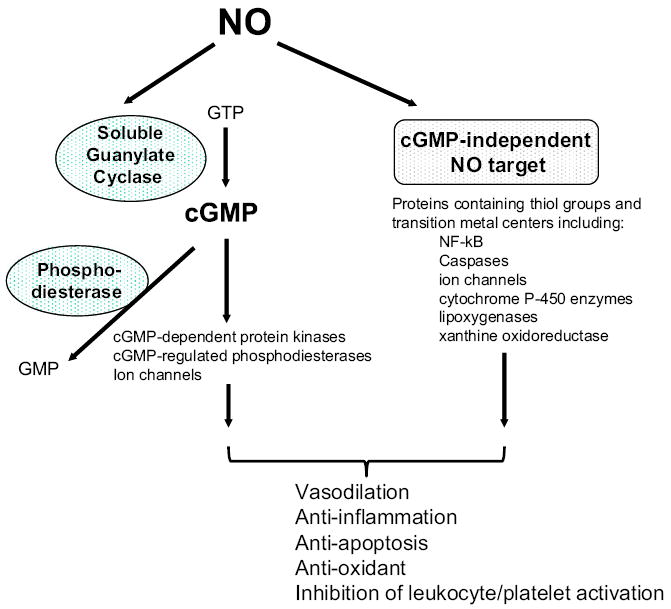
Nitric oxide signaling pathway. cGMP, cyclic guanosine monophosphate; GTP, guanosine triphosphate; GMP, guanosine monophosphate.
NO exerts a number of effects that would be expected to be beneficial during I/R injury (Bloch et al., 2007). For example, NO is a potent vasodilator which inhibits platelet and leukocyte activation and adhesion, inhibits reactive oxygen species (ROS)-producing enzymes, and directly scavenge ROS (Kubes et al., 1991). Given the vasodilating effects of NO, earlier studies in the setting of cardiac arrest examined the effects of pharmacological NOS inhibition on outcomes after CA/CPR with conflicting results. Chemical inhibitors that inhibit all NOS have been reported to improve (Krismer et al., 2001), worsen (Adams et al., 2007), or not change (Zhang et al., 2005) short term outcomes in swine models of CA/CPR. In contrast, studies using mice genetically deficient for NOS3 have consistently demonstrated salutary roles of NOS3 in I/R. Deficiency of NOS3 has been shown to aggravate I/R injury in brain and heart (Huang et al., 1996; Jones et al., 1999). Along these lines, we reported that deficiency of NOS3 or sGCα1 worsened outcomes of CA/CPR, whereas cardiomyocyte-specific overexpression of NOS3 rescued NOS3-deficient mice from myocardial and neurological dysfunction and death after CA/CPR (Nishida et al., 2009). Beneficial role of NOS3/sGC after CA was further supported by Beiser and colleagues who reported that poor cardiovascular outcomes and survival in NOS3-deficient mice after CA/CPR are associated with decreased myocardial cGMP levels (Beiser et al., 2011).
The salutary effects of NO in I/R appear to be mediated via multiple mechanisms. Dezfulian and colleagues showed that systemic administration of nitrite, which is converted in vivo to NO, improves outcome in mice 24 h after CA/CPR by reducing pathological cardiac mitochondrial oxygen consumption resulting from reactive oxygen species formation (Dezfulian et al., 2009). Administration of nitrite prevented oxidative enzymatic injury via reversible specific inhibition of respiratory chain complex I after CA/CPR. Beneficial effects of nitrite were associated with increased levels of S-nitrosothiols in the heart and brain (Dezfulian et al., 2012, 2009).
Based on the evidence that support protective effects of NO after I/R injury including CA/CPR, several treatment strategies that increase NO content in post-ischemic tissues have been examined with varying degrees of success. Unfortunately, the therapeutic use of NO-donor compounds after I/R injury appears to be hindered by their systemic vasodilator effects potentially leading to hypotension and further compromise of tissue perfusion that is already tenuous after I/R.
Inhaled NO is a selective pulmonary vasodilator that does not produce systemic hypotension when inhaled at concentrations up to 80 ppm in multiple species, including man (Ichinose et al., 2004). The absence of systemic vasodilation during NO inhalation is due to the rapid scavenging of NO by hemoglobin in the blood. Inhaled NO has been approved for the treatment of neonatal hypoxemia with acute pulmonary hypertension (Griffiths and Evans, 2005). However, breathing NO also has systemic effects (Hogman et al., 1993). For example, breathing NO was shown to reduce I/R injury of extrapulmonary organs in a variety of animal models (Fox-Robichaud et al., 1998; Guery et al., 1999; Hataishi et al., 2006; Liu et al., 2007; Nagasaka et al., 2008). The ability of inhaled NO to reduce I/R injury was subsequently reproduced in “proof-of-principle” human studies (Lang et al., 2007; Mathru et al., 2007). Based on these observations, we hypothesized that NO inhalation could improve outcomes after CA/CPR.
To examine effects of NO inhalation on the outcome of CA/ CPR in a clinically relevant manner, we have developed and thoroughly characterized a murine model of CA/CPR, in which mice exhibit poor neurological outcomes and survival after resuscitation from CA (Kida et al., 2012; Minamishima et al., 2009, 2011; Nishida et al., 2009). Briefly, after instrumentation under general anesthesia, CA was induced by an i.v. injection of potassium chloride (KCl). After 7.5 min of arrest time, CPR was performed with chest compression (~350/min), mechanical ventilation, and continuous i.v. infusion of epinephrine. Mice were weaned from mechanical ventilation and extubated at 1 h after successful CPR. Mice were then randomized to breath air alone or air supplemented with 40 ppm NO for 23 h in custom-made chambers.
There was no difference between treatment groups in the CPR time to return of spontaneous circulation (ROSC), the total epinephrine dose, blood pressure, and heart rate 1 h after CPR. The partial pressure of oxygen (PaO2) and oxygen saturation (SaO2) of arterial blood samples obtained at 2 h after CPR (1 h after initiation of air or NO breathing) did not differ between mice that breathed air or air supplemented with NO. There was no difference in core body temperature between mice that breathed air or air supplemented with NO for the first 24 h after CA/CPR. While only 4 out of 13 mice that breathed air survived 10 days after CPR, 11 out of 13 mice that breathed NO for 23 h starting 1 h after CPR survived for 10 days (P=0.003, Fig. 2).
Fig. 2.
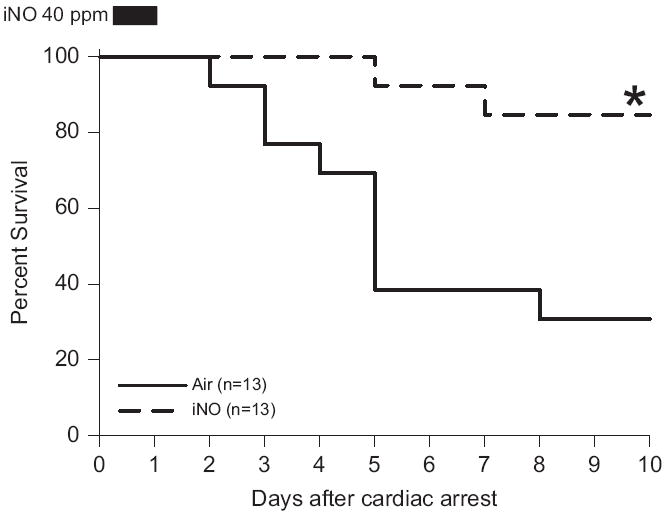
Survival rate of wild-type mice during the first 10 days after cardiac arrest and CPR. Air, mice subjected to CA/CPR and breathed air for 23 h starting 1 h after CPR. iNO, mice subjected to CA/CPR and breathed air supplemented with NO for 23 h starting 1 h after CPR. *P=0.003 vs. Air by Log-rank test (Minamishima et al., 2011).
Mice that were successfully resuscitated from 7.5 min of CA and breathed air exhibited a marked abnormality in water diffusion in the hippocampus, caudoputamen, and cortex 24 h after CPR (Fig. 3) (Minamishima et al., 2011). The presence of abnormal DWI signals in the vulnerable regions of the brain 1 day after CA/CPR correlated with worse neurological function and increased apoptosis of hippocampal neurons 4 days after CPR, as well as decreased rate of survival at 10 days. In contrast, NO breathing markedly attenuated the development of abnormality in water diffusion in the brain and improved neurological outcomes and survival rate. These observations are consistent with a recent clinical study that showed that diffuse cortical abnormalities in DWI are associated with poor outcomes in patients resuscitated from CA (Wijman et al., 2009). Hyperintense DWI signals indicate the presence of brain edema presumably due to disruption of ion pump function and membrane failure. The current observations, therefore, suggest that NO inhalation after successful CPR can preserve ion pump homeostasis and membrane integrity early after CA/CPR.
Fig. 3.
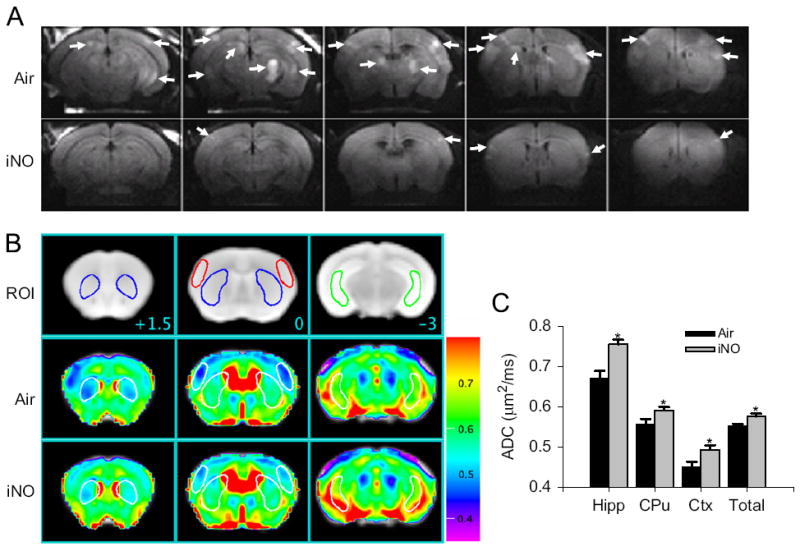
(A) Representative diffusion-weighted image (DWI) of mice brain 24 h after CA/CPR that breathed air (Air) or air supplemented with NO (iNO). White arrows indicate areas of hyperintense DWI. (B) Representative MR images showing three brain slices containing regions of interest [ROI]. Slice positions are identified in millimeters (+1.5, 0, or −3 mm) with respect to bregma in the coordinate space of the Allen Mouse Brain Atlas. Colored outlines indicate portions of ROI (blue, caudoputamen; red, lateral cortex; green, ventral lateral hippocampus) that intersect with these slice planes. Average ADC values of the slice plane for mice that breathed Air after CA/CPR [Air]. Average ADC values of the slice plane for mice that breathed NO after CA/CPR [iNO]. Color bar on the right side indicates color-code for ADC values (μm2/ms). (C) Average ADC values of each three-dimensional ROI (Hipp, ventral lateral hippocampus; CPu, caudoputamen; Cortex, lateral cortex; total, total brain) across all planes in mice that breathed air (Air, n = 6) or NO (iNO, n = 7) after CA/CPR. *P<0.05 vs. Air (Minamishima et al., 2011).
Neuroinflammation triggered by the whole-body IR injury associated with CA/CPR hinder the neurological recovery from prolonged CA. We observed that CA/CPR markedly upregulated the expression of genes encoding inflammatory cytokines and NADPH oxidase in the brain of WT mice that breathed air, but not in WT mice that breathed air supplemented with NO. These observations suggest that NO inhalation prevents neuroinflammation after CA/CPR. Furthermore, these results demonstrate a correlation between neuroinflammation, neurological dysfunction, and mortality after CA/CPR.
NO elicits biological effects via sGC-dependent and/or - independent mechanisms. To determine the role of sGC in the protective effects of inhaled NO on the outcome of CA/ CPR, we studied sGCα1−/− mice. We observed that sGCα1-deficiency increased the early mortality rate (in the first 2 h after CPR) when compared to WT mice after CA/CPR, consistent with our previous report (Nishida et al., 2009). While the cause of these early deaths is unknown, we previously reported that sGCα1 deficiency markedly exacerbated LV dysfunction early after CA/CPR (Nishida et al., 2009). After excluding the mice that died early after CPR, sGCα1−/− mice that breathed air had 10-day survival rate comparable to that in WT mice that breathed air after CA/CPR. These observations suggest that sGC activity is critically important for initial recovery after CA/CPR but may not be necessary for long-term survival after CA/CPR. In contrast, sGCα1-deficiency abolished the ability of NO inhalation to inhibit the induction of inflammatory cytokines in the brain and to improve neurological function and 10-day survival rate after CA (Minamishima et al., 2011). These observations suggest that protective effects of inhaled NO on the outcome of CA/ CPR are largely mediated via sGC-dependent mechanisms.
Our data does not exclude the possibility that sGC-independent mechanisms could contribute to the protective effects of inhaled NO on peripheral organs after CA/CPR. It is conceivable that NO modifies functions of enzymes and ion channels in a sGC-independent manner (Dezfulian et al., 2009; Kohr et al., 2011). For example, ischemic preconditioning has been shown to protect cardiomyocytes from subsequent IR injury by preventing Ca2+ overload via S-nitrosylation-mediated inhibition of L-type Ca2+ channel α1 subunit (Sun et al., 2007).
The mechanisms whereby inhaled NO exerts systemic effects are incompletely defined. It is conceivable that circulating cells are directly exposed to NO as they pass through the pulmonary capillaries and may be “pacified” in an sGC-dependent manner before they reach the reperfused peripheral tissues including brain and heart (Fig. 4). It has been reported that poor survival after CA/CPR is associated with marked leukocyte infiltration in brain, heart, lung, liver, and kidney in mice.11 Along these lines, we previously reported that neutrophils are required for inhaled NO to reduce MI size in WT mice subjected to transient left coronary artery occlusion (Hataishi et al., 2006). In a recent study, we observed that NO breathing markedly decreased MI size in WT but not in sGCα1−/− mice (Nagasaka et al., 2011). Furthermore, breathing NO decreased MI size in chimeric sGCα1−/− mice carrying WT BM generated by BM transplantation. These results raise the possibility that the neuroprotective effects of inhaled NO after CA/CPR may be mediated by BM-derived cells in a sGC-dependent manner.
Fig. 4.
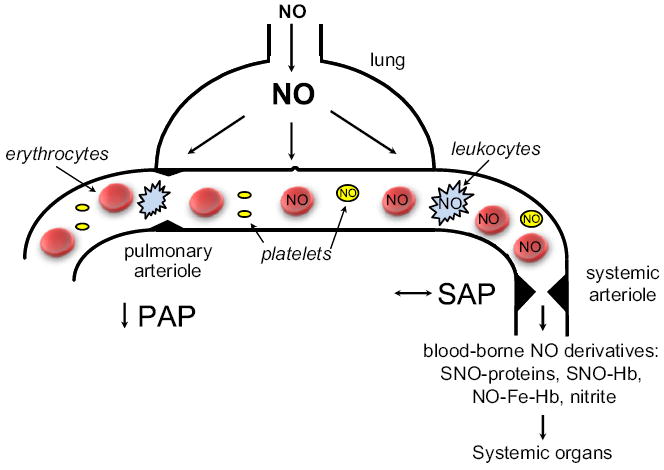
Inhaled NO is a selective pulmonary vasodilator with actions on the systemic vasculature. A schematic of an alveolar-capillary unit is presented highlighting the ability of inhaled NO to dilate pulmonary arterioles and reduce pulmonary artery pressure (PAP). Although inhaled NO does not dilate systemic arterioles or alter systemic arterial pressure (SAP) under normal conditions, inhaled NO does have systemic effects which are described in the text and may be mediated by circulating cells exposed to NO in the lungs and blood-borne NO derivatives: SNO-proteins — S-nitroso proteins including SNO-albumin; SNO-Hb — S-nitroso-hemoglobin (nitrosylated on Cys93 of the β chain); NO-Fe-Hb – nytrosyl-hemoglobin; and nitrite.
Alternatively, some NO, once inhaled, may escape scavenging by hemoglobin and be converted to relatively stable NO metabolites (e.g., nitrite and S-nitrosothiols) that can regenerate NO in the periphery (Cannon et al., 2001) (Fig. 4). In fact, beneficial effects of NO inhalation to reduce MI size in mice were associated with marked increase of NO metabolites (Nagasaka et al., 2008). Similarly, we found that NO inhalation markedly increased plasma nitrite levels in mice after CA/CPR (Minamishima et al., 2011). Regenerated NO in the periphery may exert vasodilating effects. For example, a recent study by Terpolilli et al. (2011) showed that NO inhalation prevented ischemic brain injury in mice and sheep by selective dilation of collateral arterioles. The cerebral vasodilating effects of NO inhalation were associated with increased levels of nitrite and S-nitroso-hemoglobin in arterial blood and were abolished by sGC inhibition.
From the viewpoint of translating our results into clinical benefit, it is of particular importance that NO inhalation started 1 h after successful CPR and continued for 23 h markedly improves neurological and myocardial function and survival rate 10 days after CA/CPR. For example, NO inhalation can be started after patients are transferred to hospital and informed consent obtained. To date, TH is the only therapeutic approach that is proven to improve outcomes after CA/CPR when applied hours after successful CPR (Bernard et al., 2002; The Hypothermia after Cardiac Arrest Study Group, 2002). Since body temperature of mice were allowed to decrease to ~30 °C during NO inhalation in the first 24 h after CA/CPR in our recent study, the data suggests that NO breathing may confer protection in the setting of mild hypothermia. Nonetheless, effects of combination of inhaled NO with TH, compared to either alone, on outcomes after CA/CPR remains to be formally determined in future studies.
In summary, our study revealed robust protective effects of NO inhalation on the outcome of CA/CPR in mice. Breathing NO at 40 ppm for 23 h starting 1 h after successful CPR markedly improved myocardial and neurological function and survival rate 10 days after CA/CPR, at least in part, via sGC-dependent mechanisms. The ability of “delayed” NO breathing to prevent the post-CA brain injury and promote survival in mice, if extrapolated to human beings, is highly clinically relevant and may serve as the experimental basis for future clinical trials in which effects of inhaled NO on the outcome after CA/CPR are examined. We anticipate that the established safety profile of NO inhalation (Ichinose et al., 2004) will enable the rapid translation of findings in animal models to patients suffering not only from the cardiac arrest but also from variety of disease states caused by ischemia and reperfusion.
Acknowledgments
This work was supported by funding from NIH R01 grants HL101930 and HL110378 and a sponsored research agreement from IKARIA Inc.
References
- Adams JA, Wu D, Bassuk J, Arias J, Lozano H, Kurlansky P, et al. Nitric oxide synthase isoform inhibition before whole body ischemia reperfusion in pigs: Vital or protective? Resuscitation. 2007;74:516–25. doi: 10.1016/j.resuscitation.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Beiser DG, Orbelyan GA, Inouye BT, Costakis JG, Hamann KJ, McNally EM, et al. Genetic deletion of NOS3 increases lethal cardiac dysfunction following mouse cardiac arrest. Resuscitation. 2011;82:115–21. doi: 10.1016/j.resuscitation.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. New England Journal of Medicine. 2002;346:557–63. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Bloch KD, Ichinose F, Roberts JD, Jr, Zapol WM. Inhaled NO as a therapeutic agent. Cardiovascular Research. 2007;75:339–48. doi: 10.1016/j.cardiores.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon RO, 3rd, Schechter AN, Panza JA, Ognibene FP, Pease-Fye ME, Waclawiw MA, et al. Effects of inhaled nitric oxide on regional blood flow are consistent with intravascular nitric oxide delivery. Journal of Clinical Investigation. 2001;108:279–87. doi: 10.1172/JCI12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezfulian C, Alekseyenko A, Dave KR, Raval AP, Do R, Kim F, et al. Nitrite therapy is neuroprotective and safe in cardiac arrest survivors. Nitric Oxide. 2012;26:241–50. doi: 10.1016/j.niox.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezfulian C, Shiva S, Alekseyenko A, Pendyal A, Beiser DG, Munasinghe JP, et al. Nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves cardiac and neurological function, and enhances survival via reversible inhibition of mitochondrial complex I. Circulation. 2009;120:897–905. doi: 10.1161/CIRCULATIONAHA.109.853267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox-Robichaud A, Payne D, Hasan SU, Ostrovsky L, Fairhead T, Reinhardt P, et al. Inhaled NO as a viable antiadhesive therapy for ischemia/reperfusion injury of distal microvascular beds. Journal of Clinical Investigation. 1998;101:2497–505. doi: 10.1172/JCI2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe A, Koesling D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circulation Research. 2003;93:96–105. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Taoka T, Matsuo Y, Mishima K, Ogoshi K, Kondo Y, et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Annals of Neurology. 2003;54:732–47. doi: 10.1002/ana.10751. [DOI] [PubMed] [Google Scholar]
- Griffiths MJ, Evans TW. Inhaled nitric oxide therapy in adults. New England Journal of Medicine. 2005;353:2683–95. doi: 10.1056/NEJMra051884. [DOI] [PubMed] [Google Scholar]
- Guery B, Neviere R, Viget N, Foucher C, Fialdes P, Wattel F, et al. Inhaled NO preadministration modulates local and remote ischemia-reperfusion organ injury in a rat model. Journal of Applied Physiology. 1999;87:47–53. doi: 10.1152/jappl.1999.87.1.47. [DOI] [PubMed] [Google Scholar]
- Hataishi R, Rodrigues AC, Neilan TG, Morgan JG, Buys E, Shiva S, et al. Inhaled nitric oxide decreases infarction size and improves left ventricular function in a murine model of myocardial ischemia-reperfusion injury. American Journal of Physiology – Heart and Circulatory Physiology. 2006;291:H379–84. doi: 10.1152/ajpheart.01172.2005. [DOI] [PubMed] [Google Scholar]
- Hogman M, Frostell C, Arnberg H, Hedenstierna G. Bleeding time prolongation and NO inhalation. Lancet. 1993;341:1664–5. doi: 10.1016/0140-6736(93)90802-n. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. Journal Cerebral Blood Flow Metabolism. 1996;16:981–7. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- Ichinose F, Roberts JD, Jr, Zapol WM. Inhaled nitric oxide: a selective pulmonary vasodilator: current uses and therapeutic potential. Circulation. 2004;109:3106–11. doi: 10.1161/01.CIR.0000134595.80170.62. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nature Cell Biology. 2001;3:193–7. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jones SP, Girod WG, Palazzo AJ, Granger DN, Grisham MB, Jourd’Heuil D, et al. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. AJP – Heart and Circulatory Physiology. 1999;276:H1567–73. doi: 10.1152/ajpheart.1999.276.5.H1567. [DOI] [PubMed] [Google Scholar]
- Kida K, Minamishima S, Wang H, Ren J, Yigitkanli K, Nozari A, et al. Sodium sulfide prevents water diffusion abnormality in the brain and improves long term outcome after cardiac arrest in mice. Resuscitation. 2012 doi: 10.1016/j.resuscitation.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, et al. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circulation Research. 2011;108:418–26. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krismer AC, Lindner KH, Wenzel V, Rainer B, Mueller G, Lingnau W. Inhibition of nitric oxide improves coronary perfusion pressure and return of spontaneous circulation in a porcine cardiopulmonary resuscitation model. Critical Care Medicine. 2001;29:482–6. doi: 10.1097/00003246-200103000-00003. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proceedings of the National Academy of Sciences of the USA. 1991;88:4651–5. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang JD, Jr, Teng X, Chumley P, Crawford JH, Isbell TS, Chacko BK, et al. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. Journal of Clinical Investigation. 2007;117:2583–91. doi: 10.1172/JCI31892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Medicine. 2004;30:2126–8. doi: 10.1007/s00134-004-2425-z. [DOI] [PubMed] [Google Scholar]
- Liu X, Huang Y, Pokreisz P, Vermeersch P, Marsboom G, Swinnen M, et al. Nitric oxide inhalation improves microvascular flow and decreases infarction size after myocardial ischemia and reperfusion. Journal of the American College of Cardiology. 2007;50:808–17. doi: 10.1016/j.jacc.2007.04.069. [DOI] [PubMed] [Google Scholar]
- Mathru M, Huda R, Solanki DR, Hays S, Lang JD. Inhaled nitric oxide attenuates reperfusion inflammatory responses in humans. Anesthesiology. 2007;106:275–82. doi: 10.1097/00000542-200702000-00015. [DOI] [PubMed] [Google Scholar]
- Minamishima S, Bougaki M, Sips PY, De Yu J, Minamishima YA, Elrod JW, et al. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009;120:888–96. doi: 10.1161/CIRCULATIONAHA.108.833491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamishima S, Kida K, Tokuda K, Wang H, Sips PY, Kosugi S, et al. Inhaled nitric oxide improves outcomes after successful cardiopulmonary resuscitation in mice. Circulation. 2011;124:1645–53. doi: 10.1161/CIRCULATIONAHA.111.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaka Y, Buys ES, Spagnolli E, Steinbicker AU, Hayton SR, Rauwerdink KM, et al. Soluble guanylate cyclase-1 is required for the cardioprotective effects of inhaled nitric oxide. American Journal of Physiology — Heart and Circulatory Physiology. 2011;300:H1477–83. doi: 10.1152/ajpheart.00948.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaka Y, Fernandez BO, Garcia-Saura MF, Petersen B, Ichinose F, Bloch KD, et al. Brief periods of nitric oxide inhalation protect against myocardial ischemia-reperfusion injury. Anesthesiology. 2008;109:675–82. doi: 10.1097/ALN.0b013e318186316e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumar RW. Molecular mechanisms of ischemic neuronal injury. Annals of Emergency Medicine. 2000;36:483–506. doi: 10.1067/mem.2000.110995. [DOI] [PubMed] [Google Scholar]
- Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, Inter-American Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation. 2008;118:2452–83. doi: 10.1161/CIRCULATIONAHA.108.190652. [DOI] [PubMed] [Google Scholar]
- Nishida T, Yu JD, Minamishima S, Sips PY, Searles RJ, Buys ES, et al. Protective effects of nitric oxide synthase 3 and soluble guanylate cyclase on the outcome of cardiac arrest and cardiopulmonary resuscitation in mice. Critical Care Medicine. 2009;37:256–62. doi: 10.1097/CCM.0b013e318192face. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peberdy MA, Callaway CW, Neumar RW, Geocadin RG, Zimmerman JL, Donnino M, et al. Part 9: Post-cardiac arrest care: 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2010;122:S768–86. doi: 10.1161/CIRCULATIONAHA.110.971002. [DOI] [PubMed] [Google Scholar]
- Roger VrL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics-2012 update. Circulation. 2012;125:e2–220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roine RO, Kajaste S, Kaste M. Neuropsychological sequelae of cardiac arrest. Journal of the American Medical Association. 1993;269:237–42. [PubMed] [Google Scholar]
- Sharma HS, Miclescu A, Wiklund L. Cardiac arrest-induced regional blood–brain barrier breakdown, edema formation and brain pathology: a light and electron microscopic study on a new model for neurodegeneration and neuroprotection in porcine brain. Journal of Neural Transmission. 2011;118:87–114. doi: 10.1007/s00702-010-0486-4. [DOI] [PubMed] [Google Scholar]
- Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–6. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circulation Research. 2007;101:1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Terpolilli NA, Kim SW, Thal SC, Kataoka H, Zeisig V, Nitzsche B, et al. Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circulation Research. 2011 doi: 10.1161/CIRCRESAHA.111.253419. [DOI] [PubMed] [Google Scholar]
- The Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. New England Journal of Medicine. 2002;346:549–56. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Wijman CA, Mlynash M, Caulfield AF, Hsia AW, Eyngorn I, Bammer R, et al. Prognostic value of brain diffusion-weighted imaging after cardiac arrest. Annals of Neurology. 2009;65:394–402. doi: 10.1002/ana.21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Boddicker KA, Rhee BJ, Davies LR, Kerber RE. Effect of nitric oxide synthase modulation on resuscitation success in a swine ventricular fibrillation cardiac arrest model. Resuscitation. 2005;67:127–34. doi: 10.1016/j.resuscitation.2005.03.015. [DOI] [PubMed] [Google Scholar]