

Abstract
Vascular Ca2+-activated K+ channels (KCa) are important effector proteins in the control of arterial tone and alterations in KCa channel functions have been reported to contribute to a wide range of cardiovascular pathologies. In the arterial endothelium KCa channels of the KCa3.1 and KCa2.3 subtypes have been proposed to mediate membrane hyperpolarisation and thus initiate the endothelium-derived hyperpolarizing factor (EDHF)-mediated vasodilator response, the third major endothelial dilator system next to nitric oxide and prostacyclin (PGI2). In a variety of cardiovascular disease states such as hypertension, diabetes, renal insufficiency, and endothelial dysfunction following angioplastic interventions and by-pass surgery, diminished functions of KCa3.1 and KCa2.3 channels in the endothelium have been proposed to contribute to defective vasoregulation. Moreover, up-regulation of KCa has been shown to drive proliferation of arterial smooth muscle cells and thus trigger restenosis after angioplasty and atherosclerosis. In this review, we summarize current knowledge about vascular KCa3.1 and KCa2.3 channels, their molecular and pharmacological properties and their specific roles in cardiovascular pathologies. In this review, we highlight the therapeutic potential of KCa3.1/KCa2.3 modulators as novel endothelial specific antihypertensive drugs as well as the usefulness of KCa3.1-blockers for treating cardiovascular diseases characterized by excessive cell proliferation.
Introduction
Cardiovascular diseases are a major reason for the increasing morbidity in modern societies and their socioeconomic consequences are tremendous. Essential hypertension, primary hypertension or secondary hypertension (e.g., caused by diabetes, renal disease, or unhealthy life style and poor nutrition) is one of, if not the major risk factor for myocardial infarction and stroke. Hypertension is often associated with endothelial dysfunction, athero- and arteriosclerosis, and pathological vascular remodeling [1]. At the cellular and molecular level, hypertension is accompanied by a variety of alterations such as changes in gene expression, and altered secretion of vasodilating and vasocontracting factors [1–3]. These alterations lead to dysfunctional regulation of smooth muscle contractility (vascular tone) and thus blood pressure, as well as to abnormal vascular remodeling. Pathological remodeling of the vascular wall also occurs after by-pass surgery, angioplastic interventions and vascular injury [4]. Here, excessive proliferation of smooth muscle cells leads to the formation of a neointima, which results in vessel narrowing and restenosis after balloon catheter interventions. A dysfunctional or damaged endothelium is considered to be one of the main reasons for the development of these vasculopathies.
The vascular endothelium plays a pivotal role in numerous physiological functions such as angiogenesis, inflammation, platelet aggregation, the regulation of vascular permeability, and vascular tone, and thereby blood pressure. Moreover, the endothelium tightly controls vessel integrity and growth by regulating extracellular matrix synthesis or degradation and the proliferative activity of the underlying smooth muscle cells. For blood pressure regulation, the endothelium releases relaxing and contracting factors and thereby adjusts smooth muscle contractility. Endothelial dysfunction, as present in most cardiovascular pathologies, leads to severe imbalance in vessel regulation resulting e.g. in diminished synthesis of vasodilating autacoids and excessive smooth muscle proliferation, ultimately leading to increased vascular tone and hypertension as well as to media hypertrophy and neointima formation. In this review, we intend to convey that endothelial and smooth muscle ion channels, in particular Ca2+-activated K+ channels (KCa), are pathomechanistically involved in the molecular mechanism underlying excessive smooth muscle proliferation and neointima formation as well as endothelial dysfunction in cardiovascular pathologies. We will further discuss the potential of modulators of KCa as novel anti-hypertensives and as a therapeutic strategy to halt neointima formation and atherosclerosis. The authors wish to indicate that the present review is a partial extension of a recent review by our group [20].
1. The EDHF-dilator system
Next to nitric oxide (NO) [5,6] and prostacyclin (PGI2) [7,8], the endothelium possess a third vasodilator system, the endothelium-derived hyperpolarizing factor (EDHF) [9,10]. While the role of endothelial NO and PGI2 for local and systemic blood pressure control is well established, much less is known about the role of EDHF. However, an increasing body of experimental evidence suggests that EDHF could be as important as NO in controlling blood pressure [10–13].
Per definition EDHF-type dilations are endothelium-dependent dilations which are not mediated by NO or and PGI2 but just by hyperpolarization of the underlying smooth muscle cell layer. This hyperpolarisation leads to smooth muscle relaxation by closing voltage-gated Ca2+-channels and thus decreasing [Ca2+]i by reducing Ca2+ influx (see scheme in Figure 1). In contrast to NO and PGI2 induced dilations, the molecular characteristics of EDHF and particularly the underlying signalling pathways are currently not completely understood. On the one hand, EDHF-dilator responses have been attributed to the actions of various diffusible factors, including various arachidonic acids metabolites [14–16], NO [17] and hydrogen peroxide (H2O2 [18]). For in-depth reviews of possible EDHFs see [3]. On the other hand, there is also good evidence that direct electrical coupling between the endothelium and smooth muscle underlies EDHF-type dilations [19] (Figure 1). According to this hypothesis, activation of the endothelial Ca2+-activated K+-channels first induces hyperpolarization of the endothelium which is then transferred through myo-endothelial gap-junctions to the underlying smooth muscle layer [10,11,20]. Alternatively, the K+-release through the channels has been proposed to activate smooth muscle inwardly-rectifying K+-channels and/or Na+/K+ ATPases (Na+/K+ pump), which then hyperpolarize the smooth muscle cells [21] (Figure 1).
Figure 1.
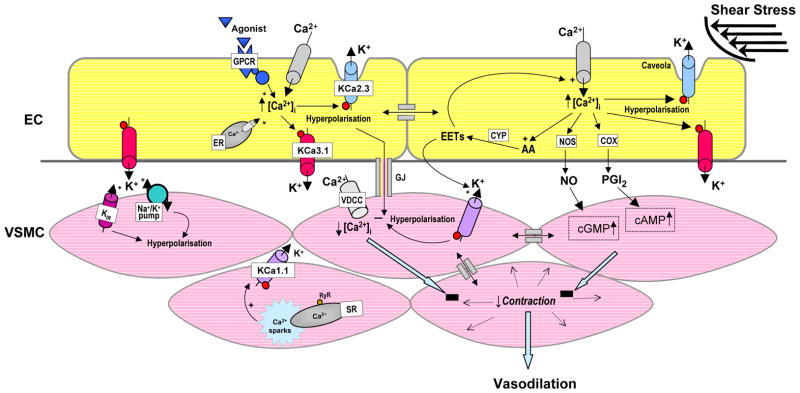
EDHF-signaling pathways involving endothelial and smooth muscle ion channel functions. AA, arachidonic acid; ACh, acetylcholine, KCa1.1, large-conductance Ca2+-activated K+ channel; CYP, cytochrome P450 epoxygenase; EC, endothelial cell; VSMC, vascular smooth muscle cell; EETs, epoxyeicosatrienoic acids; ER, endoplasmic reticulum; GPCR, G-protein coupled receptor, KCa3.1, intermediate-conductance Ca2+-activated K+ channel; Kir, inwardly-rectifying K+ channel; KCa2.3, small-conductance Ca2+-activated K+ channel subtype 3; GJ, myoendothelial gap-junction; SR, sarcoplasmic reticulum; RyR, ryanodine receptor; VDCC, voltage-dependent Ca2+ channel. Note, that this figure is an adapted version of a figure in a recent review by our group [20].
However, whatever EDHF may be, endothelial Ca2+-activated K+-channels are considered as major players in initiating EDHF dilator responses in vivo. In this review, we first summarize the evidence for this by discussing the alterations in EDHF-dilator responses and arterial blood pressure observed in mice deficient of endothelial KCa as well as in cardiovascular pathologies. We then highlight recent findings suggesting that pharmacological manipulation of the EDHF pathway by activators of endothelial KCa could represent a novel treatment option for correcting or improving the endothelial dysfunction in cardiovascular pathologies such as hypertension.
2. Endothelial Ca2+-activated K+-channels and the EDHF dilator response
The human genome contains eight Ca2+-activated K+ channels, which can be divided into two groups based on their genetic relationship, their single channel conductance, and their gating mechanisms. The most widely studied and therefore best known member of the first group, which contains KCa1.1, KCa4.1, KCa4.2 and KCa5.1, is the large-conductance Ca2+-activated K+ channel KCa1.1 (a.k.a. BK or maxiK), which is activated by both increases in intracellular Ca2+ and changes in membrane voltage. The other four KCa channels form the so-called KCa2/3 subgroup, which is further subdivided into the intermediate-conductance KCa3.1 channel (a.k.a. SK4, IK1, or simply IK) and the three small-conductance channels KCa2.1 (SK1), KCa2.2 (SK2) and KCa2.3 (SK3) [22–24]. In contrast to KCa1.1, which is binding Ca2+ in a negatively charged segment in the C-terminus, KCa2 and KCa3.1 channels do not directly bind Ca2+ but are instead gated by calmodulin, which is constitutively associated with their intracellular C-termini in a 1:1 stoichiometry and confers submicromolar Ca2+ sensitivity to these channels [25]. In the context of EDHF-type dilations, it is important to understand that KCa2 and KCa3.1 channels are voltage-independent, which means that they do not inactivate at negative membrane potentials (unlike KCa1.1 channel and classical KV channels) and can thus produce strong membrane hyperpolarisation towards values near the K+ equilibrium potential of −89 mV in physiological K+ gradients.
The two KCa channel subtypes that are predominantly expressed in the vascular endothelium of rodents, humans and pigs are KCa3.1 and KCa2.3 (for extensive review see [9,20,26,27]. Both channels have been shown to activate and mediate EDHF-dilator responses after Ca2+-release from the endoplasmic reticulum following stimulation of the endothelium with classical agonist such as acetylcholine and bradykinin [20,27]. Accordingly, combinations of KCa2.3- and KCa3.1-blockers (apamin (KCa2.3) and charybdotoxin (KCa3.1), or UCL1684 (KCa2.3) and TRAM-34 (KCa3.1) have been reported to suppress endothelial hyperpolarisation and EDHF-type dilations in numerous studies by various investigators (for extensive review see [9,10,20]). The pivotal role of these channels for initiation of EDHF-dilations is further supported by our recent studies in mice deficient of KCa3.1 or of both KCa3.1 and KCa2.3 which showed that genetically encoded deletion of the KCa3.1-gene severely impaired endothelial hyperpolarisation and EDHF-dilations to acetylcholine in carotid arteries and in the cremaster microcirculation in vivo [28]. In contrast, drastically reduced expression of endothelial KCa2.3 in KCa2.3T/T mice alone had a minor or no impact on acetylcholine-induced EDHF-dilations [13]. Deficiency of both KCa3.1 and KCa2.3 channels the KCa3.1−/−/KCa2.3T/T-mice virtually abolished hyperpolarisation and EDHF-responses to acetylcholine in these vascular beds [13]. This severe defect could be overcome by strong over-expression of endothelial KCa2.3 in KCa3.1-deficient KCa2.3T/T mice, but interestingly only partially. Notably, over-expression of KCa2.3 in KCa2.3T/T mice expressing also KCa3.1 failed to enhance EDHF-dilations to acetylcholine. Thus, these findings indicate that over-expressed KCa2.3 can contribute to EDHF-dilations to acetylcholine and exerts rescuing effects in the absence of KCa3.1. Interestingly, over-expression of KCa2.3 in KCa2.3T/T model has also been shown to produce tonic endothelial membrane hyperpolarisation in mesenteric arteries and to reduce myogenic and phenylephrine-induced tone and enlarged arterial diameter of the mesenteric vasculature [29].
In contrast to the apparently more important role of KCa3.1 for acetylcholine-induced EDHF-dilations, activation of KCa2.3 has recently been shown to be required for EDHF-dilations upon shear stress-stimulation as these dilations are severely impaired in KCa2.3-deficient mice but not in KCa3.1-deficient mice [13]. Thus, these two channels contribute in a stimulus-dependent but differential fashion to EDHF-signalling. The cellular basis for this can be explained by their distinct subcellular compartmentation. KCa2.3 has been proposed to be localized in caveolae or caveosomes in which it is perhaps a part of multi-protein signalling complexes (e.g. containing eNOS and Ca2+-influx channels of the TRP-gene family) [30,31]. KCa3.1 in contrast has been reported to be expressed in close proximity to the endoplasmic reticulum and at the endothelial projections towards the smooth muscle [32,33]. Thus, the two channels may use spatially different Ca2+-signalling pathways for activation, i.e. Ca2+-influx for KCa2.3 versus Ca2+-release from the endoplasmic reticulum for KCa3.1.
3. Role of EDHF in systemic blood pressure control
While the importance of the NO system for mediating vasodilations decreases with decreasing vessel diameter along the arterial tree, EDHF-type dilations are particularly important in small-sized arteries and resistance-sized arterioles [12]. This predominant role of EDHF in the small-sized arteries and arterioles suggested that this system could be involved in blood pressure control by influencing total peripheral resistance, but the exact contribution of the KCa2.3/KCa3.1-EDHF-dilator system to blood pressure control remained unclear for a considerable time. However, evidence from KCa3.1/KCa2.3-transgenic mice now shows that the KCa2.3/KCa3.1-EDHF-dilator system is indeed involved in blood pressure control in vivo. For instance, deficiency of KCa2.3 increased systemic blood pressure in KCa2.3T/T-mice [13,29] and KCa2.3-overexpression in these animals reversed this blood pressure increase. Similar to KCa2.3-deficient mice, the loss of KCa3.1 also increases blood pressure to a comparable level [13,28].
Combined deficiency of both KCa3.1 and KCa2.3 in KCa3.1−/−/KCa2.3T/T mice caused only a small additional elevation of blood pressure [13], indicating that the blood pressure increases caused by lack of either channel are not additive and that the loss of one channel can not be compensated for by the other channel, or, worth to mention, by any of the other endothelial vasodilator systems, such as NO and PGI2. Interestingly, in either KCa3.1- or KCa2.3-deficiency, or deficiency of both, the increase in blood pressure was mainly present during locomotor activity suggesting that the KCa3.1/KCa2.3-EDHF-system is needed for adequate vasoregulation and blood pressure control during physical activity, perhaps by counterbalancing the higher vascular tone due to the increased sympathetic activity during physical activity. Albeit this needs further clarification, these findings clearly support the idea that endothelial KCa3.1 and KCa2.3 are major components of the EDHF-signalling pathway in vivo and thereby contribute to adequate blood pressure control.
Other potential EDHF-system candidates, e.g. EETs as diffusible EDHFs [14,15], have also been proposed to contribute to blood pressure control, as concluded from the lower systolic blood pressure (≈ −12 mmHg) in mice deficient of the EET-degrading enzyme, the soluble epoxide hydrolase [34]. Interestingly, the lower blood pressure was found only in males but not in females.
4. Function of EDHF in different cardiovascular disease states
Many cardiovascular pathologies are coupled to endothelial dysfunction [1,2,35] and this endothelial dysfunction cannot always be explained by defects of NO-production or NO-availability, but in some cases also by defects of particularly the KCa3.1/KCa2.3-EDHF-dilator system [20,36] (for extensive review of cardiovascular pathologies associated with defective EDHF-system(s) and, particularly, with disturbed KCa3.1/KCa2.3-EDHF-dilator system see [20,26]). For example, impairments of EDHF-dilator responses appear to be caused by diminished functions/mRNA-expressions of KCa3.1 and/or KCa2.3 channels, i.e. in uremic 5/6 nephrectomized rats [37] and in regenerated endothelium after balloon catheter injury [38]. In diabetic ZDF-rats, a loss of KCa2.3 but not of KCa3.1 functions has been found which were unrelated to changes of KCa2.3-mRNA-expression levels [39]. Interestingly, in the later, activation of KCa3.1 seems to be disturbed due to reduced expression of the CaR-receptor [40]. Compromised KCa3.1 and/or KCa2.3 functions and dilator responses have also been reported in human cardiovascular pathologies, e.g. in skeletal muscle arterioles and the coronary microcirculation following cardiopulmonary bypass surgery [41,42] and in omental arteries of subjects with essential hypertension [43]. Moreover, hypercholesterolemic as well as aged patients showed disturbed KCa3.1 functions and EDHF-dilations in gastroepiploic arteries and distal microvessels [44].
Taken together, these disturbances of the KCa3.1/KCa2.3 EDHF-system can contribute to endothelial dysfunction in a considerable number of cardiovascular disease states.
5. Smooth muscle KCa1.1 and blood pressure control
Although not clearly an endothelial KCa and therefore not the main focus of this review, KCa1.1 (a.k.a. maxi K, BK or slo1) channels in smooth muscle are also involved in blood pressure control and have been implicated in alternative EDHF-dilator responses, i.e. KCa1.1 channels are the presumed target of diffusible EDHFs (e.g. EETs, H2O2 and NO) (for review see [14,26]). The importance of KCa1.1 in blood pressure control is evidenced by studies in genetic animal models and in a variety of hypertensive animal models showing on the one hand that deficiency of the channel causes mild hypertension and on the other hand that alterations in KCa1.1 function or expression parallel hypertensive disease states. For instance, smooth muscle cells from mice lacking the gating-modifying β1-subunit of the KCa1.1 channel show defects in the spontaneous transient outward currents (STOCs) and thus have a lower resting membrane potential, as a consequence of which smooth muscle contractility is enhanced and systemic arterial blood pressure is increased in these mice [45,46]. Mice deficient of pore-forming α-subunit also show increased systemic blood pressure, which can be explained by the loss of STOCs facilitating membrane depolarization, and by primary hyperaldosteronism [47]. Interestingly, the KCa1.1-deficient mice exhibit significant erectile dysfunction and show reduced responses to the phosphodiesterase-5 (PDE-5) inhibitor sildenafil [48,49]. Moreover, diminished KCa1.1-functions in smooth muscle have been reported in SHR [50] and angiotensin II-hypertensive rats [51] and have been related to a down-regulation of the expression of the β1-subunit. On the other hand, rats with pulmonary hypertension and L-nitro-arginine-treated hypertensive rats show a decreased expression of the pore-forming α-subunit in smooth muscle [52,53] while aged rats exhibit lower expression levels of α- and β-subunits [54]. In contrast, increased KCa1.1 functions in smooth muscle have been reported in several models of hypertension and up-regulation of KCa1.1α-subunits could thus represent a mechanism to counteract hypertension (for review see [26]).
Intriguingly, a genetic polymorphism in the β1-subunit (E65K) of the human KCa1.1 channel causing enhanced channel activity (gain-of-function mutation) has been reported to be protective against diastolic hypertension in post-menopausal women and has been considered as one of the strongest genetic factors associated to date with protection against myocardial infarction and stroke [55].
Thus, these findings clearly support a significant role of smooth muscle KCa1.1 in blood pressure regulation and demonstrate that this channel represents a similarly attractive target for blood pressure lowering therapy in hypertension.
6. Pharmacological modulators of vascular Ca2+-activated K+-channels
Based on the crucial roles of KCa channels in both vascular endothelium (KCa2.3 and KCa3.1) and vascular smooth muscle (KCa1.1), KCa channels have been postulated to constitute new drug targets for alternative or specific treatments of cardiovascular diseases. Fortunately, KCa-channels have a well-developed pharmacology and can be both activated and inhibited by a number of relatively potent and selective peptides and small molecules (for in-depth review see [56,57]). Of these compounds, the small molecules are of particular interest because of their suitability for in vivo applications as pharmacological tools compounds and as potential drug candidates for clinical development.
Both KCa2 and KCa3.1 channels are activated with very similar potencies by a number of relatively simple heterocyclic benzimidazolones and benzothiazoles, which increase the Ca2+ sensitivity of these Ca2+/calmodulin-gated channels. These “activators”, which should not be termed “openers” because they are ineffective in the absence of Ca2+ and therefore do not “open” the channel per se, potentially constitute a new antihypertensive principle based on their ability to increase EDHF-type vasodilations (see left panel of Figure 2 for chemical structures and EC50 values). 1-EBIO [58], its 10-fold more potent dichloro-substituted derivative DC-EBIO [59] and the oxim NS309 [60] activate KCa2 and KCa3.1 channels with potencies ranging from 100 μM (EBIO) to 30 nM (NS309) and all display a roughly 3–5 fold selectivity for KCa3.1 over KCa2.3. However, a disadvantage of DC-EBIO and NS309 is that they are rather non-selective and also block L-type Ca2+-channels [61] and in the case of NS309 the cardiac hERG (Kv11.1) channel. A structurally somewhat related compound is the “old” neuroprotectant riluzole, which activates both KCa3.1 and KCa2.3 channels with EC50s of 2 and 12 μM [62] and which we recently used as a template for the synthesis of SKA-31 [63]. SKA-31 has an EC50 of 250 nM for KCa3.1, is more selective than DC-EBIO and NS309 and suitable for in vivo use because of its relatively long plasma-half life of 12 hours. The Danish company Neurosearch AS recently described the first selective KCa2 channel activator and demonstrated that it is indeed feasible to design subtype-specific KCa2 channel activators [64]. The aminopyrimidine CyPPA increases KCa2.3 and KCa2.2 channel activity with EC50 values of 6 and 14 μM but does not affect KCa2.1 or KCa3.1 channels at concentrations up to 100 μM.
Figure 2.

Molecular and pharmacological characteristics of KCa3.1, KCa2.X, and KCa1.1 channels. A: Activators of KCa3.1 and KCa2.1–3 channels. B: Openers of the KCa1.1 channel. Molar concentrations above structures are the reported EC50s. 1-EBIO, 1-ethyl-2-benzimidazolinone; BMS-204352, ([3S]-[+]-[5-chloro-2-methoxyphenyl]-1,3-dihydro-3-fluoro-6-[trifluoromethyl]-2H-indol-2-one); CyPPA, cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine; DC-EBIO, 5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazole-2-one; DHS-1, dehydrosoyasaponin-1; NS11021 1-(3,5-bis-trifluoromethyl-phenyl)-3-[4-bromo-2-(1H-tetrazol-5-yl)-phenyl]-thiourea; NS1608 (N-(3-trifluoromethyl)phenyl) N′-(2-hydroxy-5-chlorophenyl) urea; NS1619, 1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzimidazol-2-one; NS309, 3-oxime-6,7-dichloro-1H-indole-2,3-dione; SKA-31, naphtho[1,2-d]thiazol-2-amine.
KCa1.1 (maxi K+) is activated by a large number of both natural and synthetic compounds (see right panel of Figure 2 for chemical structures and EC50s of the different openers). The two KCa1.1 activators that are most commonly used as pharmacological tools are NS1619 (EC50 ~20 μM) and the more potent NS1608 (EC50 2 μM), in which the benzimidazolone ring if NS1619 was opened to a diphenylurea. Both compounds were discovered at NeuroSearch A/S [65] and activate KCa1.1 channels by shifting the voltage-dependency of KCa1.1’s α-subunit to more negative values causing the channel to open at less depolarized potentials and to deactivate more slowly. Both compounds do not require intracellular Ca2+ for their effects and can therefore truly be termed “openers”. Unfortunately, NS1619 also blocks voltage-gated calcium channels and KCa3.1 channel [66], which makes it a problematic tool compound for vascular preparations. The more recently reported tetrazole substitute thiourea NS11021 [67] and BMS-204352 [68], both with potencies in the upper nanomolar range, currently seem to be the most selective compounds. For example, NS11021 activates KCa1.1 with an EC50 of 400 nM and in contrast to NS1609 exerts no effect on L- or T-type Ca2+ channels at 30 μM. Another KCa1.1 channel opener, that is sometimes used in the cardiovascular field, is the triterpenoid glycoside dehydrosoyasaponin I (DHS-1), which stimulates KCa1.1 via its regulatory β-subunit [69]. However, DHS-1 is difficult to study despite its low nanomolar potency because it is poorly membrane-permeable and needs to be applied intracellularly in electrophysiological experiments.
7. Openers of KCa2.3 and KCa3.1 as drugs for the treatment of endothelial dysfunction and hypertension?
Considering the importance of the two channels for EDHF-dilator responses in vivo, openers of KCa2.3 and KCa3.1 channels should lower blood pressure by enhancing EDHF-dilations. That this is in principle possible was demonstrated by our recent study with the preferentially KCa3.1-activating compound SKA-31 [63]. Nanomolar concentrations of SKA-31 potentiated acetylcholine-induced EDHF-dilator responses in carotid arteries, an effect which strictly depended on the expression of the channel as this potentiation was not seen in vessels from KCa3.1-deficient mice. A subsequent in vivo treatment revealed that SKA-31 was capable of lowering blood pressure in normotensive KCa3.1-expressing mice but not in KCa3.1-deficient mice and, intriguingly, reduced blood pressure in angiotensin-II-infused hypertensive mice [63]. Equally promising results have been obtained with the NeuroSearch A/S compound NS309 which caused KCa2.3 and KCa3.1-mediated and endothelium-dependent hyperpolarisations in guinea pigs carotid arteries [70] and has been recently reported to lower blood pressure in SHR rats [71]. Notably, NS309 has also been reported to have additional positive effects on NO formation in porcine arteries, presumably through the potentiation of KCa2.3 [72,73]. Whether the KCa2.3 channel activator CyPPA is also able to enhance EDHF-type dilations and to lower blood pressure has not been tested or published so far. Taken together, the results from recent studies suggest that openers of KCa2.3 and KCa3.1 channels may serve as blood pressure lowering drugs.
8. Openers of KCa1.1 as antihypertensive drugs?
Without any doubts, opening of KCa1.1 causes robust vasodilation in isolated vessels and a large body of experimental evidence suggests that KCa1.1 contributes to various cardiovascular disease states such as hypertension, stroke and erectile dysfunction [26]. However, up to now, almost all clinical trials involving KCa1.1 openers have been discontinued because of a lack of efficacy or other published or unpublished side effects precluding their therapeutic usage. For example, the Bristol-Myers-Squibb compound BMS-204352 has been shown to have neuroprotective properties in stroke models [74] presumably by inducing neural hyperpolarisation, which then prevents Ca2+-overload by closing voltage-gated Ca2+-channels and NMDA receptors and thus “excitotoxic” cell death. However, after Phase-1 and Phase-2 clinical trials revealed good in vivo tolerance and safety, the Phase-3 trial failed to prove efficacy in improving neurological outcomes after ischemic stroke [75]. Reasons for the failure could be the weak potency or the late administration (within 48 hours) of BMS-204352 after the stroke. At present, KCa1.1 openers are only considered beneficial for the treatment of bronchial asthma and chronic obstructive pulmonary diseases [76]. Interestingly, a first small scale clinical trial employing gene transfer of the hSlo1α subunit to patients with severe erectile dysfunction gave promising results [77]. Conversely, inhibiting KCa1.1 in smooth muscle has been shown to be beneficial in the treatment of hypotension during hemorrhagic shock in rats [78].
Thus, at present there is not enough data available to conclusively decide whether KCa1.1 openers could be of potential therapeutic utility in the treatment of hypertension or of other cardiovascular diseases.
9. Targeting KCa3.1 to treat restenosis disease and other cardiovascular diseases characterized by abnormal cell proliferation
Healthy contractile vascular smooth muscle cells do not express KCa3.1 or KCa2 channels in considerable amounts. Instead, the predominant KCa conductance in these cells is carried by KCa1.1 channels, which mediate membrane repolarization and thus counterbalance depolarization by L-type Ca2+ channels and possibly avoid vasospasm. In addition, KCa1.1 contributes to the setting of the resting membrane potential by producing spontaneous transient outward currents [79] activated by highly localized Ca2+-signals, known as Ca2+ sparks (Figure 1).
However, the expression pattern of KCa channels changes dramatically if smooth muscle cells undergo phenotypic modulation and mitogenesis. For instance, mitogenically stimulated proliferation of smooth muscle cells is accompanied by an up-regulation of KCa3.1 as determined by de novo mRNA and protein expression of this channel [80–82]. This expression of KCa3.1 is mediated through activation of the ras/raf/MEK/ERK MAP kinase signaling cascade [83] and has also been related to the decreased expression of the REST-transcription factor, which normally acts as a suppressor of KCa3.1 in these cells [84]. Moreover, up-regulation of KCa3.1 is paralleled by a down-regulation of KCa1.1 expression as the typical KCa in the differentiated contractile smooth muscle cells [81], further supporting the notion that alterations of the KCa-expression pattern are indicative of phenotypic modulation.
Strikingly, induction of KCa3.1 was also seen in proliferating neointimal smooth muscle cells in vivo, e.g., in balloon catheterized carotid arteries of rats and coronary arteries of swine. Notably, induction of KCa3.1 was also detected in neointima developing coronary bypass vessels from patients, suggesting that induction of KCa3.1 in abnormal smooth muscle growth is a common feature and indicator of a phenotypic switch and, more importantly, of potential clinical relevance [85].
The up-regulation of KCa3.1 has been shown to be required for cellular activation based on the fact that KCa3.1 inhibition with the selective KCa3.1-blocker TRAM-34 inhibits human, rodent and swine smooth muscle mitogenesis and migration in vitro [81,82,85]. More intriguingly, in vivo treatment with TRAM-34 by either i.p. injections or via TRAM-34-coated balloons during the angioplasty were effective in suppressing neointima formation in two different models of post-angioplasty restenosis e.g. in balloon catheterized carotid arteries from rats and coronary arteries from swine, respectively [81,82].
However, up-regulation/induction of KCa3.1 is not unique for proliferating smooth muscle cells and has been found also in activated T-cells, macrophages, fibroblasts, several cancer cell lines, and endothelial cells [56,57]. Based on its expression in proliferating smooth muscle and immune cells, KCa3.1 blockers might be particularly useful for the treatment of atherosclerosis. Aortas of apoE−/−mice with atherosclerosis have been shown to strongly express KCa3.1 in vascular smooth muscle cells that proliferated and migrated into plaques, and in macrophages and T lymphocytes that infiltrated these atherosclerotic lesions [85]. Treatment of apoE−/− mice, which were on a high cholesterol diet, with TRAM-34 significantly prevented atherosclerosis development in these animals by reducing vascular smooth muscle cells proliferation as well as infiltration of plaques by macrophages and T lymphocytes.
Interestingly, a higher KCa3.1-mRNA expression was also found in mesenteric endothelium of colon cancer patients [86] and such an up-regulation could be mimicked in cultured endothelial cells by stimulation with pro-angiogenic factors (VEGF and bFGF) [87]. Moreover, the KCa3.1-blocker TRAM-34 also effectively reduces endothelial cell proliferation in vitro and vascularization of matrigel plugs in vivo. This suggests that, besides its role in the mechanism of endothelium-dependent vasodilation and EDHF-signalling as outlined above, KCa3.1 also contributes to endothelial mitogenesis and phenotypic modulation of endothelial cells [87] and could thus be a novel target for prevention of neo-angiogenesis due to malignancies.
Expert opinion
The recent progress in the EDHF-field has provided substantial evidence that the KCa3.1/KCa2.3-EDHF-dilator system is a fully emancipated endothelial dilator system, next to the classical NO and PGI2-systems and other presumed EDHF-systems. The recent data furthermore suggest that KCa3.1 and KCa2.3 channels serve distinct as well as overlapping functions, by either contributing predominantly to EDHF-dilator responses elicited perhaps by stimulation of G-protein-coupled receptors and mediated by KCa3.1 and those induced by shear stress stimulation and mediated by KCa2.3. Moreover, the current data corroborate the idea that the KCa3.1/KCa2.3-EDHF system acts independently and is non-compensable by other endothelium-dependent vasodilator systems like NO and PGI2. Importantly, the KCa3.1/KCa2.3 EDHF-dilator system appears to be impaired in cardiovascular disease states such as hypertension, diabetes, and perhaps in atherosclerosis and this defect may thus contribute to the overall endothelial dysfunction present in these cardiovascular pathologies.
The recent advances in the development of selective KCa3.1/KCa2.3 activators further suggest that KCa3.1 and KCa2.3 channels may represent novel attractive targets for the development of alternative antihypertensive drugs that target the EDHF-dilator system and the endothelium. Mixed KCa3.1 and KCa2.3 activators, which preferentially activate KCa3.1, as exemplified by SKA-31 [63], might prove valuable tool compounds to explore the therapeutic potential of pharmacologically increasing EDHF responses and could, eventually, be developed into drugs for the treatment of various cardiovascular pathologies. Activators of KCa2.3 such as CyPPA could also constitute interesting drug candidates because they would not only improve EDHF-dilator response but also potentially improve NO-synthesis [72]. Also, NS309 [60] has been proposed to be useful [70,72] however, its short plasma half-life and unspecific blocking effects on L-type channels and the cardiac hERG-channels raise doubts about its clinical usefulness. An indication for both KCa2.3 and KCa3.1 activators could be different forms of hypertension, including essential hypertension or even more rare forms such as pulmonary hypertension, and cardiovascular disease states which are linked to a diminished function of KCa3.1 and KCa2.3 channels or compromised NO-formation or NO availability. A more specific indication could be arterial dysfunction following coronary by-pass surgery, which is associated with endothelial dysfunction due to a defect in the KCa3.1/KCa2.3 EDHF-dilator system. Of note, blockers of KCa3.1 may also be of therapeutic usefulness in cardiovascular pathologies characterized by abnormal cell proliferations. This is evidenced by studies showing that inhibition of KCa3.1 in proliferating smooth muscle halted neointima formation and prevented atherosclerosis development. Moreover, the increased expression of this channel in T-cells, cancer cell lines, endothelial cells, atherosclerotic lesions and in fibroblast, strongly suggests KCa3.1 blockers as novel therapeutics for the treatment of autoimmune disease, malignancies and tumor angiogenesis, athero- and arterioscleosis, and organ fibrosis. In this context, we would like to point out that the triarylmethane ICA-17043, which is structurally similar to TRAM-34, has been tested in phase III clinical trials for sickle cell anemia, where it failed after having been found to be both safe and effective in phase II clinical trials. Interestingly, dose-escalating studies with ICA-17043 in 28 otherwise healthy patients with sickle cell disease showed that the compound did not increase blood pressure or lead to electrocardiogram changes [88,89].
In general, activators of endothelial KCa3.1 channels appear to be more attractive as potential novel antihypertensives than KCa2.3 activators because KCa3.1 is not considerably expressed in neurons or the brain. KCa2.3 in contrast is widely expressed in the central nervous system and activators of the channel could lead to substantial sedation by reducing neuronal firing. For this reason, KCa2 channel activators have actually been suggested as potential therapeutics for ataxia and epilepsy. However, activators of KCa3.1 may give rise to other side effects since this channel is also expressed in intestinal and bronchial epithelia, erythrocytes, T-cells and proliferating smooth cells and cancer cells.
First in vivo treatments with SKA-31 did not indicate general toxicity of the compound. However, SKA-31 is currently only a good pharmacological tool and attractive lead compound, but further efforts are required to develop SKA-31 or a more potent and selective derivative into a clinically useful drug. At present we are, however, not aware of any pharmaceutical activities disclosed publicly to develop KCa2.3. and/or KCa3.1 activators for antihypertensive therapy. A general concern with developing a channel activator is of course the possibility that permanent channel activation might lead to a compensatory down-regulation of the targeted KCa channels. However, we believe that this will need to tested in various animal models of hypertension and would like to point out here that Kv7.2/7.3 (KCNQ2/3) channel activation for the treatment of epilepsy does not seem to lead to tolerance development in either animals or humans.
Considering other current antihypertensive therapy strategies, KCa-openers seem to use the same principle as classical Ca2+-channel blockers such as dihydropyridines (nifedipine) and benzothiazepine (diltiazem), i.e. reducing smooth muscle intracellular Ca2+. However, dihydropyridines and benzothiazepine are known to exert a variety of non-selective detrimental side effects such as e.g. edema (nifedipine), bradycardia and constipation (diltiazem), headache, nausea, and erectile dysfunction. Thus under conditions in which Ca2+-antagonist cause unwanted side effects or are contraindicated (unstable angina pectoris, acute myocardial infarction, pregnancy) KCa-openers may provide alternative treatment options. Albeit this therapeutic hypothesis still needs further exploration and remains speculative at this stage.
Other drug therapies such as angiotensin converting enzyme inhibitors, antagonists of angiotensin receptors and PDE-3 inhibitors as well as other therapeutic options such as dietary supplements (estrogens, omega-3 polyunsaturated fatty acids, polyphenol derivatives) have been shown to improve cardiovascular pathologies which can at least in part be explained by a beneficial effect on the EDHF-system [26]. Whether KCa-openers may become more effective than these therapies remains to be proven. Other targets with beneficial effects on the EDHF system and blood pressure may include openers of endothelial Ca2+-permeable transient receptor potential channels (TRP) like TRPV4 and vascular inwardly rectifying K+ channels (KIR). However regarding TRPV4, a recently described opener from GlaxoSmithKline, GSK1016790A, has been shown to not only cause vasodilation by activating the NO-system as well as the EDHF-system, but also to induce endothelial damage with fatal consequences in mice [90].
In summary, a general consensus is slowly emerging that small molecule modulators of endothelial KCa3.1 and KCa2.3 channels can provide novel treatment options for a variety of cardiovascular pathologies as well as for other diseases. However, additional development of these compounds to improve their specificity and bioavailability is desirable and would further validate these channels as novel drug targets in cardiovascular diseases.
Figure 3.


Modulation of endothelial KCa3.1 and KCa2.3 channel alters systemic blood pressure. A: Schematic illustration of the deleterious impact of KCa3.1/KCa2.3-deficiency on EDHF-dilations and blood pressure. B: Schematic illustration of the proposed blood pressure lowering effects of enhanced mRNA-expression of KCa2.3 or of pharmacological potentiation of KCa3.1 by SKA-31.
List of abbreviations
- EDHF
endothelium-derived hyperpolarising factors
- EDCF
endothelium-derived contracting factors
- EETs
eicosatetraenoic acids
- KCa
calcium activated potassium channels
- KCa1.1
large-conductance KCa
- KCa2.3
small-conductance KCa subtype 3
- KCa3.1
intermediate-conductance KCa
- PGI2
prostacyclin
- sEH
soluble epoxide hydrolase
References
- 1.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–92. doi: 10.1097/01.ASN.0000132474.50966.DA. [DOI] [PubMed] [Google Scholar]
- 2.Spieker LE, Flammer AJ, Luscher TF. The vascular endothelium in hypertension. Handb Exp Pharmacol. 2006:249–83. doi: 10.1007/3-540-36028-x_8. [DOI] [PubMed] [Google Scholar]
- 3.Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol Heart Circ Physiol. 2006;291:H985–1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- 4.Zargham R. Preventing restenosis after angioplasty: a multistage approach. Clin Sci (Lond) 2008;114:257–64. doi: 10.1042/CS20070228. [DOI] [PubMed] [Google Scholar]
- 5.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 6.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–6. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 7.Moncada S, Gryglewski R, Bunting S, et al. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263:663–5. doi: 10.1038/263663a0. [DOI] [PubMed] [Google Scholar]
- 8.De Mey JG, Claeys M, Vanhoutte PM. Endothelium-dependent inhibitory effects of acetylcholine, adenosine triphosphate, thrombin and arachidonic acid in the canine femoral artery. J Pharmacol Exp Ther. 1982;222:166–73. [PubMed] [Google Scholar]
- 9.Busse R, Edwards G, Feletou M, et al. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–80. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 10.Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol. 2006;26:1215–25. doi: 10.1161/01.ATV.0000217611.81085.c5. [DOI] [PubMed] [Google Scholar]
- 11.Köhler R, Hoyer J. The endothelium-derived hyperpolarizing factor: insights from genetic animal models. Kidney Int. 2007;72:145–50. doi: 10.1038/sj.ki.5002303. [DOI] [PubMed] [Google Scholar]
- 12.Shimokawa H, Yasutake H, Fujii K, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–11. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Brähler S, Kaistha A, Schmidt VJ, et al. Genetic Deficit of SK3 and IK1 Channels Disrupts the Endothelium-Derived Hyperpolarizing Factor Vasodilator Pathway and Causes Hypertension. Circulation. 2009 Apr 20; doi: 10.1161/CIRCULATIONAHA.108.846634. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 14.Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension. 2007;49:590–6. doi: 10.1161/01.HYP.0000255173.50317.fc. [DOI] [PubMed] [Google Scholar]
- 15.Fisslthaler B, Popp R, Kiss L, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–7. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 16.Faraci FM, Sobey CG, Chrissobolis S, et al. Arachidonate dilates basilar artery by lipoxygenase-dependent mechanism and activation of K(+) channels. Am J Physiol Regul Integr Comp Physiol. 2001;281:R246–53. doi: 10.1152/ajpregu.2001.281.1.R246. [DOI] [PubMed] [Google Scholar]
- 17.Bolotina VM, Najibi S, Palacino JJ, et al. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–3. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 18.Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol. 2005;39:725–32. doi: 10.1016/j.yjmcc.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol. 2004;141:881–903. doi: 10.1038/sj.bjp.0705698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grgic I, Kaistha BP, Hoyer J, et al. Endothelial Ca(2+)-activated K(+) channels in normal and impaired EDHF-dilator responses - relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009 doi: 10.1111/j.1476-5381.2009.00132.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards G, Dora KA, Gardener MJ, et al. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–72. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 22.Wei AD, Gutman GA, Aldrich R, et al. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev. 2005;57:463–72. doi: 10.1124/pr.57.4.9. [DOI] [PubMed] [Google Scholar]
- 23.Ishii TM, Silvia C, Hirschberg B, et al. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci U S A. 1997;94:11651–6. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Köhler M, Hirschberg B, Bond CT, et al. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–14. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- 25.Fanger CM, Ghanshani S, Logsdon NJ, et al. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J Biol Chem. 1999;274:5746–54. doi: 10.1074/jbc.274.9.5746. [DOI] [PubMed] [Google Scholar]
- 26.Feletou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol. 2009;156:545–62. doi: 10.1111/j.1476-5381.2009.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–59. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 28.Si H, Heyken WT, Wölfle SE, et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res. 2006;99:537–44. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- 29.Taylor MS, Bonev AD, Gross TP, et al. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–31. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- 30.Weston AH, Absi M, Ward DT, et al. Evidence in favor of a calcium-sensing receptor in arterial endothelial cells: studies with calindol and Calhex 231. Circ Res. 2005;97:391–8. doi: 10.1161/01.RES.0000178787.59594.a0. [DOI] [PubMed] [Google Scholar]
- 31.Saliez J, Bouzin C, Rath G, et al. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–74. doi: 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
- 32.Dora KA, Gallagher NT, McNeish A, et al. Modulation of endothelial cell KCa3. 1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008;102:1247–55. doi: 10.1161/CIRCRESAHA.108.172379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ledoux J, Taylor MS, Bonev AD, et al. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–32. doi: 10.1073/pnas.0801963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinal CJ, Miyata M, Tohkin M, et al. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–10. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 35.Yildiz O. Vascular smooth muscle and endothelial functions in aging. Ann N Y Acad Sci. 2007;1100:353–60. doi: 10.1196/annals.1395.038. [DOI] [PubMed] [Google Scholar]
- 36.Feletou M, Vanhoutte PM. EDHF: new therapeutic targets? Pharmacol Res. 2004;49:565–80. doi: 10.1016/j.phrs.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 37.Köhler R, Eichler I, Schonfelder H, et al. Impaired EDHF-mediated vasodilation and function of endothelial Ca-activated K channels in uremic rats. Kidney Int. 2005;67:2280–7. doi: 10.1111/j.1523-1755.2005.00331.x. [DOI] [PubMed] [Google Scholar]
- 38.Köhler R, Brakemeier S, Kuhn M, et al. Impaired hyperpolarization in regenerated endothelium after balloon catheter injury. Circ Res. 2001;89:174–9. doi: 10.1161/hh1401.093460. [DOI] [PubMed] [Google Scholar]
- 39.Burnham MP, Johnson IT, Weston AH. Impaired small-conductance Ca2+-activated K+ channel-dependent EDHF responses in Type II diabetic ZDF rats. Br J Pharmacol. 2006;148:434–41. doi: 10.1038/sj.bjp.0706748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weston AH, Absi M, Harno E, et al. The expression and function of Ca(2+)-sensing receptors in rat mesenteric artery; comparative studies using a model of type II diabetes. Br J Pharmacol. 2008;154:652–62. doi: 10.1038/bjp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng J, Liu Y, Clements RT, et al. Calcium-activated potassium channels contribute to human coronary microvascular dysfunction after cardioplegic arrest. Circulation. 2008;118:S46–51. doi: 10.1161/CIRCULATIONAHA.107.755827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Sellke EW, Feng J, et al. Calcium-activated potassium channels contribute to human skeletal muscle microvascular endothelial dysfunction related to cardiopulmonary bypass. Surgery. 2008;144:239–44. doi: 10.1016/j.surg.2008.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Zhou Z, Jiang DJ, et al. Reduction of NO- and EDHF-mediated vasodilatation in hypertension: role of asymmetric dimethylarginine. Clin Exp Hypertens. 2007;29:489–501. doi: 10.1080/10641960701616194. [DOI] [PubMed] [Google Scholar]
- 44.Urakami-Harasawa L, Shimokawa H, Nakashima M, et al. Importance of endothelium-derived hyperpolarizing factor in human arteries. J Clin Invest. 1997;100:2793–9. doi: 10.1172/JCI119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenner R, Perez GJ, Bonev AD, et al. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–6. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 46.Plüger S, Faulhaber J, Furstenau M, et al. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87:E53–60. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- 47.Sausbier M, Arntz C, Bucurenciu I, et al. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation. 2005;112:60–8. doi: 10.1161/01.CIR.0000156448.74296.FE. [DOI] [PubMed] [Google Scholar]
- 48.Werner ME, Zvara P, Meredith AL, et al. Erectile dysfunction in mice lacking the large-conductance calcium-activated potassium (BK) channel. J Physiol. 2005;567:545–56. doi: 10.1113/jphysiol.2005.093823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Werner ME, Meredith AL, Aldrich RW, et al. Hypercontractility and impaired sildenafil relaxations in the BKCa channel deletion model of erectile dysfunction. Am J Physiol Regul Integr Comp Physiol. 2008;295:R181–8. doi: 10.1152/ajpregu.00173.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amberg GC, Santana LF. Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ Res. 2003;93:965–71. doi: 10.1161/01.RES.0000100068.43006.36. [DOI] [PubMed] [Google Scholar]
- 51.Amberg GC, Bonev AD, Rossow CF, et al. Modulation of the molecular composition of large conductance, Ca(2+) activated K(+) channels in vascular smooth muscle during hypertension. J Clin Invest. 2003;112:717–24. doi: 10.1172/JCI18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonnet S, Savineau JP, Barillot W, et al. Role of Ca(2+)-sensitive K(+) channels in the remission phase of pulmonary hypertension in chronic obstructive pulmonary diseases. Cardiovasc Res. 2003;60:326–36. doi: 10.1016/s0008-6363(03)00527-3. [DOI] [PubMed] [Google Scholar]
- 53.Bratz IN, Dick GM, Partridge LD, et al. Reduced molecular expression of K(+) channel proteins in vascular smooth muscle from rats made hypertensive with N{omega}-nitro-L-arginine. Am J Physiol Heart Circ Physiol. 2005;289:H1277–83. doi: 10.1152/ajpheart.01052.2004. [DOI] [PubMed] [Google Scholar]
- 54.Nishimaru K, Eghbali M, Lu R, et al. Functional and molecular evidence of MaxiK channel beta1 subunit decrease with coronary artery ageing in the rat. J Physiol. 2004a;559:849–62. doi: 10.1113/jphysiol.2004.068676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Senti M, Fernandez-Fernandez JM, Tomas M, et al. Protective effect of the KCNMB1 E65K genetic polymorphism against diastolic hypertension in aging women and its relevance to cardiovascular risk. Circ Res. 2005;97:1360–5. doi: 10.1161/01.RES.0000196557.93717.95. [DOI] [PubMed] [Google Scholar]
- 56.Wulff H, Zhorov BS. K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem Rev. 2008;108:1744–73. doi: 10.1021/cr078234p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wulff H, Kolski-Andreaco A, Sankaranarayanan A, et al. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14:1437–57. doi: 10.2174/092986707780831186. [DOI] [PubMed] [Google Scholar]
- 58.Devor DC, Singh AK, Frizzell RA, et al. Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca(2+)-dependent K+ channel. Am J Physiol. 1996;271:L775–84. doi: 10.1152/ajplung.1996.271.5.L775. [DOI] [PubMed] [Google Scholar]
- 59.Singh S, Syme CA, Singh AK, et al. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2001;296:600–11. [PubMed] [Google Scholar]
- 60.Strobaek D, Teuber L, Jorgensen TD, et al. Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime) Biochim Biophys Acta. 2004;1665:1–5. doi: 10.1016/j.bbamem.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 61.Morimura K, Yamamura H, Ohya S, et al. Voltage-dependent Ca2+-channel block by openers of intermediate and small conductance Ca2+-activated K+ channels in urinary bladder smooth muscle cells. J Pharmacol Sci. 2006;100:237–41. doi: 10.1254/jphs.sc0060011. [DOI] [PubMed] [Google Scholar]
- 62.Grunnet M, Jespersen T, Angelo K, et al. Pharmacological modulation of SK3 channels. Neuropharmacology. 2001;40:879–87. doi: 10.1016/s0028-3908(01)00028-4. [DOI] [PubMed] [Google Scholar]
- 63.Sankaranarayanan A, Raman G, Busch C, et al. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3. 1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol. 2009;75:281–95. doi: 10.1124/mol.108.051425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hougaard C, Eriksen BL, Jørgensen S, et al. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br J Pharmacol. 2007;51:655–65. doi: 10.1038/sj.bjp.0707281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strobaek D, Christophersen P, Holm NR, et al. Modulation of the Ca(2+)-dependent K+ channel, hslo, by the substituted diphenylurea NS 1608, paxilline and internal Ca2+ Neuropharmacology. 1996;35:903–14. doi: 10.1016/0028-3908(96)00096-2. [DOI] [PubMed] [Google Scholar]
- 66.Cai S, Garneau L, Sauve R. Single-channel characterization of the pharmacological properties of the K(Ca2+) channel of intermediate conductance in bovine aortic endothelial cells. J Membr Biol. 1998;163:147–58. doi: 10.1007/s002329900379. [DOI] [PubMed] [Google Scholar]
- 67.Bentzen BH, Nardi A, Calloe K, et al. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K+ channels. Mol Pharmacol. 2007;72:1033–44. doi: 10.1124/mol.107.038331. [DOI] [PubMed] [Google Scholar]
- 68.Gribkoff VK, Starrett JE, Jr, Dworetzky SI, et al. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med. 2001;7:471–7. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]
- 69.McManus OB, Harris GH, Giangiacomo KM, et al. An activator of calcium-dependent potassium channels isolated from a medicinal herb. Biochemistry. 1993;32:6128–33. doi: 10.1021/bi00075a002. [DOI] [PubMed] [Google Scholar]
- 70.Leuranguer V, Gluais P, Vanhoutte PM, et al. Openers of calcium-activated potassium channels and endothelium-dependent hyperpolarizations in the guinea pig carotid artery. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:101–9. doi: 10.1007/s00210-008-0267-x. [DOI] [PubMed] [Google Scholar]
- 71.Feletou M. K+ channels and endothelial dysfunction - therapeutic option? Fundamen Clin Pharmacol. 2008;22:9. doi: 10.1111/j.1476-5381.2009.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dalsgaard T, Kroigaard C, Bek T, et al. Role of calcium-activated potassium channels with small conductance in bradykinin-induced vasodilation of porcine retinal arterioles. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.08-3168. [DOI] [PubMed] [Google Scholar]
- 73.Stankevicius E, Hiughes AD, Simonsen U. K+ channels and release of nitric oxide. Fundam Clin Pharmacol. 2008;22:9. [Google Scholar]
- 74.Starrett JE, PH, Ortiz AA, et al. Synthesis, pharmacokinetic analysis and MCAO stroke activity of the maxi-K opener BMS-204352. Proceedings of the Keystone symposium: potassium channels: structure, function and therapeutic utilities; Keystone Colorado. 2000. [Google Scholar]
- 75.Jensen BS. BMS-204352: a potassium channel opener developed for the treatment of stroke. CNS Drug Rev. 2002;8:353–60. doi: 10.1111/j.1527-3458.2002.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nardi A, Olesen SP. BK channel modulators: a comprehensive overview. Curr Med Chem. 2008;15:1126–46. doi: 10.2174/092986708784221412. [DOI] [PubMed] [Google Scholar]
- 77.Melman A, Bar-Chama N, McCullough A, et al. hMaxi-K gene transfer in males with erectile dysfunction: results of the first human trial. Hum Gene Ther. 2006;17:1165–76. doi: 10.1089/hum.2006.17.1165. [DOI] [PubMed] [Google Scholar]
- 78.Zhao G, Zhao Y, Pan B, et al. Hypersensitivity of BKCa to Ca2+ sparks underlies hyporeactivity of arterial smooth muscle in shock. Circ Res. 2007;101:493–502. doi: 10.1161/CIRCRESAHA.107.157271. [DOI] [PubMed] [Google Scholar]
- 79.Pedarzani P, Stocker M. Molecular and cellular basis of small--and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell Mol Life Sci. 2008;65:3196–217. doi: 10.1007/s00018-008-8216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Neylon CB, Lang RJ, Fu Y, et al. Molecular cloning and characterization of the intermediate-conductance Ca(2+)-activated K(+) channel in vascular smooth muscle: relationship between K(Ca) channel diversity and smooth muscle cell function. Circ Res. 1999;85:e33–43. doi: 10.1161/01.res.85.9.e33. [DOI] [PubMed] [Google Scholar]
- 81.Köhler R, Wulff H, Eichler I, et al. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;108:1119–25. doi: 10.1161/01.CIR.0000086464.04719.DD. [DOI] [PubMed] [Google Scholar]
- 82.Tharp DL, Wamhoff BR, Wulff H, et al. Local delivery of the KCa3. 1 blocker, TRAM-34, prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis. Arterioscler Thromb Vasc Biol. 2008;28:1084–9. doi: 10.1161/ATVBAHA.107.155796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Si H, Grgic I, Heyken WT, et al. Mitogenic modulation of Ca2+-activated K+ channels in proliferating A7r5 vascular smooth muscle cells. Br J Pharmacol. 2006;148:909–17. doi: 10.1038/sj.bjp.0706793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cheong A, Bingham AJ, Li J, et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol Cell. 2005;20:45–52. doi: 10.1016/j.molcel.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 85.Toyama K, Wulff H, Chandy KG, et al. The intermediate-conductance calcium-activated potassium channel KCa3. 1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025–37. doi: 10.1172/JCI30836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Köhler R, Degenhardt C, Kuhn M, et al. Expression and function of endothelial Ca2+-activated K+ channels in human mesenteric artery: A single-cell reverse transcriptase-polymerase chain reaction and electrophysiological study in situ. Circ Res. 2000;87:496–503. doi: 10.1161/01.res.87.6.496. [DOI] [PubMed] [Google Scholar]
- 87.Grgic I, Eichler I, Heinau P, et al. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2005;25:704–9. doi: 10.1161/01.ATV.0000156399.12787.5c. [DOI] [PubMed] [Google Scholar]
- 88.Ataga KI, Smith WR, De Castro LM, et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111:3991–97. doi: 10.1182/blood-2007-08-110098. [DOI] [PubMed] [Google Scholar]
- 89.Ataga KI, Orringer EP, Styles L, et al. Dose-escalation study of ICA-17043 in patients with sickle cell disease. Pharmacotherapy. 2006;26:557–64. doi: 10.1592/phco.26.11.1557. [DOI] [PubMed] [Google Scholar]
- 90.Willette RN, Bao W, Nerurkar S, et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther. 2008;326:443–52. doi: 10.1124/jpet.107.134551. [DOI] [PubMed] [Google Scholar]