

Abstract
Glutamatergic neurotransmission in the central nucleus of the amygdala (CeA) plays an important role in many behaviors including anxiety, memory consolidation and cardiovascular responses. While these behaviors can be modulated by corticotropin releasing factor (CRF) and catecholamine signaling, the mechanism(s) by which these signals modify CeA glutamatergic neurotransmission remains unclear. Utilizing whole-cell patch-clamp electrophysiology recordings from neurons in the lateral subdivision of the CeA (CeAL), we show that CRF, dopamine (DA) and the β-adrenergic receptor agonist isoproterenol (ISO) all enhance the frequency of spontaneous excitatory postsynaptic currents (sEPSC) without altering sEPSC kinetics, suggesting they increase presynaptic glutamate release. The effect of CRF on sEPSCs was mediated by a combination of CRFR1 and CRFR2 receptors. While previous work from our lab suggests that CRFRs mediate the effect of catecholamines on excitatory transmission in other subregions of the extended amygdala, blockade of CRFRs in the CeAL failed to significantly alter effects of DA and ISO on glutamatergic transmission. These findings suggest that catecholamine and CRF enhancement of glutamatergic transmission onto CeAL neurons occurs via distinct mechanisms. While CRF increased spontaneous glutamate release in the CeAL, CRF caused no significant changes to optogenetically evoked glutamate release in this region. The dissociable effects of CRF on different types of glutamatergic neurotransmission suggest that CRF may specifically regulate spontaneous excitatory transmission.
Keywords: Extended Amygdala, excitatory transmission, norepinephrine, dopamine, CRF
Introduction
Glutamatergic neurotransmission in the central nucleus of the amygdala (CeA) is important for many behaviors and physiologic processes. Extracellular glutamate levels increase in the CeA in response to acute stressors (Reznikov et al., 2007) and CeA glutamate activity has been suggested to play a critical role in the expression of anxiety-like behaviors (Kalin et al., 2004), fear conditioning (Samson and Pare, 2005), and conditioned place aversion (Watanabe et al., 2002). Furthermore, inactivation of the CeA is associated with disruptions to multiple forms of learning (Robledo et al., 1996; Lingawi and Balleine, 2012), cardiovascular regulation (Roozendaal et al., 1991; Saha, 2005), decreased pain sensitivity (Li and Neugebauer, 2004) and reductions in enhanced ethanol drinking during withdrawal (Roberts et al., 1996). While CeA glutamate signaling appears to be fundamentally important to a variety of functions, a clear understanding of the mechanisms regulating CeA glutamatergic transmission is currently lacking.
Corticotropin Releasing Factor (CRF) signaling plays an important role in many of the CeA-mediated behaviors described above (Fu and Neugebauer, 2008; Koob, 2009; Pitts et al., 2009; Skorzewska et al., 2009) and can modulate CeA excitability (Ji and Neugebauer, 2007; Liu et al., 2004). Furthermore, deletion of CRF type 1 receptors (CRFR1) specifically in forebrain glutamatergic neurons reduces anxiety-like behaviors (Refojo et al., 2011), suggesting a critical role of CRF in the regulation of glutamate transmission in the amygdala. In addition, catecholamine signaling may also play a role in the regulation of CeA glutamatergic transmission. For example, enhanced dopamine (DA) signaling within the CeA is associated with fear conditioning (Guarraci et al., 1999), drug preference/seeking (Rezayof et al., 2002; Thiel et al., 2010; Weiss et al., 2000), and conditioned stress paradigms (Coco et al., 1992). Enhanced norepinephrine (NE) signaling has been shown to play a role in immobilization stress (Pacak et al., 1993) drug withdrawal and reinstatement (Watanabe et al., 2003; Yamada and Bruijnzeel, 2011), and pain sensitivity (Ortiz et al., 2007). CeA NE signaling, particularly via β-adrenergic receptor (β-AR) activation, is also important in drug-withdrawal induced conditioned place aversion (Watanabe et al., 2003) and in memory consolidation (Ellis and Kesner, 1983; Liang et al., 1986; Roozendaal et al., 1993). However, the mechanisms by which CRF and catecholamines may alter CeA glutamatergic neurotransmission have yet to be fully clarified.
Anatomical (Asan et al., 2005; Rudoy et al., 2009) and behavioral (Li et al., 1998) evidence suggests that catecholamines may directly influence the activity of CRF producing neurons in the CeA, which are mainly found in the lateral subdivision of the CeA (CeAL) (Asan et al., 2005; Eliava et al., 2003; Swanson et al., 1983; Treweek et al., 2009). These findings may suggest that catecholamine actions in the CeAL could require CRF signaling to enhance glutamatergic activity, a mechanism similar to that shown in a related subregion of the extended amygdala, the bed nucleus of the stria terminalis (BNST) (Kash et al., 2008; Nobis et al., 2011; Silberman et al., 2013). Therefore, we sought to determine if catecholamine and CRF signaling mechanisms interact to enhance CeAL glutamatergic transmission. Surprisingly, our findings indicate that DA, β-AR and CRF agonists all enhance spontaneous glutamatergic transmission in the CeAL through non-overlapping mechanisms. Furthermore, we also show that the effect of CRF on spontaneous glutamatergic transmission is distinct from that of evoked transmission in this brain region.
2. Methods
2.1 Animals and Brain Slice Preparation
Seven-to-14 week old, male wild-type C57BL/6J mice (Jackson Laboratories) were used for most studies. In a subset of studies, 7–14 week old, male Thy1-ChR2 mice [B6.Cg-Tg(Thy1-COP4/EYFP)18Gfng/J; Jackson Laboratories] were used for optogenetic stimulation of glutamatergic afferents in the CeAL. In this transgenic mouse line, the light activated channel rhodopsin receptor (ChR2) is expressed in neurons under the control of the mouse thymus cell antigen 1 (Thy1) promoter. Expression of the transgenic ChR2 protein is detected predominantly in layer 5 cortical neurons, CA1 and CA3 pyramidal neurons of the hippocampus, cerebellar mossy fibers, and neurons in the thalamus, midbrain and brainstem (Wang et al., 2007). All mice were group housed throughout these studies. Food and water were available ad libitum. All procedures were approved by the Animal Care and Use Committee at Vanderbilt University. Brain slices (300 μm) containing the CeAL were prepared as previously described (Silberman et al., 2013). Following dissection, slices were transferred to a holding chamber where they were heated (27°–30°C) and were allowed to equilibrate for at least 1 hour before being transferred to a submerged perfusion chamber (also heated to 27°–30°C) for electrophysiology studies.
2.2 Electrophysiology
All electrophysiology recordings were made using Clampex 9.2 and analyzed using Clampfit 10.2 (Molecular Devices, Sunnyvale, California). Whole-cell voltage-clamp recordings of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor-mediated spontaneous excitatory postsynaptic currents (sEPSCs) and optically-evoked excitatory postsynaptic currents (oEPSCs) were made at –70 mV and pharmacologically isolated by the addition of 25 μM picrotoxin to the artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl (124), KCl (4.4), CaCl2 (2), MgSO4 (1.2), 1 NaH2PO4 (1), glucose (10), and NaHCO3 (26). Electrode placement was limited to be within the CeAL. Cells were allowed to equilibrate to whole-cell configuration for 3–5 min before recordings began. Recording electrodes (3–6 MΩ) were pulled on a Flaming/Brown Micropipette Puller (Sutter Instruments, Novato, CA) using thin-walled borosilicate glass capillaries and filled with (in mM): CsOH (118), D-gluconic acid (117), NaCl (5), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 10), ethylene glycol tetra-acetic acid (EGTA, 0.4), MgCl2 (2), Tetraethylammonium chloride (5), adenosine triphosphate (ATP, 4), guanosine triphosphate (GTP, 0.3), pH 7.2–7.3, 280–290 mOsmol. sEPSC recordings were acquired and analyzed in 2-min gap-free blocks. Access resistance was monitored between blocks of sEPSC recordings. oEPSCs were evoked every 30 sec by a 1msec TTL pulse to activate a LED light driver (Thorlabs, Newton, NJ) passed through a EN-GFP filter cube (Olympus) to produce blue wavelength light. Access resistance was monitored continuously. Experiments in which access resistance changed by more than 20% were not included in the data analyses.
2.3 Statistical analyses
Statistical analyses were performed using Microsoft Excel 2010 and GraphPad Prism 5, while figures were finalized in Coreldraw 12. Specifically, when determining if a compound had a significant effect, a Student’s paired t test was used, comparing the baseline value to the experimental value. One-way ANOVA was used to compare the effects of drugs between groups, followed by Tukey’s post-test to determine the significance of specific comparisons. All values given for drug effects throughout the study are presented as average ± SEM typically expressed as a normalized percentage of baseline where baseline levels are set as 100%.
2.4 Drugs
Isoproterenol, CRF, Stressin, Astressin-2B and NBI27914 were purchased from Tocris. All other compounds and experimental drugs were purchased from Sigma-Aldrich unless otherwise noted in the text. All experimental drugs were bath applied at their final concentrations as noted in the text. Dimethylsulfoxide (DMSO) was the solvent used for stock solutions of NBI27914 and picrotoxin where the maximum final concentration of DMSO in ACSF was 0.02% by volume.
3. Results
3.1 Effect of CRF receptor activation on sEPSCs in the CeAL
We first assessed whether CRF can enhance sEPSCs in the CeAL. A 6 min bath application of 300nM CRF significantly enhanced sEPSC frequency from baseline levels (175.3±21.3%, n=11, p<0.05, Fig 1), without causing any significant changes to sEPSC amplitude (102.5±7.8%, p>0.05), area (101.6±9.2%, p>0.05), rise time (101.0±6.8%, p>0.05) or decay time (100.1±4.2%, p>0.05). To determine the CRF receptor subtype required for CRF mediated enhancement of CeAL glutamatergic activity, we next assessed the effect of Stressin, a CRFR1 selective agonist (Rivier et al., 2007), on sEPSCs. Bath application of 100nM Stressin for 9 min significantly increased sEPSC frequency (148.2±17.6%, n=6, p<0.05; Fig. 2) without causing significant changes to sEPSC amplitude (94.2±7.0%, p>0.05), area (95.9±7.3%, p>0.05), rise time (104.9±7.0%, p>0.05) or decay time (98.8±3.9%, p>0.05). Previous work from our lab has shown that CRFR1 activation increases sEPSC frequency in the BNST (Kash et al., 2008; Nobis et al., 2011; Silberman et al., 2013), a brain region closely related to the CeA. Therefore, as a positive control, we tested the effect of 100nM Stressin on sEPSCs in the BNST and found that Stressin enhanced sEPSC frequency (157.7±14.5%, n=7, p<0.05) without altering sEPSC kinetics. Together, these data suggest that the effect of CRFR1 activation on glutamatergic neurotransmission is similar in the CeAL compared to the BNST.
Figure 1. CRF enhances spontaneous glutamatergic transmission via a presynaptic mechanism in the CeAL.
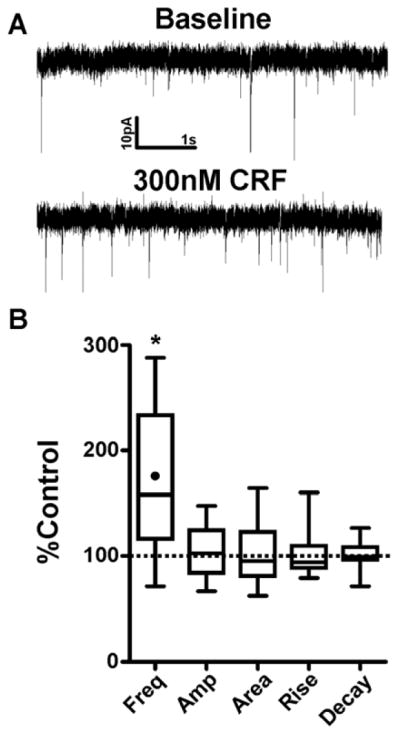
(A) Example sEPSC traces during baseline and after 6 min bath application of CRF. (B) Box-and-whisker plots summarizing of effects of CRF on sEPSC frequency and kinetics. Dotted line indicates normalized baseline values. * indicates significant difference from baseline (p<0.05). Filled circle inside the box plot indicates the mean effect of CRF on sEPSC frequency.
Figure 2. CRFR1 agonist, Stressin, enhances spontaneous glutamatergic transmission via a presynaptic mechanism in the CeAL.
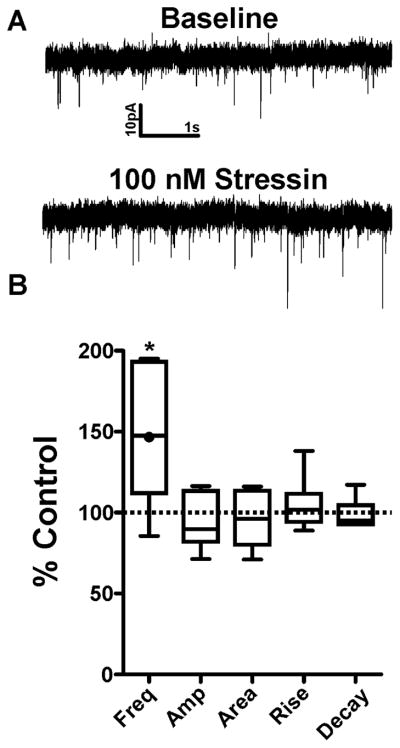
(A) Example sEPSC traces during baseline and after 9 min bath application of Stressin. (B) Box-and-whisker plots summarizing of effects of Stressin on sEPSC frequency and kinetics. Dotted line indicates normalized baseline values. * indicates significant difference from baseline (p<0.05). Filled circle inside the box plot indicates the mean effect of Stressin on sEPSC frequency.
3.2 Effect of CRF on evoked glutamatergic neurotransmission in the CeAL
The above findings suggest that CRF can enhance spontaneous glutamatergic neurotransmission in the CeAL via a presynaptic mechanism. However, these studies cannot determine the presynaptic source of glutamate that is altered by CRF. To begin to address this question, we recorded optically evoked EPSCs (oEPSCs) in CeAL neurons from Thy1-ChR2 mice. These mice harbor ChR2 predominantly in glutamatergic neurons in the cortex and hippocampus, which are known to send projections to the CeA, as well as in neurons of the thalamus, midbrain, brain stem and cerebellum (Wang et al., 2007). Therefore, many of the glutamatergic afferents to the CeA that were enhanced in sEPSC experiments may also be activated by light stimulation of the CeA in Thy1-ChR2 mice. oEPSCs in CeAL were not modulated by picrotoxin but had a reversal potential near 0mV (data not shown) and were almost completely inhibited by a 10 min bath application of 10 μM NBQX, a AMPA receptor antagonist, (3.7±1.0% of baseline, n=4. p<0.05, Fig 3). Interestingly, although CRF enhanced sEPSC in the CeAL, bath application of 300nM CRF did not significantly alter oEPSCs compared to baseline (92.1±7.6%, n=5, p>0.05, Fig 3).
Figure 3. CRF has no effect on optically evoked EPSCs in the CeAL.

(A) Example traces of optically evoked EPSC amplitude during baseline, after CRF application or after application of the AMPA receptor antagonist NBQX. Each trace is an average of 10 consecutive sweeps taken from the time course shown in B. Dotted line indicates peak EPSC amplitude during baseline. Inverted triangle indicates onset of optical stimulation. (B). Time course from exemplar cell shown in A indicating a lack of effect of CRF on optically evoked EPSC amplitude. Traces in A correspond to the lower case letters shown in B. Black bars on x-axis indicate time-points during which drugs were applied. (C) Bar graph summarizing the effect of CRF and NBQX on optically evoked EPSC amplitude. Dotted line indicates normalized baseline values. * indicates significant difference from baseline (p<0.05).
3.3 CRFR1 and CRFR2 are required for CRF effects on sEPSCs in the CeAL
To further determine the CRF receptor subtype mediating the effect of exogenous CRF on sEPSC frequency, we pretreated CeA slices with CRFR1 and CRF type 2 receptor (CRFR2) subtype specific antagonists either alone or in combination for a minimum of 12 minutes prior to bath application of CRF (Fig. 4). Interestingly, pretreating CeA slices with CRF antagonists resulted in significant increases in basal sEPSC frequency when recording from neurons in the CeAL compared to cells recorded under normal conditions (Fig 4A, ANOVA: F=4.822, p<0.05,). However, even though basal sEPSC frequency was higher following pretreatment with CRFR antagonists, bath application of CRF was still able to produce significant further increases in sEPSC frequency in the presence of either a CRFR1 antagonist [1 μM NBI27914 (NBI); basal sEPSC frequency = 6.4±0.6 Hz, n= 27; effect of 300nM CRF in NBI treated cells: 201.1±35.4%, n=9, p<0.05] or in the presence of a CRFR2 antagonist [100nM Astressin2B (Ast2B); basal sEPSC frequency = 7.1±1.5 Hz, n=8; effect of 300nM CRF in Ast2B treated cells: 166.8±26.7%, n=8, p<0.05]. The effect of 300nM CRF was only significantly reduced when slices were pretreated with both NBI and Ast2B (NBI+Ast2B; basal frequency = 5.9±1.0 Hz, n=20; effect of 300nM CRF in NBI+Ast2B treated cells: 113.8±7.5%, n=6, p>0.05; Fig 4B).
Figure 4. Pretreatment of both CRFR1 and CRFR2 antagonists are required for blockade of CRF effect on spontaneous glutamatergic transmission in the CeAL.

(A) Bar graph summarizing the effects of CRFR1 and CRFR2 antagonist pretreatment on basal sEPSC frequency. (B) Line-graph summarizing the effect of CRFR1 and R2 antagonist pretreatment on subsequent effect of CRF. Note that only combined pretreatment with CRFR1 and CRFR2 antagonists could significantly reduce the effect of CRF on sESPC frequency. Dotted line indicates normalized baseline values. * indicates significant difference between groups (p<0.05).
3.4 Catecholamines enhance CeAL spontaneous glutamatergic transmission
We next tested the effects of DA and the β–AR agonist Isoproterenol (ISO) on sEPSCs in CeAL neurons using concentrations previously found to alter sEPSCs in the other extended amygdala regions (Kash et al., 2008; Nobis et al., 2011). We found that a 6 min bath application of 1 μM DA significantly enhanced the frequency of CeAL sESPC from baseline levels (161.0 ± 19.3%, n=10, p<0.05, Fig 5A), while causing no significant changes to sEPSC amplitude (84.6±8.0%, p>0.05), area (83.4±7.8%, p>0.05), rise time (104.9±3.4%, p>0.05) or decay time (92.8±3.3%, p>0.05). In a separate group of neurons, we found that a 10 min bath application of 3 μM ISO also significantly enhanced sEPSC frequency from baseline levels (220.5±28.8%, n=8, p<0.05, Fig 5B) without causing any significant changes to sEPSC amplitude (112.2±16.0%, p>0.05), area (110.2±10.4%, p>0.05), rise time (97.1±7.9%, p>0.05), or decay time (105.2±4.2%, p>0.05).
Figure 5. Dopamine and Isoproterenol enhance spontaneous glutamatergic transmission via a presynaptic mechanism in the CeAL.

(A) top: example sEPSC traces during baseline and after 6 min bath application of dopamine. Bottom: Box-and-whisker plots summarizing of effects of DA on sEPSC frequency and kinetics. (B) top: example sEPSC traces during baseline and after 10 min bath application of the β-AR agonist, isoproterenol. Bottom: Box-and-whisker plots summarizing of effects of isoproterenol on sEPSC frequency and kinetics. (C) top: example sEPSC traces during baseline and after co- application of dopamine and isoproterenol. Bottom: Box-and-whisker plots summarizing of effects of DA+ISO on sEPSC frequency and kinetics. Dotted line indicates normalized baseline values. * indicates significant difference from baseline (p<0.05). Filled circle inside the box plot indicates the mean effect of drugs on sEPSC frequency.
Previous work from our lab showed that DA and ISO can additively enhance sEPSC frequency in the BNST (Nobis et al., 2011). We therefore wanted to determine if DA and ISO can work in combination to enhance sEPSC frequency in the CeAL as well. To that end, we bath applied ISO for 4 minutes and then co-applied ISO and DA for 6 minutes to ensure we time-matched the peak drug effects. Co-application of ISO+DA resulted in a significant increase in sEPSC frequency from baseline levels (252.5±37.6%, n=7, p<0.05, Fig 5C) with no significant changes to sEPSC amplitude (97.0±5.7%, p>0.05), area (96.2±3.2%, p>0.05), rise time (92.5±3.8%, p>0.05), or decay time (103.4±2.6%, p>0.05). One-way ANOVA revealed that the effect of ISO+DA on sEPSC frequency was not significantly different from the effects of DA or ISO alone (F=2.9, p=0.07).
Catecholamine mediated enhancement of sEPSC frequency in the BNST is dependent on CRFR1 activation (Kash et al., 2008; Nobis et al., 2011; Silberman et al., 2013). Since anatomical evidence suggests that catecholaminergic afferents likely innervate CeA-CRF neurons, and since catecholamine receptor and CRFR agonists all increase sEPSC frequency, it was hypothesized that the effect of catecholamines in the CeAL may also be dependent on CRFR activation. Therefore, we pretreated CeA slices with CRFR1 and CRFR2 antagonists either alone or combined before application of either DA or ISO (Fig 6). Interestingly pretreatment of CRFR antagonists did not significantly alter the potentiating effects of DA (ANOVA: F=1.352, p=0.28) or ISO (ANOVA: F=1.166, p=0.33) on sEPSC frequency in CeAL neurons.
Figure 6. Pretreatment of both CRFR1 and CRFR2 antagonists does not block the effect of catecholamine receptor activation on spontaneous glutamatergic transmission in the CeAL.
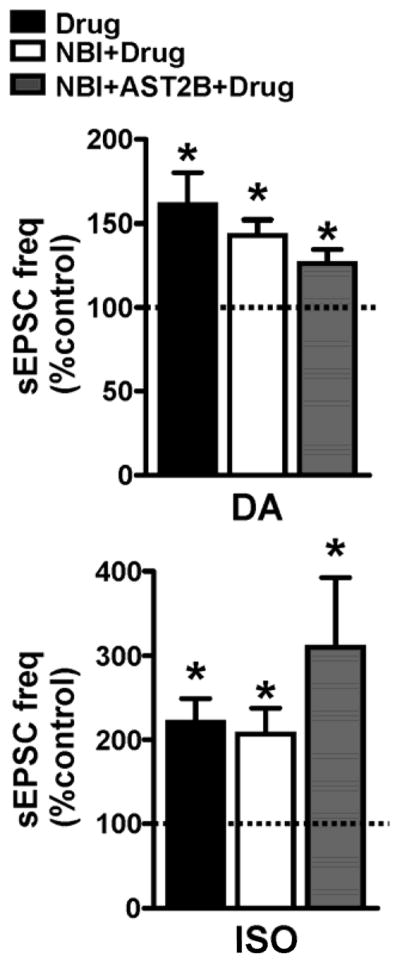
Bar graph showing that pretreatment with a CRFR1, CRFR2 antagonist or combination of antagonists does not block the potentiating effects DA or ISO on sEPSC frequency. Dotted line indicates normalized baseline values. * indicates significant difference from baseline (p<0.05).
4. Discussion
Behavioral data indicate that CRF and catecholamine signaling play an important role in CeA dependent behaviors via enhancement of CeA activity, likely via modulation of glutamatergic transmission. Here we sought to determine the mechanisms by which CRF and catecholamines may modulate glutamatergic transmission in the CeAL. Our findings indicate that although CRF, DA, and β–adrenergic receptor activation can all enhance spontaneous presynaptic glutamatergic release in the CeAL, these effects not occur through separable common mechanism. Intriguingly, although CRF can enhance spontaneous glutamate release, CRF has no effect on glutamatergic transmission evoked by optical stimulation of ChR2 containing glutamatergic afferents to the CeAL, suggesting selective effects of CRF on specific glutamatergic afferents or specific to spontaneous transmission (see below).
The evidence presented here shows that CRF can enhance presynaptic glutamatergic neurotransmission in the CeAL, findings in accordance to previous results indicating enhanced extracellular glutamate levels in the CeA after ICV injections of CRF (Skorzewska et al., 2009). These findings suggest that elevated CRF after stress exposure likely results in enhanced CeA excitability to coordinate behavioral responses. It should be noted that the mice used in this study were not exposed to any prior behavioral manipulations. This is an important consideration as previous work shows that CRF has predominantly inhibitory effects on evoked glutamatergic transmission in the lateral capsular subdivision of the CeA in naïve animals (Liu et al., 2004), while CRF induces long-term potentiation in the lateral capsular CeA after animals have been exposed to daily handling and saline injections, an effect that is even more pronounced following daily cocaine exposures (Krishnan et al., 2010). Therefore, CeA CRF may be highly malleable even by minimal stressors and increasingly pertinent in time of high stress load. Recent evidence suggests that spontaneous neurotransmitter release is important in stabilizing synaptic function (Sutton et al., 2006). Together, these findings suggest that CRF enhancement of sEPSCs could possibly be an initial permissive factor for synaptic plasticity in the CeA that may first stabilize basal synaptic function and further allows for enhanced plasticity following stress, drug exposure, or conditioning paradigms.
Although it is not fully understood what roles CRF receptor subtypes play in CeA-dependent behaviors, these results indicate that CRF receptor modulation of CeAL function is much more complicated than previously thought. The effect of CRF in these studies was mimicked by a CRFR1 selective agonist, Stressin, suggesting that CRFR1 activation alone can enhance glutamatergic transmission in the CeA. However, the effect of exogenous CRF could only be fully blocked by a combination of CRFR1 and CRFR2 antagonists. Since CRFR1 has a higher affinity for CRF than does CRFR2 (Bale and Vale, 2004), these findings may suggest that CRF can enhance glutamatergic neurotransmission in the CeA via CRFR2 even after CRFR1 becomes saturated. Interestingly, our data indicate that pretreatment with either CRFR1 or CRFR2 antagonists alone or together resulted in significant increases in basal sEPSC frequency. These findings are similar to previous results indicating that CRFR1 (Liu et al., 2004) or CRFR2 (Fu and Neugebauer, 2008) antagonists can enhance CeA glutamatergic transmission in rats. This may indicate that CRFR1 and CRFR2 may both tonically regulate CeAL glutamatergic transmission in a balanced fashion and that blockade of one or both of these receptors may disrupt homeostatic glutamatergic transmission in this brain region. Future studies will be needed to determine the role of CRF receptor subtypes in CeA-dependent behaviors and pathologies.
Previous studies show that CRF receptor activation can either inhibit or enhance CeA glutamatergic transmission depending on the model or stimulation procedure used (Fu and Neugebauer, 2008; Krishnan et al., 2010; Liu et al., 2004). Even within this study CRF had variable effects based on the type of recording performed, as CRF enhanced sEPSC frequency while having no effect on oEPSCs. These findings suggest that CRF modulation of CeAL excitability may be pathway specific. However, since sEPSCs do not rely on stimulation of specific glutamate afferents, it is difficult to determine from these findings which source of glutamate may be enhanced by CRF. Our finding that CRF did not modulate oEPSCs may suggest that CRF does not modulate inputs that express ChR2 under the Thy1 promoter (include that same citation again). Conversely, the lack of effect of CRF on oEPSCs could be due to the fact that a wide variety of cells express ChR2 in this mouse line. Therefore, CRF may have enhanced some inputs while inhibiting others to result in an overall neutral effect. Future experiments using more targeted delivery of ChR2 to specific brain regions that project to the CeA will be necessary to fully determine the role of CRF on enhancement of sEPSC from specific afferents. On the other hand, recent evidence indicates that spontaneous transmitter release relies on separable vesicular pools compared to that released during evoked transmission (Ramirez et al., 2012). Therefore, the lack of effect of CRF on oEPSCs, which theoretically should activate similar glutamatergic afferents in the CeAL as those being recorded in sEPSC experiments, could suggest that CRF may have preferential effects on stimulus-independent glutamate release pools which could function to secure synaptic homeostasis in the CeAL (Sutton et al., 2006). Future work will be needed to explore these intriguing possibilites.
One caveat to consider is that the complex nature of CeA circuitry makes it difficult to interpret many electrophysiology studies and reconcile them with what would be expected from behavioral studies. BLA → CeA microcircuitry has been the focus of a number of elegant studies in the past few years (Ciocchi et al., 2010; Haubensak et al., 2010; Tye et al., 2011). These studies have shown that the BLA can modulate the behavioral output of the CeA via direct projections to the medial subnucleus of the CeA and also through an indirect pathway that drives feed-forward inhibition of the CeM via activation of CeAL GABAergic neurons. Although the mechanism by which CRF may modulate the BLA- CeAL -CeM pathway is not yet clear, the findings presented here indicate that increased CRF in the CeAL may enhance glutamatergic transmission from a number of sources. This may then modulate excitatory transmission from BLA afferents either in a synergistic fashion or by functional occlusion, as glutamate transmission from the BLA may be less salient. Ultimately, CRF mediated enhancement of CeAL excitation would be predicted to increase feed-forward inhibition of the CeM and in turn decrease fear/stress related behaviors. Such a mechanism would be inconsistent with what would be expected from behavioral data described in the introduction suggesting that enhanced CeA excitability would lead to enhanced fear/stress related behaviors. However, the CeA contains a number of neuronal subtypes based on co-transmitter content or intracellular protein kinases (Haubensak et al., 2010) suggesting that CRF may have differential effects on CeAL neuronal subtypes in a pathway specific manner to modulate specific forms of CeA-dependent behaviors. Future studies will be needed to explore this hypothesis.
The CeAL receives dense catecholaminergic afferents (Asan et al., 2005; Eliava et al., 2003; Freedman and Cassell, 1994) and, as described in the introduction, enhanced catecholamine signaling has been shown to be a critical modulator for a wide variety of CeA-dependent behaviors. Since many of these behaviors are thought to be related to enhanced neuroplasticity in the CeA (Duvarci et al., 2011; Gilpin and Roberto, 2012; Krishnan et al., 2011), these findings suggests that catecholamine receptor activation may enhance excitatory neurotransmission in the CeA. In agreement with this hypothesis, the present study indicates that DA and the β–AR agonist, ISO, can increase presynaptic glutamate release in the CeAL. This enhanced glutamatergic signaling by catecholamines in the CeAL could result in enhanced neuroplasticity in this region, as has been shown in previous studies indicating DA receptors to be important mediators of long-term potentiation in the lateral capsular subdivision of the CeA (Krishnan et al., 2010). DA has also been shown to decrease GABAergic transmission in the CeA (Naylor et al., 2010). Therefore the combination of increased excitation and reduced inhibition of CeA networks by DA could be an important factor in CeA neuroplasticity and in the regulation of CeA-dependent behaviors.
On the other hand, noradrenergic stimulation of CeA glutamatergic transmission has been much less well characterized. A limited number of electrophysiological studies show that exogenous NE may actually inhibit CeA excitability via presynaptic α2-AR activation (Delaney et al., 2007). α2-AR activation in the CeA has been shown to decrease behavioral outcomes typically associated with increased CeA activation (Ortiz et al., 2007; Yamada and Bruijnzeel, 2011), suggesting that these receptors likely limit the overall effect of extracellular NE in the CeA. However, numerous studies indicate that extracellular NE levels increase in the CeA following a stressor (Pacak et al., 1993; Watanabe et al., 2003) and that stress and fear conditioning can increase markers of CeA activation (Honkaniemi et al., 1992; Radulovic et al., 1998). This suggests that enhanced NE levels may lead to enhanced neuronal activation in the CeA, possibly via β–AR activation (Liang et al., 1986; Watanabe et al., 2003). Consistent with this hypothesis, here we show that ISO can enhance presynaptic glutamate release in the CeAL suggesting the β–AR enhancement of CeAL excitability could be important in the regulation of many of the CeA-dependent behaviors described above.
Anatomical and electrophysiological studies combine to suggest that catecholamine and CRF signaling interactions play an important role in increasing neuronal excitability in the amygdala (Asan et al., 2005; Kash et al., 2008; Krishnan et al., 2010; Nobis et al., 2011). In the BNST, a component of the extended amygdala, catecholamine receptor activation directly depolarizes CRF producing neurons which may release CRF to then enhance glutamatergic transmission onto BNST projection neurons (Silberman et al., 2013). Since our data shows that exogenous CRF enhances CeA glutamatergic transmission in a similar manner as DA and ISO, it seemed plausible that catecholamine signaling might enhance glutamatergic transmission in the CeAL via modulation of CRF signaling. However, this did not seem to be the case as blockade of CRFR1 and CRFR2 receptors in the CeAL did not modulate the facilitatory effects of DA or ISO on glutamatergic transmission. The anatomical evidence suggests clear interaction between catecholaminergic afferents and CRF cell bodies in the CeAL, but it remains unclear how this interaction might modulate CeAL excitability. Recent studies indicate that CRF might induce DA release to increase long-term potentiation in the CeA (Krishnan et al., 2010), a mechanism of catecholamine-CRF interactions that would be the inverse of what would be expected from anatomical data. The source of extracellular CRF in the CeAL is also unclear as CRF producing neurons in this brain region are known to project to other brain regions (Erb et al., 2001; Rodaros et al., 2007; Wu et al., 1999) and much of the CRF produced by these neurons is likely to be transported to downstream brain regions (Asan et al., 2005). Indeed we have recently found that CeA CRF neurons have distinct electrophysiological characteristics than BNST CRF neurons (Silberman et al., 2013). Thus one possibility is that a substantial population of CRF-containing neurons in the BNST may be interneurons, while the predominant CRF neurons in the CeA may be projection type. Together these findings suggest that other brain regions may be major sources of extracellular CRF in the CeA (Uryu et al., 1992). Other evidence indicates that volume transmission plays an important role in neuronal signaling in the CeA (Perez et al., 2008). Although this hypothesis has yet to be tested specifically for CeA CRF signaling, potential volume transmission of CRF does bring up the intriguing possibility that CeA CRF neurons may release transmitter from dendritic sites in a mechanism similar to other peptides in various brain regions (Bergquist and Ludwig, 2008; Kennedy and Ehlers, 2011). Clearly, future work will be needed to fully elucidate the source(s) of elevated extracellular CRF in the CeA during stress responses.
In summary, although CRF signaling in the CeA is important to a wide variety of behaviors, the underlying mechanism of CRF action on CeA glutamatergic excitability is complex. Future work will be necessary to fully determine the role of CRF receptor subtypes in the CeA glutamatergic function and its relationship to behavior. However, while anatomical evidence predicts interaction between catecholamine and CRF signaling in the CeAL, the potentiating effect of catecholamines on CeAL glutamatergic transmission does not appear to be regulated by CRF receptors. It is important to note that this finding does not rule out the possibility that catecholamine activation of CeAL CRF neurons can modulate CRF release at target sites. Overall, catecholamine and CRF signaling in the CeAL increase glutamatergic neurotransmission via distinct mechanisms and each one could result in novel targets for pharmacotherapies targeting CeA-dependent behavioral abnormalities.
Highlights.
CRFR1 and CRFR2 enhance spontaneous glutamate release in the CeAL
CRF effects on spontaneous release is distinct from effects on evoked transmission
DA and β-AR activation enhances spontaneous glutamate release in the CeAL
These three receptor systems appear to function independently
Findings suggests dissociable systems for CeA-targeted pharmacotherapeutics
Acknowledgments
We thank Dr. Amy C. Arnold and Dr. Tiffany Wills for their help with manuscript revisions. Funded by NIH grants AA19455 (DGW), DA19112 (DGW), and AA20140 (YS).
Abbreviations
- ISO
isoproterenol
- CRF
corticotropin releasing factor
- CRFR1 and CRFR2
CRF receptor type 1 or type 2
- CeAL
lateral subdivision of the central nucleus of the amygdala
- ChR2
channel rhodopsin
- sEPSCs
spontaneous excitatory postsynaptic currents
- oEPSCs
optically evoked excitatory postsynaptic currents
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Asan E, Yilmazer-Hanke DM, Eliava M, Hantsch M, Lesch KP, Schmitt A. The corticotropin-releasing factor (CRF)-system and monoaminergic afferents in the central amygdala: investigations in different mouse strains and comparison with the rat. Neuroscience. 2005;131:953–967. doi: 10.1016/j.neuroscience.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Bergquist F, Ludwig M. Dendritic transmitter release: a comparison of two model systems. J Neuroendocrinol. 2008;20:677–686. doi: 10.1111/j.1365-2826.2008.01714.x. [DOI] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, Muller C, Luthi A. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- Coco ML, Kuhn CM, Ely TD, Kilts CD. Selective activation of mesoamygdaloid dopamine neurons by conditioned stress: attenuation by diazepam. Brain Res. 1992;590:39–47. doi: 10.1016/0006-8993(92)91079-t. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Crane JW, Sah P. Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron. 2007;56:880–892. doi: 10.1016/j.neuron.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Popa D, Pare D. Central amygdala activity during fear conditioning. J Neurosci. 2011;31:289–294. doi: 10.1523/JNEUROSCI.4985-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliava M, Yilmazer-Hanke D, Asan E. Interrelations between monoaminergic afferents and corticotropin-releasing factor-immunoreactive neurons in the rat central amygdaloid nucleus: ultrastructural evidence for dopaminergic control of amygdaloid stress systems. Histochem Cell Biol. 2003;120:183–197. doi: 10.1007/s00418-003-0557-9. [DOI] [PubMed] [Google Scholar]
- Ellis ME, Kesner RP. The noradrenergic system of the amygdala and aversive information processing. Behav Neurosci. 1983;97:399–415. doi: 10.1037//0735-7044.97.3.399. [DOI] [PubMed] [Google Scholar]
- Erb S, Salmaso N, Rodaros D, Stewart J. A role for the CRF-containing pathway from central nucleus of the amygdala to bed nucleus of the stria terminalis in the stress-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2001;158:360–365. doi: 10.1007/s002130000642. [DOI] [PubMed] [Google Scholar]
- Freedman LJ, Cassell MD. Distribution of dopaminergic fibers in the central division of the extended amygdala of the rat. Brain Res. 1994;633:243–252. doi: 10.1016/0006-8993(94)91545-8. [DOI] [PubMed] [Google Scholar]
- Fu Y, Neugebauer V. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci. 2008;28:3861–3876. doi: 10.1523/JNEUROSCI.0227-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Roberto M. Neuropeptide modulation of central amygdala neuroplasticity is a key mediator of alcohol dependence. Neurosci Biobehav Rev. 2012;36:873–888. doi: 10.1016/j.neubiorev.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarraci FA, Frohardt RJ, Young SL, Kapp BS. A functional role for dopamine transmission in the amygdala during conditioned fear. Ann N Y Acad Sci. 1999;877:732–736. doi: 10.1111/j.1749-6632.1999.tb09312.x. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, Fanselow MS, Luthi A, Anderson DJ. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkaniemi J, Kainu T, Ceccatelli S, Rechardt L, Hokfelt T, Pelto-Huikko M. Fos and jun in rat central amygdaloid nucleus and paraventricular nucleus after stress. Neuroreport. 1992;3:849–852. doi: 10.1097/00001756-199210000-00007. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Differential effects of CRF1 and CRF2 receptor antagonists on pain-related sensitization of neurons in the central nucleus of the amygdala. J Neurophysiol. 2007;97:3893–3904. doi: 10.1152/jn.00135.2007. [DOI] [PubMed] [Google Scholar]
- Kalin NH, Shelton SE, Davidson RJ. The role of the central nucleus of the amygdala in mediating fear and anxiety in the primate. J Neurosci. 2004;24:5506–5515. doi: 10.1523/JNEUROSCI.0292-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash TL, Nobis WP, Matthews RT, Winder DG. Dopamine enhances fast excitatory synaptic transmission in the extended amygdala by a CRF-R1-dependent process. J Neurosci. 2008;28:13856–13865. doi: 10.1523/JNEUROSCI.4715-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Mechanisms and function of dendritic exocytosis. Neuron. 2011;69:856–875. doi: 10.1016/j.neuron.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Brain stress systems in the amygdala and addiction. Brain Res. 2009;1293:61–75. doi: 10.1016/j.brainres.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan B, Centeno M, Pollandt S, Fu Y, Genzer K, Liu J, Gallagher JP, Shinnick-Gallagher P. Dopamine receptor mechanisms mediate corticotropin-releasing factor-induced long-term potentiation in the rat amygdala following cocaine withdrawal. Eur J Neurosci. 2010;31:1027–1042. doi: 10.1111/j.1460-9568.2010.07148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan B, Genzer KM, Pollandt SW, Liu J, Gallagher JP, Shinnick-Gallagher P. Dopamine-induced plasticity, phospholipase D (PLD) activity and cocaine-cue behavior depend on PLD-linked metabotropic glutamate receptors in amygdala. PLoS One. 2011;6:e25639. doi: 10.1371/journal.pone.0025639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Takeda H, Tsuji M, Liu L, Matsumiya T. Antagonism of central CRF systems mediates stress-induced changes in noradrenaline and serotonin turnover in rat brain regions. Methods Find Exp Clin Pharmacol. 1998;20:409–417. doi: 10.1358/mf.1998.20.5.485702. [DOI] [PubMed] [Google Scholar]
- Li W, Neugebauer V. Block of NMDA and non-NMDA receptor activation results in reduced background and evoked activity of central amygdala neurons in a model of arthritic pain. Pain. 2004;110:112–122. doi: 10.1016/j.pain.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Liang KC, Juler RG, McGaugh JL. Modulating effects of posttraining epinephrine on memory: involvement of the amygdala noradrenergic system. Brain Res. 1986;368:125–133. doi: 10.1016/0006-8993(86)91049-8. [DOI] [PubMed] [Google Scholar]
- Lingawi NW, Balleine BW. Amygdala central nucleus interacts with dorsolateral striatum to regulate the acquisition of habits. J Neurosci. 2012;32:1073–1081. doi: 10.1523/JNEUROSCI.4806-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu B, Neugebauer V, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher JP. Corticotropin-releasing factor and Urocortin I modulate excitatory glutamatergic synaptic transmission. J Neurosci. 2004;24:4020–4029. doi: 10.1523/JNEUROSCI.5531-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor JC, Li Q, Kang-Park MH, Wilson WA, Kuhn C, Moore SD. Dopamine attenuates evoked inhibitory synaptic currents in central amygdala neurons. Eur J Neurosci. 2010;32:1836–1842. doi: 10.1111/j.1460-9568.2010.07457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobis WP, Kash TL, Silberman Y, Winder DG. beta-Adrenergic receptors enhance excitatory transmission in the bed nucleus of the stria terminalis through a corticotrophin-releasing factor receptor-dependent and cocaine-regulated mechanism. Biol Psychiatry. 2011;69:1083–1090. doi: 10.1016/j.biopsych.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz JP, Heinricher MM, Selden NR. Noradrenergic agonist administration into the central nucleus of the amygdala increases the tail-flick latency in lightly anesthetized rats. Neuroscience. 2007;148:737–743. doi: 10.1016/j.neuroscience.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak K, Palkovits M, Kvetnansky R, Fukuhara K, Armando I, Kopin IJ, Goldstein DS. Effects of single or repeated immobilization on release of norepinephrine and its metabolites in the central nucleus of the amygdala in conscious rats. Neuroendocrinology. 1993;57:626–633. doi: 10.1159/000126417. [DOI] [PubMed] [Google Scholar]
- Perez dlM, Jacobsen KX, Crespo-Ramirez M, Flores-Gracia C, Fuxe K. Wiring and volume transmission in rat amygdala. Implications for fear and anxiety. Neurochem Res. 2008;33:1618–1633. doi: 10.1007/s11064-008-9722-9. [DOI] [PubMed] [Google Scholar]
- Pitts MW, Todorovic C, Blank T, Takahashi LK. The central nucleus of the amygdala and corticotropin-releasing factor: insights into contextual fear memory. J Neurosci. 2009;29:7379–7388. doi: 10.1523/JNEUROSCI.0740-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Kammermeier J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Khvotchev M, Trauterman B, Kavalali ET. Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron. 2012;73:121–134. doi: 10.1016/j.neuron.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, Dedic N, Schumacher M, von WG, Avrabos C, Touma C, Engblom D, Schutz G, Nave KA, Eder M, Wotjak CT, Sillaber I, Holsboer F, Wurst W, Deussing JM. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–1907. doi: 10.1126/science.1202107. [DOI] [PubMed] [Google Scholar]
- Rezayof A, Zarrindast MR, Sahraei H, Haeri-Rohani AH. Involvement of dopamine D2 receptors of the central amygdala on the acquisition and expression of morphine-induced place preference in rat. Pharmacol Biochem Behav. 2002;74:187–197. doi: 10.1016/s0091-3057(02)00989-9. [DOI] [PubMed] [Google Scholar]
- Reznikov LR, Grillo CA, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J. Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci. 2007;25:3109–3114. doi: 10.1111/j.1460-9568.2007.05560.x. [DOI] [PubMed] [Google Scholar]
- Rivier J, Gulyas J, Kunitake K, DiGruccio M, Cantle JP, Perrin MH, Donaldson C, Vaughan J, Million M, Gourcerol G, Adelson DW, Rivier C, Tache Y, Vale W. Stressin1-A, a potent corticotropin releasing factor receptor 1 (CRF1)-selective peptide agonist. J Med Chem. 2007;50:1668–1674. doi: 10.1021/jm0613875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Cole M, Koob GF. Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol Clin Exp Res. 1996;20:1289–1298. doi: 10.1111/j.1530-0277.1996.tb01125.x. [DOI] [PubMed] [Google Scholar]
- Robledo P, Robbins TW, Everitt BJ. Effects of excitotoxic lesions of the central amygdaloid nucleus on the potentiation of reward-related stimuli by intra-accumbens amphetamine. Behav Neurosci. 1996;110:981–990. doi: 10.1037//0735-7044.110.5.981. [DOI] [PubMed] [Google Scholar]
- Rodaros D, Caruana DA, Amir S, Stewart J. Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience. 2007;150:8–13. doi: 10.1016/j.neuroscience.2007.09.043. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Koolhaas JM, Bohus B. Attenuated cardiovascular, neuroendocrine, and behavioral responses after a single footshock in central amygdaloid lesioned male rats. Physiol Behav. 1991;50:771–775. doi: 10.1016/0031-9384(91)90016-h. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Koolhaas JM, Bohus B. Posttraining norepinephrine infusion into the central amygdala differentially enhances later retention in Roman high-avoidance and low-avoidance rats. Behav Neurosci. 1993;107:575–579. doi: 10.1037//0735-7044.107.4.575. [DOI] [PubMed] [Google Scholar]
- Rudoy CA, Reyes AR, Van Bockstaele EJ. Evidence for beta1-adrenergic receptor involvement in amygdalar corticotropin-releasing factor gene expression: implications for cocaine withdrawal. Neuropsychopharmacology. 2009;34:1135–1148. doi: 10.1038/npp.2008.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S. Role of the central nucleus of the amygdala in the control of blood pressure: descending pathways to medullary cardiovascular nuclei. Clin Exp Pharmacol Physiol. 2005;32:450–456. doi: 10.1111/j.1440-1681.2005.04210.x. [DOI] [PubMed] [Google Scholar]
- Samson RD, Pare D. Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci. 2005;25:1847–1855. doi: 10.1523/JNEUROSCI.3713-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Matthews RT, Winder DG. A corticotropin releasing factor pathway for ethanol regulation of the ventral tegmental area in the bed nucleus of the stria terminalis. J Neurosci. 2013;33:950–960. doi: 10.1523/JNEUROSCI.2949-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorzewska A, Bidzinski A, Hamed A, Lehner M, Turzynska D, Sobolewska A, Szyndler J, Maciejak P, Wislowska-Stanek A, Plaznik A. The effect of CRF and alpha-helical CRF((9-41)) on rat fear responses and amino acids release in the central nucleus of the amygdala. Neuropharmacology. 2009;57:148–156. doi: 10.1016/j.neuropharm.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Thiel KJ, Wenzel JM, Pentkowski NS, Hobbs RJ, Alleweireldt AT, Neisewander JL. Stimulation of dopamine D2/D3 but not D1 receptors in the central amygdala decreases cocaine-seeking behavior. Behav Brain Res. 2010;214:386–394. doi: 10.1016/j.bbr.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treweek JB, Jaferi A, Colago EE, Zhou P, Pickel VM. Electron microscopic localization of corticotropin-releasing factor (CRF) and CRF receptor in rat and mouse central nucleus of the amygdala. J Comp Neurol. 2009;512:323–335. doi: 10.1002/cne.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Okumura T, Shibasaki T, Sakanaka M. Fine structure and possible origins of nerve fibers with corticotropin-releasing factor-like immunoreactivity in the rat central amygdaloid nucleus. Brain Res. 1992;577:175–179. doi: 10.1016/0006-8993(92)90554-m. [DOI] [PubMed] [Google Scholar]
- Wang H, Peca J, Matsuzaki M, Matsuzaki K, Noguchi J, Qiu L, Wang D, Zhang F, Boyden E, Deisseroth K, Kasai H, Hall WC, Feng G, Augustine GJ. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci U S A. 2007;104:8143–8148. doi: 10.1073/pnas.0700384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Nakagawa T, Yamamoto R, Maeda A, Minami M, Satoh M. Involvement of glutamate receptors within the central nucleus of the amygdala in naloxone-precipitated morphine withdrawal-induced conditioned place aversion in rats. Jpn J Pharmacol. 2002;88:399–406. doi: 10.1254/jjp.88.399. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Nakagawa T, Yamamoto R, Maeda A, Minami M, Satoh M. Involvement of noradrenergic system within the central nucleus of the amygdala in naloxone-precipitated morphine withdrawal-induced conditioned place aversion in rats. Psychopharmacology (Berl) 2003;170:80–88. doi: 10.1007/s00213-003-1504-0. [DOI] [PubMed] [Google Scholar]
- Weiss F, Maldonado-Vlaar CS, Parsons LH, Kerr TM, Smith DL, Ben-Shahar O. Control of cocaine-seeking behavior by drug-associated stimuli in rats: effects on recovery of extinguished operant-responding and extracellular dopamine levels in amygdala and nucleus accumbens. Proc Natl Acad Sci U S A. 2000;97:4321–4326. doi: 10.1073/pnas.97.8.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JS, Ku YH, Li LS, Lu YC, Ding X, Wang YG. Corticotropin releasing factor and substance P mediate the nucleus amygdaloideus centralis-nucleus ventromedialis-nucleus dorsomedialis pressor system. Brain Res. 1999;842:392–398. doi: 10.1016/s0006-8993(99)01862-4. [DOI] [PubMed] [Google Scholar]
- Yamada H, Bruijnzeel AW. Stimulation of alpha2-adrenergic receptors in the central nucleus of the amygdala attenuates stress-induced reinstatement of nicotine seeking in rats. Neuropharmacology. 2011;60:303–311. doi: 10.1016/j.neuropharm.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]