

Abstract
Drugs active at G protein-coupled receptors (GPCRs) can differentially modulate either canonical or non-canonical signaling pathways via a phenomenon known as functional selectivity or biased signaling. We report biochemical studies that show that the hallucinogen lysergic acid diethylamide (LSD), its precursor ergotamine (ERG) and related ergolines display strong functional selectivity for β-arrestin signaling at the 5-hydroxytryptamine (5-HT) receptor 5-HT2B, while being relatively unbiased at the 5-HT1B receptor. To investigate the structural basis for biased signaling, we determined the crystal structure of the human 5-HT2B receptor bound to ERG, and compared it with the 5-HT1B/ERG structure. Given the relatively poor understanding of GPCR structure-function to date, insight into different GPCR signaling pathways are important to better understand both adverse and favorable therapeutic activities.
Apart from canonical G protein mediated signaling, G protein-coupled receptors (GPCRs) also activate non-canonical G protein-independent pathways, frequently mediated by β-arrestins (1, 2). So-called ‘biased’ GPCR agonists differentially activate both signaling pathways with distinct efficacies and potencies as compared to unbiased agonists that activate both pathways equally (3). This preferential activation of one pathway over the other has been termed “functional selectivity” or “signaling bias” (2–5). Depending on the receptor, biased signaling patterns are key for mediating inflammation (6), apoptosis (7), and many other processes (2). Biased ligands have been proposed to stabilize receptor conformations that are distinct from those induced by unbiased ligands, and selectively change the propensity of GPCR coupling to either G proteins or β-arrestin (2).
Agonist-induced changes in “trigger motifs” of GPCRs (8) near the binding pocket facilitate large-scale helical movements that are accompanied by rearrangements in highly conserved residues called “micro-switches” (9) that prime GPCRs for subsequent G protein binding and activation (10). The structural features of a signaling biased receptor state remain elusive, and although complexes of two β-arrestin-biased ligands with the β1 adrenergic receptor (β1AR) have been recently solved (11), they did not reveal activation-related changes in the receptor.
To elucidate molecular and structural details of biased signaling, we characterized G protein- and β-arrestin-mediated signaling at G protein-coupled serotonin (5-HT; 5-hydroxytryptamine) receptors with several representative ergolines like LSD and ERG, and solved the crystal structure of the 5-HT2B receptor in complex with ERG, which was identified as a highly biased agonist for the 5-HT2B receptor (12).
To investigate potential differences of ergoline signaling at 5-HT receptors, we examined three prototypical serotonin receptors that interact with distinct G proteins. The 5-HT1B receptor inhibits cyclic adenosine monophosphate (cAMP) production through Gi, the 5-HT2B receptor mediates phospholipase C activation through Gq, and the 5-HT7A receptor stimulates cAMP production through Gs (13). We compared G protein- and β-arrestin-mediated signaling at cloned human 5-HT1B and 5-HT2B receptors, and G protein-mediated signaling at 5-HT7A receptors stimulated by selective and non-selective ligands in HEK293 cells (Fig. 1, table S1) (14).
Fig. 1.

Distinct signaling properties of lysergic acid diethylamide (LSD) and ergotamine (ERG) at 5-HT1B, 5-HT7A and 5-HT2B receptors. We used luminescence-based assays to measure 5-HT1B receptor mediated Gi activation and cAMP production; fluorescence-based calcium mobilization assays to measure 5-HT2B receptor mediated Gq activation and β-arrestin translocation-dependent luciferase reporter assays to measure 5-HT1B and 5-HT2B receptors mediated β-arrestin recruitment, all in HEK293 derived cells. (A) Normalized concentration-response studies for LSD and ERG at human cloned 5-HT1B receptor-mediated activation of Gi and non-canonical (Arrestin) signaling. (B) Normalized concentration-response studies for LSD and ERG at human cloned 5-HT7A receptor-mediated activation of Gs signaling in the presence and absence of 5-Hydroxytryptamine (5-HT). (C) Normalized concentration-response studies for LSD and ERG at human cloned 5-HT2B receptor-mediated activation of Gq and non-canonical (Arrestin) signaling. The solid circles for LSD-Gs signals are superimposed by the solid squares for ERG-Gs signals and thus, not visible. (D) Mean β-arrestin bias factors were calculated for serotonergic agonists (dihydroergotamine (DHE), methylergonovine (MTE), pergolide (PER) and cabergoline (CAB)), at 5-HT2B and 5-HT1B receptors. Concentration-responses curves were fit to the Black and Leff operational model to obtain transduction coefficients [Log(τ/KA)] for each ligand at each corresponding pathway. The ΔLog(τ/KA) was then calculated with 5-HT as a reference agonist for each pathway, and the ΔΔLog(τ/KA) was calculated between two pathways for each ligand. The bias factor is unit-less and defined as 10ΔΔLog(τ/KA) (28). Compounds with values close to one represent unbiased agonists while compounds with large numerical values, typically >100, represent extremely biased agonists. *p<0.0001 via two-way ANOVA comparing 5-HT2B vs 5-HT1B bias factors; N=3–6 separate experiments. ERG, DHE and, to a lesser extent, LSD, MTE and PER show strong β-arrestin bias at 5-HT2B receptor, but not 5-HT1B receptor.
LSD, and especially ERG displayed bias for β-arrestin signaling at 5-HT2B (Bias factors 101 and 228 respectively; Fig. 1D), minimal bias at 5-HT1B (Bias factors 5 and 25 respectively; Fig. 1D), and G protein antagonism at 5-HT7A receptors (Fig. 1B, table S1). We also found significant β-arrestin signaling bias for other ergolines, such as dihydroergotamine (DHE), methylergonovine (MTE), pergolide (PER), and cabergoline (CAB) at the 5-HT2B receptor, whereas all other evaluated compounds showed no significant bias (Fig. 1D). ERG and DHE, both of which contain a large tripeptide moiety substitution at the amide scaffold, displayed more extreme signaling bias at the 5-HT2B receptor compared to LSD.
To investigate the molecular details responsible for biased signaling, we crystallized an engineered 5-HT2B receptor construct in complex with ERG, solved its structure at 2.7 Å (figure S1, S2, table S3), and compared it to the structure of 5-HT1B/ERG reported in the companion manuscript (15), as well as to other known unbiased active-state GPCR structures.
Residues P5.50, I3.40 and F6.44 (16, 17), the “P-I-F” motif, form an interface between helix V, helix III and helix VI near the base of the ligand binding pocket in the β2 adrenergic receptor (β2AR) and many other aminergic receptors, including all 5-HT GPCRs. In the active-state structures of β2AR (8, 18), a chain of conformational rearrangements occur in the P-I-F residues, in which an inward shift of helix V residue P2115.50 is coupled with: (i) a rotamer switch in I1213.40, (ii) a large movement of the F2826.44 side chain, and (iii) a corresponding rotation of helix VI on the cytoplasmic side (8). The 5-HT1B and 5-HT2B receptor structures display two different conformations of the P-I-F motif (Fig. 2). For the 5-HT1B receptor, we observe that the P-I-F configuration is essentially identical to that of the active-state of β2AR (β2AR-R*, PDB ID: 3SN6, (18)) (Fig. 2B). Whereas the 5-HT2B receptor adopts a similar conformation of P2295.50 and I1433.40, the side-chain conformation of F3336.44 was similar to that observed in the inactive β2AR (β2AR-R, PDB ID: 2RH1, (19)) (Fig. 2C, figure S2B). The P-I-F motif, therefore, appears to be in an active-like conformation in the 5-HT1B structure, but only in an intermediate active conformation in the 5-HT2B receptor structure.
Fig. 2.
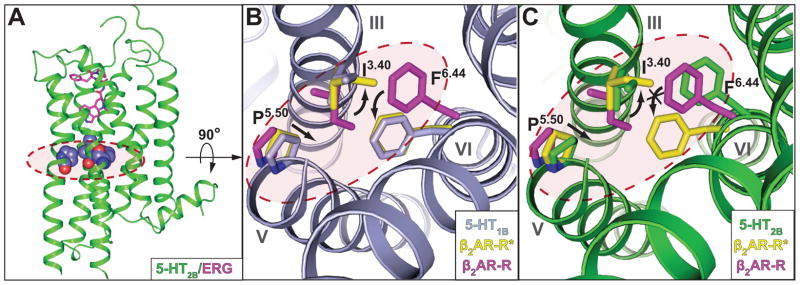
Trigger motif P-I-F displays active- and intermediate active-state for 5-HT1B and 5-HT2B receptors, respectively. Residues of the P-I-F motif are highlighted in a dashed red circle. (A) Overall architecture of the 5-HT2B receptor (green) bound to ERG (magenta); residues of the P5.50 I3.40 F6.44 motif are illustrated in space-filling representation. (B) Alignment between 5-HT1B receptor (grey), β2AR-R (magenta; PDB ID: 2RH1) and β2AR-R*(yellow; PDB ID: 3SN6) indicates an activated P-I-F motif in the 5-HT1B/ERG structure. Black arrows indicate likely rearrangements upon 5-HT1B receptor activation according to analysis of β2AR. (C) Alignment between 5-HT2B receptor (green), β2AR-R (magenta) and β2AR-R* (yellow) suggests intermediate active-state of the P-I-F motif in the 5-HT2B/ERG structure, with F6.44 in an inactive conformation. Black arrows indicate likely rearrangements upon 5-HT2B receptor activation according to analysis of β2AR.
At the cytoplasmic side of the receptors, GPCR activation is generally characterized by the displacement of helices V, VI and VII (10). The magnitude of the helical motions depends on the activation-state; for example, the outward displacement of the intracellular tip of helix VI ranges between 3 and 14 Å, whereas helix VII shifts between 3 to 5 Å towards the receptor core (10, 18–22). These concerted rearrangements induce an opening of the helical bundle at the cytoplasmic side, which facilitates the binding and subsequent activation of G proteins. Analysis of the 5-HT1B and 5-HT2B receptor structures shows that the conformation in the intracellular half of the helical bundle is notably shifted towards that seen in active-state GPCR structures (Fig. 3, figures S3 and S4), and distinct from those of inactive-state GPCR structures (Fig. 3, figures S5 and S6). In the 5-HT1B and 5-HT2B receptor structures, the helix VI intracellular part is located at least 2 to 4 Å further away from the receptor core than in the inactive-state structures of other aminergic GPCRs (figure S5). This conformation of helix VI is close to the one observed in active-state structures of the A2A adenosine receptor (A2AAR) (figure S3), and rhodopsin (Rho) (figure S4), though the outward shift of the helix is smaller in magnitude compared to that of the G protein-bound β2AR (Fig. 3A, 3C). The only difference that we observed in helix VI between 5-HT receptor subtypes was a small clockwise rotation in the 5-HT1B receptor towards the active-state that is absent in the 5-HT2B structure (Fig. 2B, 2C, 3A, 3C). Helix VII in both 5-HT receptor structures also displays intermediate active-states when compared to β2AR (Fig. 3B, 3D), where it is shifted further towards the receptor core than in inactive-state structures of other aminergic GPCRs (figure S6). Whereas the 5-HT2B/ERG receptor structure shows less pronounced active-like changes in helix VI, helix VII appears to be in a more active conformation than it is in the 5-HT1B/ERG receptor structure (Fig. 3B, 3D).
Fig. 3.
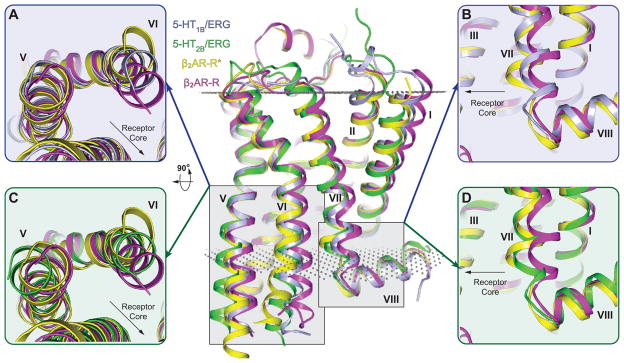
Structural alignment with β2AR-R and β2AR-R* reveals distinct active-state 7TM conformations of the 5-HT1B and 5-HT2B receptor structures. All structures were analyzed based on the last membrane embedded residue to minimize the effect of G proteins and fusion partners on the relative helix positions (see supplementary section). Center panel shows overall 7TM configuration of β2AR-R (magenta; PDB ID: 2RH1), β2AR-R*(yellow; PDB ID: 3SN6), 5-HT1B (grey) and 5-HT2B (green) receptors aligned through helices I–IV. Membrane boundaries are indicated by grey dots according to the OPM database (29). (A, C) Intracellular view of helix V and helix VI in β2AR-R, β2AR-R* compared to (A) the 5-HT1B, or (C) the 5-HT2B receptor. Residues on the intracellular side of the membrane have been removed for better comparison of ligand-induced helical rearrangements (see main text). Helix VI of the 5-HT1B and 5-HT2B receptors is in an intermediate active-state compared to β2AR. (B, D) Side view of helix VII and helix VIII in β2AR-R, β2AR-R* and (B) 5-HT1B receptor, or (D) 5-HT2B receptor. Helix VII of the 5-HT1B and 5-HT2B receptors is in an intermediate active-state compared to β2AR, with more pronounced activation features for the 5-HT2B receptor.
Another important aspect of GPCR activation are rearrangements of side chains in highly conserved motifs D(E)/RY (helix III) and NPxxY (helix VII), which are referred to as “micro-switches” (9). Thus, the D(E)/RY motif of all inactive-state and most active-state GPCR structures shows an intact salt bridge between the side chains of D(E)3.49 and R3.50. Importantly, this salt bridge is broken only in the active-state structures of β2AR-Gαβγ (18) and Opsin-GαCT (21), where R3.50 interacts instead with the G protein and Gα peptide, respectively. The salt bridge is preserved between the side chains of R1533.50 and D1523.49 in the 5-HT2B receptor structure, but it is broken in the 5-HT1B receptor structure (Fig. 4A, 4B, 4D, figure S2C). In the 5-HT1B receptor structure, the D1463.49 side chain forms a hydrogen bond to Y157 in ICL2, and the R1473.50 side chain interacts with the main chain carbonyl in a loop of the fusion protein BRIL (residue L1048), which is partially inserted into the G protein binding crevice (Fig. 4A). Thus, the conformation of the D(E)RY motif mimics the active state of β2AR in the 5-HT1B structure, but resembles the inactive state in the 5-HT2B structure (Fig. 4A, 4C and figure S7).
Fig. 4.

Activation state of the D(E)/RY and NPxxY motifs in 5-HT1B and 5-HT2B receptors compared to β2AR-R and β2AR-R*. Configuration of the D(E)/RY motif in (A) the 5-HT1B receptor (grey), (B) the 5-HT2B receptor (green), (C) β2AR-R* (yellow; PDB ID: 3SN6) and (D) β2AR-R (magenta; PDB ID: 2RH1). Residues of the G protein in β2AR-R* (C) and G protein mimicking BRIL loop (A) are highlighted in orange. The conformation of the D(E)/RY motif in the 5-HT1B receptor is similar to that observed in β2AR-R*, whereas the configuration of the 5-HT2B receptor compares to that of β2AR-R. (E, F) Conformational states of Y7.53 of the NPxxY motif and the proceeding residue 7.54 in β2AR-R, β2AR-R* and (E) the 5-HT1B, or (F) 5-HT2B receptor. When compared to β2AR, the conformation of the NPxxY motif in the 5-HT1B receptor is in an intermediate active-state, whereas the configuration of the 5-HT2B receptor is similar to β2AR-R*.
The highly conserved NPxxY motif at the cytoplasmic end of helix VII is another key micro-switch of GPCR activation (9). Upon GPCR activation, (i) the intracellular end of helix VII moves towards the receptor core and (ii) a rotation of Y7.53 around the helical axis moves the side chain further into the 7TM bundle (10). Both 5-HT receptor structures show active-state conformations of the NPxxY motif when compared to β2AR, A2AAR and Rho (Fig. 4D–E and figures S2D and S7), with more pronounced activation features in the 5-HT2B receptor.
Our analysis indicates that the 5-HT1B/ERG structure has most of the attributes of a classical agonist-induced active-like state, consistent with our biochemical findings that ERG is a comparatively unbiased agonist at the 5-HT1B receptor. In contrast, the 5-HT2B/ERG structure exhibits conformational characteristics of both the active- and inactive-state. The structure of β2AR-Gαβγ complex, along with recent NMR and fluorescence studies of β2AR, implicate helix VI predominantly in G protein signaling, whereas conformational changes in helix VII are associated with enhanced β-arrestin signaling (18, 23, 24). Thus, an active-like state in the helix VII conformation of the 5-HT2B receptor, but only partial changes in helix VI, mirrors the strong β-arrestin bias of ERG at 5-HT2B receptors observed in pharmacological assays.
A likely structural explanation for the distinct conformational features and biased pharmacology of ERG between 5-HT1B and 5-HT2B receptors can be found in the region of the extracellular loop 2 (ECL2) junction with helix V. In the 5-HT2B receptor structure, E212-R213-F214 form an additional helical turn which is stabilized by a structured water molecule and forms a sharp angle to the extracellular tip of helix V (Fig. 5). As a result, the segment of ECL2 that connect helices III and V via the conserved disulfide bond is shortened in the 5-HT2B receptor inducing an inward shift and creating a conformational constraint on the position of the extracellular tip of helix V. Because of these rearrangements, ERG forms additional hydrophobic contacts with M2185.39, L3476.58, V3486.59, L3627.35 and K211ECL2 (figure S8A) and stabilizes a closer distance between helices V and VI, which form hydrophobic contacts between L2195.40, V3486.59 and L3496.60, along with a hydrogen bond between S2225.43 and N3446.45 (figure S8B). These extensive ligand-mediated interactions between helices V and VI in the 5-HT2B/ERG complex may be inferred to prevent rearrangements in helix VI and the corresponding rotation of F6.44 (8), observed in the 5-HT1B receptor and structures of other active-state GPCRs. The strengthened interactions of helix V and VI through ligand-mediated hydrogen bonds to both helices have also been linked to inhibition of G protein signaling at β2AR by the most efficacious inverse agonist (25).
Fig. 5.

Structural differences in extracellular configuration of helix V between 5-HT1B and 5-HT2B receptors likely explain β-arrestin functional selectivity at the 5-HT2B receptor. Side view (left) and top view (right) of 5-HT1B (grey) and 5-HT2B (green) receptors shows a kink in the extracellular end of helix V in the 5-HT2B receptor. A water molecule (red sphere) was found to stabilize the kink through hydrogen bonds (grey dashed lines) with the E2125.33 main-chain carbonyl oxygen and the main-chain nitrogens of D2165.37 and G2155.36. Main-chain atoms of both residues are shown as lines.
Ergolines predominantly signal through β-arrestin pathways at 5-HT2B receptors, whereas signaling at 5-HT1B receptors appears non-biased. The differential signaling patterns are mirrored in the crystal structures, which show features of an intermediate active-state for the 5-HT1B receptor, and a β-arrestin-biased activation-state for the 5-HT2B receptor. We propose a mechanism by which ERG stabilizes a conformation of the 5-HT2B receptor that selectively interferes with G protein signaling (figure S9). The tripeptide moiety of ERG appears to function as a negative allosteric regulator of G protein signaling at the 5-HT2B receptor, as ERG exhibits strongly increased β-arrestin signaling bias compared to LSD and MTE. Because both therapeutic (26) and adverse (27) drug effects have been associated with β-arrestin recruitment by GPCRs, identifying the features of biased GPCR states by known and yet to be discovered intracellular signaling proteins may facilitate the development of safer and more effective therapeutics with selective signaling profiles.
Supplementary Material
Acknowledgments
This work was supported by the NIGMS PSI:Biology grant U54 GM094618 for biological studies and structure production (target GPCR-4) (V.K., V.C. and R.C.S.), NIH Common Fund in Structural Biology grant P50 GM073197 for technology development (V.C. and R.C.S.); the Jay and Betty Van Andel Foundation, Amway (China), R01 DK071662, Ministry of Science and Technology (China) grants 2012ZX09301001-005 and 2012CB910403 (H.E.X.); U19 MH82441, R01 MH61887 and the NIMH Psychoactive Drug Screening Program (X.-P.H., E.V. and B.L.R.) and the Michael Hooker Chair of Pharmacology (B.L.R.). D.W. is supported by a Boehringer Ingelheim Fonds PhD Fellowship. We thank J. Velasquez for help on molecular biology; T. Trinh, K. Allin and M. Chu for help on baculovirus expression; L. N. Collins for help on initial construct selection; K. Kadyshevskaya for assistance with figure preparation; A. Walker for assistance with manuscript preparation; I. Wilson for careful review and scientific feedback on the manuscript; T. Kenakin (UNC) for helpful discussions regarding the quantification of ligand bias; J. Smith, R. Fischetti, and N. Sanishvili for assistance in development and use of the minibeam and beamtime at GM/CA-CAT beamline 23-ID at the Advanced Photon Source, which is supported by National Cancer Institute grant Y1-CO-1020 and National Institute of General Medical Sciences grant Y1-GM-1104. Use of the Advanced Photon Source was supported by the Office of Science of the United States Department of Energy. The coordinates and the structure factors of the 5-HT2B/ERG complex have been deposited in the Protein Data Bank under the accession code 4IB4.
References and Notes
- 1.Luttrell LM, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 2.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 4.Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011;51:117. doi: 10.1146/annurev-pharmtox-010510-100553. [DOI] [PubMed] [Google Scholar]
- 5.Urban JD, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 6.Gao H, et al. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell. 2004;14:303. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- 7.Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. {beta}-Arrestin-2 Mediates Anti-apoptotic Signaling through Regulation of BAD Phosphorylation. J Biol Chem. 2009;284:8855. doi: 10.1074/jbc.M808463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol Sci. 2009;30:249. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Katritch V, Cherezov V, Stevens RC. Structure-function of the g protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warne T, Edwards PC, Leslie AG, Tate CG. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20:841. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang XP, et al. Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine(2B) receptor agonists: implications for drug safety assessment. Mol Pharmacol. 2009;76:710. doi: 10.1124/mol.109.058057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Materials and methods and supplementary figures and tables are available as supplementary material on Science Online.
- 15.Wang C, et al. Structural Basis for Molecular Recognition at Serotonin Receptors. 2013. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Superscripts refer to the Ballesteros-Weinstein numbering, in which the most conserved among class A GPCRs residues in each transmenbrane helix are designated x.50, where x is the helix number.
- 17.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neuroscience. 1995;25:366. [Google Scholar]
- 18.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imamoto Y, Kataoka M, Tokunaga F, Palczewski K. Light-induced conformational changes of rhodopsin probed by fluorescent alexa594 immobilized on the cytoplasmic surface. Biochemistry. 2000;39:15225. doi: 10.1021/bi0018685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 22.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahmeh R, et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A. 2012;109:6733. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wacker D, et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen JA, et al. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A. 2011;108:18488. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 28.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3:193. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.