

Abstract
Adult neurogenesis is actively studied in part because of the potential to manipulate endogenous neural stem and progenitor cells for tissue repair. Although constitutive generation of neurons in the adult rodent olfactory bulb and hippocampal dentate gyrus is widely accepted and stroke-induced generation of striatal inhibitory neurons consistently observed, evidence supporting the generation of neurons in the neocortex after neuronal loss remains slim. Nevertheless, a few studies suggested that targeted apoptosis of neocortical glutamatergic neurons could trigger the generation of new ones in the adult brain. In light of such studies, we tested whether apoptosis of glutamatergic cortical neurons using two novel transgenic approaches in mice, an inducible Caspase-8 protein and an inducible diphtheria toxin gene, results in new neurons. After a thorough analysis, no new neurons were detected in the neocortex. Interestingly, an increase in the expression of the neuroblast marker DCX was observed in both models, in some cases in cells with morphologies previously associated with poststroke neuroblasts, but DCX+ cells coexpressed the oligodendrocyte precursor marker Olig2, suggesting caution when using DCX as a marker for neuroblasts after injury. Given that the adult neocortex lacks an innate potential to regenerate lost glutamatergic neurons, future strategies should concentrate on manipulating the differentiation potential of endogenous or exogenous precursor cells.
Introduction
Much has been learned about the proliferation, migration, and differentiation of adult neural stem and progenitor cells in the subgranular zone of the hippocampus and the mouse subventricular zone (SVZ) of the lateral ventricles (Ming and Song, 2011). Yet a question with potential clinical relevance that has not been adequately addressed is whether endogenous neural stem and progenitor cells are capable of replacing glutamatergic neocortical neurons after their loss.
In rodents, stroke increases the proliferation of SVZ and subgranular zone precursors and the numbers of neuroblasts and new neurons in the striatum (Yamashita et al., 2006; for review, see Kernie and Parent, 2010). Such findings have opened the possibility of manipulating endogenous neural stem and progenitor cells for tissue repair. Despite the consistent detection of newborn neurons in the poststroke striatum, reports identifying new neurons in the neocortex, another major target of stroke in humans, have been contradictory (Jiang et al., 2001; Arvidsson et al., 2002; Parent et al., 2002; Ohab et al., 2006). Importantly, the handful of studies that have characterized the identity of newborn neurons in the normal and poststroke rodent neocortex have described them as inhibitory neurons (Dayer et al., 2005; Inta et al., 2008; Kreuzberg et al., 2010; Ohira et al., 2010). Because regenerating only inhibitory neurons would likely prove insufficient for functional recovery, two studies showing neurogenesis of neocortical glutamatergic pyramidal neurons after targeted apoptosis were met with enthusiasm (Magavi et al., 2000; Chen et al., 2004). An additional study using the same targeted apoptosis approach showed the recruitment of neural progenitors to the area of injury (Brill et al., 2009). However, since these reports, there have been no follow-up studies showing an endogenous generation of glutamatergic projection neurons in the adult neocortex.
For endogenous neural stem and progenitor cells to be considered a therapeutic target, their in vivo potential to generate glutamatergic neurons after injury should be determined. Here we developed two independent noninvasive transgenic models for inducing apoptosis in glutamatergic neurons. A thorough confocal microscopy analysis of both models yielded no evidence of new neurons in the adult neocortex, whether in areas of high or low neuronal apoptosis. Additionally, DCX expression was increased in areas of neuronal death, but most cells coexpressed the oligodendrocyte progenitor marker Olig2. Overall, the data indicate that the adult brain does not have an innate ability to mount a regenerative response to replace lost glutamatergic neurons in the neocortex.
Materials and Methods
Mice.
Mice hemizygous for CamkCreER (Jax#012362) and heterozygous for R26DTA (Wu et al., 2006) in a predominantly Swiss Webster background were used for all experiments. To generate Camk-iCasp8 mice, a NotI fragment of the AP2Casp8 plasmid (Pajvani et al., 2005) containing the inducible human Caspase8, was placed downstream of a NotI fragment in PMM403 (CamkIIα promoter plasmid) (Nicholls et al., 2008). Hemizygous Camk-iCasp8 mice in an FVB background were used for all experiments. Two- to 4-month-old males and females were used. Tamoxifen-treated Cre-negative littermates and vehicle-treated Camk-iCasp8 or wild-type littermates served as controls for DTA or Casp8 experiments, respectively. Three animals per group were used unless otherwise indicated.
BrdU, dimerizer, and tamoxifen injections.
For CamkCreER;R26-GFP reporter experiments, mice received tamoxifen intraperitoneally (5 mg/35 g) once every other day for 5 d and were perfused 2, 4, or 6 weeks after the last injection. CamkCreER;R26-DTA mice received tamoxifen (7 mg/35 g) every other day for a total of 3 injections. Camk-iCasp8 mice received intraperitoneal injections of the AP20187 dimerizer (Clontech) (2.5 mg/kg) once per day for 2 d. BrdU (100 mg/kg) was injected twice a day starting 3 d before dimerizer or tamoxifen administration and continued as described in Figure 2A.
Figure 2.
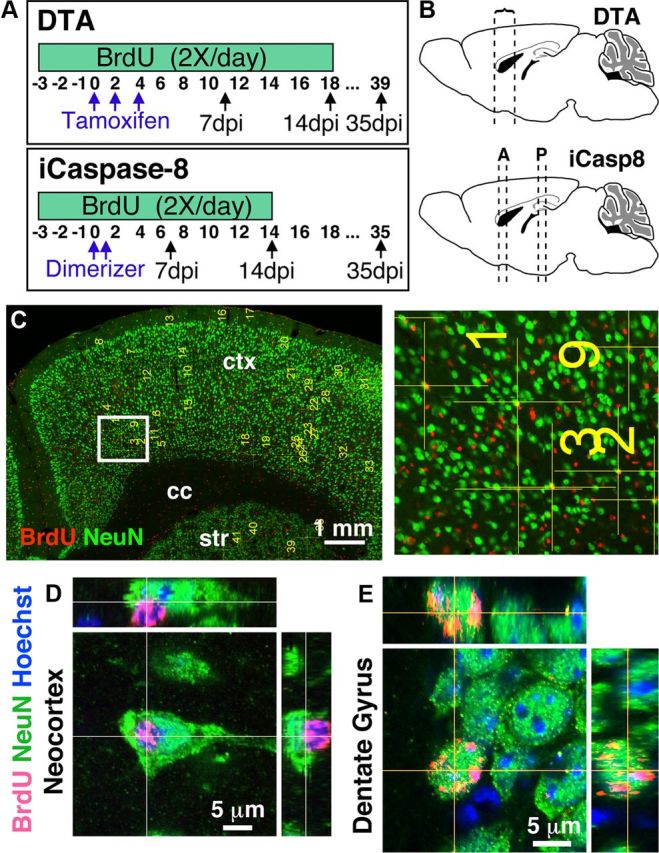
Apoptosis of glutamatergic neurons does not trigger the generation of new glutamatergic neurons. A, Injection schedules for BrdU, tamoxifen, and dimerizer in DTA and iCasp8 mice. B, Schematic showing the location of sections analyzed for BrdU/NeuN colabeling (DTA: at least four half sections/animal from bregma 1.34 to 0.5; iCasp8: at least three sections/animal from bregma 1.1 to 0.2 [anterior, A] and at least two sections/animal from bregma −1.22 to −1.82 [posterior, P]). C, Representative confocal 20× tile scan used to mark putatively colabeled BrdU+/NeuN+ cells with a higher-magnification view of boxed area. D, Example of closely appositioned BrdU+ and NeuN+ cells. A single confocal optical plane suggests colabeling, but the nuclear stain and orthogonal views from the z-stack show that the BrdU+ cell is not a neuron. E, Example of a double-labeled BrdU+/NeuN+ neuron in the DG of the same DTA mice as in D with orthogonal views.
Immunofluorescence.
Mice were perfused with 4% paraformaldehyde. Brains were collected, postfixed, sucrose protected, OCT embedded, and stored at −80°C. Antigen retrieval was performed on mounted cryosections (20 μm) by boiling the slides for 10 min in 10 mm Na citrate/0.05% Tween, pH 6.0. For double labeling with BrdU, antigen retrieval was followed by a 30 min cooling period and DNA denaturation in 2 m HCl for 30 min at room temperature. The following primary antibodies were used: mouse anti-NeuN (1:100, Millipore), rat anti-BrdU (1:100, Accurate), rabbit anti-GFAP (1:500, Dako), rabbit anti-Olig2 (1:400, Millipore), rabbit anti-Iba1 (1:200, Wako Chemicals), rabbit anti-DCX (1:1000, Abcam), guinea pig anti-DCX (1:2000, Millipore), and mouse anti-Ki67 (1:100, BD Biosciences PharMingen). Primary antibodies were incubated for 24–48 h at 4°C and Alexa-488 or Alexa-568 secondaries (1:400 Invitrogen) for 2 h, room temperature. To minimize cross-reactivity, NeuN and BrdU colabeling was performed sequentially. TUNEL with antigen retrieval and Fluoro JadeB (FJB) (Millipore) were used according to the manufacturer's protocol.
FJB analysis.
For CamkCreER-DTA mice, FJB+ cells were counted in at least three separate cortical columns spanning the thickness of the neocortex, and numbers were normalized to area. For CamK-iCasp8 mice, FJB+ cells were counted in the cingulate cortex of at least three anterior coronal sections and in the retrosplenial cortex of at least three posterior coronal sections. A line was drawn from the dorsal-most point of the cingulum above the corpus collosum to the pia, and all cells medial to that border were counted. Numbers were averaged and reported per section.
BrdU, Olig2, Iba1, and GFAP analyses.
For CamkCreER-DTA mice, at least three sections and three independent fields in the neocortex were counted per animal. For CamK-iCasp8 mice, at least two matching sections at the level of the cingulate and retrosplenial cortex were used per animal. Positions of sections are indicated in Figure 2A.
Neocortical BrdU/NeuN colabeling.
At least four coronal sections per animal were scanned at 20× using the tile function in Zeiss Meta 510 duoV2. For each section, the tile covered one hemisphere and the full thickness of the neocortex from the midline to the lateral edge of the corpus callosum. Cells were identified and analyzed as described in Results. Nuclear staining was used in reconstructions to aid in the unambiguous differentiation of very close BrdU+ and NeuN+ cells.
Neurogenesis in the SVZ and subgranular zone.
Hippocampal BrdU/NeuN-colabeled cells were detected and validated using the same method as for the neocortex (above). Tile scans covering the dentate gyrus (DG) were performed with the 63× objective. BrdU/NeuN-colabeled cells were counted from single optical slices of every 10th section (four sections/animal; three animals per experimental condition) starting at bregma −1.34 going posterior. For the SVZ, DCX/Ki67-labeled cells were identified using single optical slices (40× water-immersion objective) of every 10th section (four sections/animal) starting at bregma 1.1 mm. A cell was counted as a neuroblast if the DCX staining surrounded most or all of the Ki67 or nuclear stain.
Statistical analyses.
Data are presented as mean ± SEM with p values obtained from Student's two-tailed t tests.
Results
Inducible Caspase8 and DTA mouse models target glutamatergic neurons for degeneration
FJB, a marker for degenerating neurons (Schmued and Hopkins, 2000), TUNEL, and activated Caspase-3 (aCasp3) were used to assess cell death in both models. In CamkCreER-DTA mice, FJB was rarely detected in control mice but was widespread throughout the neocortex (and other forebrain regions) 7 d after induction of DTA expression via tamoxifen (FJB+ cells: 194 ± 36/mm2 in experimental animals and 12 ± 6 in controls, p = 0.0082; Fig. 1D–F; and data not shown).
Figure 1.

Inducible Caspase8 and DTA mouse models target glutamatergic neurons for degeneration. A, In the CamkCreER-DTA model, tamoxifen-induced Cre recombination results in DTA expression from the Rosa26 locus. B, In the Camk-iCasp8 model, binding of the dimerizer (red) to modified FKBP domains (purple) results in cleavage and activation of a membrane-bound (blue myristoylation site) Caspase8. C, Red boxes indicate the locations of panels D, E, G–I. G, H, FJB labeling marks degenerating neurons. No or few FJB+ neurons were detected in controls (D) but were clearly identified in both DTA (E) and iCasp8 (G, H) animals 7 d after tamoxifen or dimerizer treatment. F, Close-up of FJB+ neuron boxed in E, showing pyramidal morphology. In iCasp8 animals, degeneration was higher posteriorly (H) compared with more anterior areas (G) and correlated with TUNEL (I). J–L, CamKCreER mice were crossed with a R26 reporter. J, Only neurons were targeted as shown by GFP (green) and NeuN (red) colabeling. Most of these neurons were TBR1+ glutamatergic neurons (K), with very few targeted GABA+ cells (L). M, Confocal single optical plane picture showing aCasp3 and NeuN colabeling.
In Camk-iCasp8 mice, after induction with dimerizer, FJB was primarily detected in medial cortical regions (and other forebrain regions), with a graded increase from the cingulate (anterior) to retrosplenial (posterior) cortex (70.5 ± 8.4 and 123.6 ± 19.3/mm2 in induced vs 1.2 ± 0.5 and 2.9 ± 0.7 in controls, respectively, p < 0.0001 for both anterior and posterior), areas where TUNEL was also detected (Fig. 1G–I). In addition, all aCasp3-labeled cells 7 d after induction of apoptosis were positive for NeuN (n = 1141 cells in 3 animals) (Fig. 1M). These results suggest that Camk-iCasp8 and CamkCreER-DTA can be used to target neurons for degeneration.
The CamkIIα promoter should drive expression in forebrain glutamatergic cells (Mayford et al., 1996). To determine the neuron types targeted with the CamkCreER transgene, mice were crossed with an R26-lox-stop-lox-GFP reporter line. After tamoxifen administration, 100% of the GFP+ cells (n = 765 cells in 3 animals) colabeled with NeuN and 94.7 ± 0.7% of GFP+ cells colabeled with the glutamatergic marker TBR1, whereas only 2.3 ± 0.7% colabeled with GABA (Fig. 1J–L), confirming the primarily glutamatergic nature of the targeted neurons.
Apoptosis of glutamatergic neurons does not trigger the generation of new neurons
To analyze cortical neurogenesis after apoptosis of glutamatergic neurons, newborn cells were labeled by injecting BrdU for a total of 19–23 consecutive days starting 3 d before induction of cell death and brains were analyzed 35–40 d after tamoxifen or dimerizer (Fig. 2A, treatment schedules). Newly generated neurons were identified as colabeled for NeuN, a marker specific for mature neurons (Mullen et al., 1992), and BrdU.
For CamkCreER-DTA mice, a low-magnification tile scan using a confocal microscope was performed covering the cingulate, motor, and some somatosensory cortex (Fig. 2B,C). For Camk-iCasp8 mice, to control for whether differing levels of neuronal loss could affect the generation of new glutamatergic neurons, the confocal BrdU/NeuN analysis was not restricted to medial structures (cingulate and retrosplenial cortex, where death was prominent) but also included the lateral areas of the neocortex in both anterior and posterior regions, as illustrated in Figure 2B,C.
Each cell that seemed colabeled at 20× was marked and examined at 63×. If, after one or two single optical slices, a BrdU+ cell appeared to have some nuclear and/or perinuclear NeuN overlap, a z-stack followed by 3D and orthogonal reconstruction was performed. Using this method, a total of 25,481 BrdU+ cells in experimental mice (19,566 in DTA and 5915 in iCasp8) and 2413 cells in control mice (1564 DTA controls and 849 in iCasp8 controls) were scanned, of which 799 in the experimental mice (519 in DTA and 280 in iCasp8) and 250 in control mice (137 in DTA and 113 in iCasp8 controls) seemed potentially colabeled with NeuN at low magnification (20×) and were therefore reexamined at high magnification in one or two optical slices (63×). Of these cells, only 77 cells still seemed colabeled in experimental mice (58 in DTA and 19 in iCasp8) and 28 in controls (22 in DTA and 6 in iCasp8) and were further examined using z-stacks. However, after z-stacks and 3D reconstructions and aided by a nuclear stain, potentially colabeled cells were found to be closely appositioned BrdU+ and NeuN+ cells (Fig. 2D) with zero colabeled cells being identified in the neocortex of either CamkCreER-DTA or Camk-iCasp8 mice 35 d after induction of apoptosis (and zero in controls). Therefore, in two separate transgenic models of glutamatergic neuron loss, no new neurons could be identified. As positive controls for the approach, BrdU+NeuN+ cells could readily be identified in the DG of treated mice (Fig. 2E).
Most BrdU+ cells after iCaspase8 or DTA-mediated neuronal death are microglia
Given that there was an ∼10-fold increase of BrdU+ cells 7 d after inducing apoptosis in the neocortex of both iCaspase8 and DTA animals (Fig. 3F), we characterized the identity of these cells by colabeling for GFAP, Olig2, and Iba1 to identify astrocytes, oligodendrocyte precursors, and microglia, respectively, cells that represent the majority of the dividing population in other injury models (Simon et al., 2011). Consistent with previous findings, most BrdU+ cells in the neocortex of control animals were Olig2+ (Fig. 3B,F). However, after induction of apoptosis, 70–80% of the BrdU+ cells in both models were Iba+ microglia with activated morphology (Fig. 3D,F). The presence of activated microglia and hypertrophic astrocytes in our models was not necessarily expected given that neuron-specific apoptosis has been reported as noninflammatory (Macklis, 1993). Very few of the hypertrophic astrocytes incorporated BrdU (Fig. 3E,F). Thus, proliferative microglia with activated morphology constituted the majority of BrdU+ cells after targeted neuronal death after both iCaspase8 and DTA-mediated cell death of glutamatergic neurons.
Figure 3.
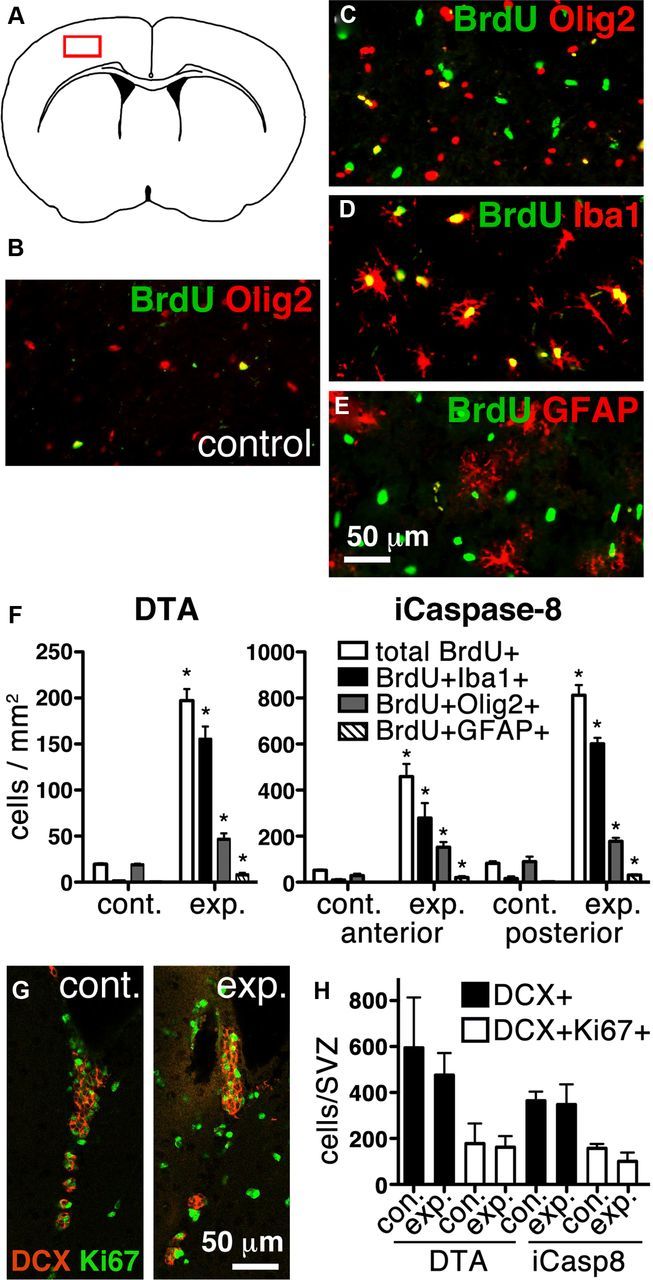
Most BrdU+ cells after iCaspase8 or DTA-mediated neuronal death are microglia. A, Schematic of a coronal section with the boxed area showing the location of B–E. B–F, A significant increase in total BrdU+ cells occurs in both DTA (p = 0.0002) and iCasp8 (anterior, p = 0.0017; posterior, p < 0.0001) experimental mice 7 d after tamoxifen or dimerizer administration, respectively, compared with controls (B–E, representative pictures; F, quantifications). After neuronal apoptosis, most of the increase in BrdU+ cells results from BrdU+Iba1+-activated microglia in both DTA (p = 0.0004) and iCasp8 (anterior, p = 0.0141; posterior, p < 0.0001) experimental mice (D,F). BrdU+GFAP+ astrocytes also increase in both DTA (p = 0.0261) and iCasp8 (anterior, p = 0.0013; posterior, p < 0.0001) experimental mice compared with controls (E,F). A small increase in BrdU+ oligodendrocyte precursors also occurs (C,F, DTA, p = 0.007; iCasp anterior, p = 0.0043; posterior, p = 0.0092). *, indicates significant difference. G, Representative single plane confocal tile scans of the SVZ of control and experimental DTA mice stained for DCX (red) and Ki67 (green). H, Despite the increase in activated microglia and gliosis, no significant difference was observed in the number of proliferating (Ki67+DCX+) (DTA, p = 0.7957; iCasp, p = 0.2488) or total (DCX+) neuroblasts (DTA, p = 0.4399; iCasp, p = 0.8745) in the SVZ of experimental versus control mice.
Positive and negative effects of inflammation with respect to neurogenesis are poorly understood. To address whether inflammation resulting from neuronal death is generally inhibitory to neurogenesis, we analyzed the DG of CamkCreER-DTA mice, which also exhibited activated astrocytes and microglia. No decrease, and instead a minor increase, in BrdU+/NeuN+ cells was observed (CamkCreER-DTA, 42 ± 3.2 BrdU+/NeuN+ cells in the DG with 24 ± 4.9 in controls, p = 0.038). In addition, the number of Ki67+ and total number of DCX+ neuroblasts in the SVZ of CamkCreER-DTA and CamK-iCasp8 mice were analyzed, and no significant differences were detected (Fig. 3G,H). These results suggest that inflammation in these animals does not inhibit endogenous neurogenesis.
DCX upregulation in Olig2+ cells in the neocortex after neuronal death
In addition to the strongly DCX+ neuroblasts in the SVZ and DG, weakly stained DCX cells were observed in other brain regions, including the neocortex, hippocampus, and septum of both control and experimental mice 7 d after inducing apoptosis. However, the expression levels of DCX appeared elevated in experimental animals in regions of extensive neuronal death compared with controls (Fig. 4), and an investigator blind to the treatment conditions correctly differentiated between all slides from experimental (both Camk-iCasp8 and CamkCreER-DTA) and control mice based on the intensity of DCX staining alone in cortical areas (n = 5 each for experimental and control). Interestingly, virtually all DCX+ cells outside the DG expressed the oligodendrocyte precursor marker Olig2 (Fig. 4).
Figure 4.

DCX upregulation in Olig2+ cells in the neocortex after neuronal death. A, DCX expression increases in cells in areas of extensive death and most or all of the DCX+ cells express Olig2. Shown are examples in the neocortex, hippocampus, and septum. Arrows point to examples of DCX+/ Olig2+ cells. Arrowhead points to a DCX+/Olig2− cell. B, Z-stack projection of a DCX+/Olig2+ cell in the septum of an iCasp8 experimental mouse 7 d after apoptosis.
Discussion
An inducible Caspase8 has been used to induce apoptosis in multiple organs outside the CNS (Wencker et al., 2003; Pajvani et al., 2005; Wang et al., 2008). Here we generated the first transgenic mice with an inducible neuron-specific Caspase8. Interestingly, although neuronal death occurred in medial cortical regions, aCasp3 was readily detected in neurons throughout the cerebral cortex (data not shown), suggesting that the caspase pathway was initiated, but that iCaspase8 is expressed at lower levels laterally or that lateral cells are more resistant to caspase-dependent apoptosis. Nevertheless, the induced death of glutamatergic neurons in the neocortex using either the iCaspase8 or DTA models did not trigger the generation of new neurons.
Induced neuronal apoptosis triggered the generation of cell types other than neurons. These were primarily activated microglia, which, together with the observed hypertrophic GFAP+ astrocytes, is indicative of an inflammatory response. An increase in BrdU+ Olig2+ cells was also observed, but the roles of these cells in response to neuronal death remain unclear. The increases in BrdU+ microglia, oligodendrocyte precursors, and, to a lesser extent, astrocytes were similar regardless of whether cell death was induced by iCaspase8 or DTA. Microglia can release proinflammatory and/or anti-inflammatory and neurogenic cytokines depending on the pathological insult (Carpentier and Palmer, 2009; Ransohoff and Perry, 2009). Despite the death of neurons in the hippocampus and the presence of activated microglia and astrocytes, hippocampal neurogenesis was not inhibited, consistent with a previous study in which hippocampal neurons were ablated with a tetracycline-inducible DTA (Yamasaki et al., 2007). Similarly, the numbers of neuroblasts in the SVZ did not decrease after neuronal death. Together, these findings indicate that inflammation does not necessarily result in a hostile environment for neurogenesis. Importantly, these results also indicate that precursors, including neuroblasts, were not directly targeted for apoptosis in either model.
Last, DCX expression was upregulated in Olig2+ cells in the neocortex and other forebrain regions after neuronal loss. Despite expressing DCX, these cells are not likely to be neuroblasts because: (1) there was no apparent increase in the number of Olig2+/DCX+ cells in the neocortex of experimental mice compared with controls; (2) no new mature neurons (BrdU+NeuN+) were observed; (3) oligodendrocyte precursors have previously been reported to express DCX (Tamura et al., 2007; Guo et al., 2010; Ehninger et al., 2011; Clarke et al., 2012); and (4) lineage tracing studies have confirmed the commitment of Olig2+ cells to glial differentiation after injury (Dimou et al., 2008). Nevertheless, further studies would be needed to establish the identity and functional significance of DCX+/ Olig2+ cells after injury. DCX expression in the uninjured and injured adult neocortex has been described in several species (Luzzati et al., 2009; Bloch et al., 2011), but whether in any of these cases the cells coexpressed Olig2 was not reported. Given the similarity of the DCX+/ Olig2+ cell morphologies observed in our models with DCX+ cells observed in the poststroke neocortex, for example (Ohab et al., 2006), caution is warranted when interpreting the nature of these cells.
Overall, the data presented here suggest that the potential of endogenous precursors to regenerate glutamatergic neurons in the neocortex is minimal or nonexistent. Although a few reports claim that, under certain experimental conditions, new cortical projection neurons can be generated in the adult (Magavi et al., 2000; Chen et al., 2004; Brill et al., 2009), our results are consistent with previous findings showing that cortical projection neurons are not generated in the adult under normal conditions or after injury (Parent et al., 2002; e.g., Arvidsson et al., 2002; Dayer et al., 2005; Bhardwaj et al., 2006; Rakic, 2006; Inta et al., 2008).
Future studies should therefore concentrate on altering the differentiation potential of endogenous precursors or on alternative approaches using cell transplantation. In humans, neurogenesis in the SVZ becomes extremely limited in adulthood, if it occurs at all (Sanai et al., 2011; Wang et al., 2011; Bergmann et al., 2012). So it is questionable whether neuronal stem or progenitor cells remain in the adult SVZ or exist elsewhere in the neocortex as a potential source of new projection neurons. The activation and reprogramming of endogenous glial progenitors may provide other strategies, given, for example, that astrocytes can be reprogrammed to neurons in culture using a single transcription factor (Berninger et al., 2007). Alternatively, transplanted progenitor cells could be used. However, these cells would need to be endowed with the ability to migrate throughout damaged or diseased areas, to generate the relevant cell types, and to become functionally integrated. Initial studies aimed at repairing basal ganglia function in mice with transplanted human progenitors suggest that such approaches may also be possible for the neocortex (Kriks et al., 2011; Ma et al., 2012).
Footnotes
This work was supported by the National Institutes of Health (Grants NS073761 and MH070596 to J.M.H.; Grant F31 NS052932 to F.D.), the Hirschl/Weill-Caulier Research Foundation (J.M.H.), and the Brain Research Foundation (J.M.H.). We thank Fiona Doetsch (Columbia University) for helpful discussions, Kostantin Dobrenis and Daniel Murphy for confocal microscopy assistance, Jean Hou for cell counts, Russell Nicholls for the CaMKIIα promoter construct, Philip Scherer and Zhao Wang (University of Texas Southwestern) for providing the human caspase 8 construct and samples used as positive controls during the initial characterization of the Camk-iCasp8 mice, Mario Capecchi (University of Utah) and Karyn Esser (University of Kentucky) for kindly providing the R26DTA mice, and the Allen Institute for Brain Science for the CamkCreER mice.
The authors declare no competing financial interests.
References
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Bergmann O, Liebl J, Bernard S, Alkass K, Yeung MS, Steier P, Kutschera W, Johnson L, Landén M, Druid H, Spalding KL, Frisén J. The age of the olfactory bulb neurons in humans. Neuron. 2012;74:634–639. doi: 10.1016/j.neuron.2012.03.030. [DOI] [PubMed] [Google Scholar]
- Berninger B, Costa MR, Koch U, Schroeder T, Sutor B, Grothe B, Götz M. Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. J Neurosci. 2007;27:8654–8664. doi: 10.1523/JNEUROSCI.1615-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Björk-Eriksson T, Nordborg C, Gage FH, Druid H, Eriksson PS, Frisén J. Neocortical neurogenesis in humans is restricted to development. Proc Natl Acad Sci U S A. 2006;103:12564–12568. doi: 10.1073/pnas.0605177103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch J, Kaeser M, Sadeghi Y, Rouiller EM, Redmond DE, Jr, Brunet JF. Double cortin-positive cells in the adult primate cerebral cortex and possible role in brain plasticity and development. J Comp Neurol. 2011;519:775–789. doi: 10.1002/cne.22547. [DOI] [PubMed] [Google Scholar]
- Brill MS, Ninkovic J, Winpenny E, Hodge RD, Ozen I, Yang R, Lepier A, Gascón S, Erdelyi F, Szabo G, Parras C, Guillemot F, Frotscher M, Berninger B, Hevner RF, Raineteau O, Götz M. Adult generation of glutamatergic olfactory bulb interneurons. Nat Neurosci. 2009;12:1524–1533. doi: 10.1038/nn.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Palmer TD. Immune influence on adult neural stem cell regulation and function. Neuron. 2009;64:79–92. doi: 10.1016/j.neuron.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Magavi SS, Macklis JD. Neurogenesis of corticospinal motor neurons extending spinal projections in adult mice. Proc Natl Acad Sci U S A. 2004;101:16357–16362. doi: 10.1073/pnas.0406795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Young KM, Hamilton NB, Li H, Richardson WD, Attwell D. Properties and fate of oligodendrocyte progenitor cells in the corpus callosum, motor cortex, and piriform cortex of the mouse. J Neurosci. 2012;32:8173–8185. doi: 10.1523/JNEUROSCI.0928-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayer AG, Cleaver KM, Abouantoun T, Cameron HA. New GABAergic interneurons in the adult neocortex and striatum are generated from different precursors. J Cell Biol. 2005;168:415–427. doi: 10.1083/jcb.200407053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Götz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28:10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Wang LP, Klempin F, Römer B, Kettenmann H, Kempermann G. Enriched environment and physical activity reduce microglia and influence the fate of NG2 cells in the amygdala of adult mice. Cell Tissue Res. 2011;345:69–86. doi: 10.1007/s00441-011-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ma J, Xu J, Horiuchi M, Miers L, Vaccarino F, Pleasure D. Pyramidal neurons are generated from oligodendroglial progenitor cells in adult piriform cortex. J Neurosci. 2010;30:12036–12049. doi: 10.1523/JNEUROSCI.1360-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inta D, Alfonso J, von Engelhardt J, Kreuzberg MM, Meyer AH, van Hooft JA, Monyer H. Neurogenesis and widespread forebrain migration of distinct GABAergic neurons from the postnatal subventricular zone. Proc Natl Acad Sci U S A. 2008;105:20994–20999. doi: 10.1073/pnas.0807059105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Gu W, Brännström T, Rosqvist R, Wester P. Cortical neurogenesis in adult rats after transient middle cerebral artery occlusion. Stroke. 2001;32:1201–1207. doi: 10.1161/01.STR.32.5.1201. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Parent JM. Forebrain neurogenesis after focal ischemic and traumatic brain injury. Neurobiol Dis. 2010;37:267–274. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzberg M, Kanov E, Timofeev O, Schwaninger M, Monyer H, Khodosevich K. Increased subventricular zone-derived cortical neurogenesis after ischemic lesion. Exp Neurol. 2010;226:90–99. doi: 10.1016/j.expneurol.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzati F, Bonfanti L, Fasolo A, Peretto P. DCX and PSA-NCAM expression identifies a population of neurons preferentially distributed in associative areas of different pallial derivatives and vertebrate species. Cereb Cortex. 2009;19:1028–1041. doi: 10.1093/cercor/bhn145. [DOI] [PubMed] [Google Scholar]
- Ma L, Hu B, Liu Y, Vermilyea SC, Liu H, Gao L, Sun Y, Zhang X, Zhang SC. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 2012;10:455–464. doi: 10.1016/j.stem.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklis JD. Transplanted neocortical neurons migrate selectively into regions of neuronal degeneration produced by chromophore-targeted laser photolysis. J Neurosci. 1993;13:3848–3863. doi: 10.1523/JNEUROSCI.13-09-03848.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–955. doi: 10.1038/35016083. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S, Kandel ER. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58:104–117. doi: 10.1016/j.neuron.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira K, Furuta T, Hioki H, Nakamura KC, Kuramoto E, Tanaka Y, Funatsu N, Shimizu K, Oishi T, Hayashi M, Miyakawa T, Kaneko T, Nakamura S. Ischemia-induced neurogenesis of neocortical layer 1 progenitor cells. Nat Neurosci. 2010;13:173–179. doi: 10.1038/nn.2473. [DOI] [PubMed] [Google Scholar]
- Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005;11:797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Rakic P. Neuroscience: no more cortical neurons for you. Science. 2006;313:928–929. doi: 10.1126/science.1131713. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, Gupta N, Berger MS, Huang E, Garcia-Verdugo JM, Rowitch DH, Alvarez-Buylla A. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:382–386. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/S0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Simon C, Götz M, Dimou L. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia. 2011;59:869–881. doi: 10.1002/glia.21156. [DOI] [PubMed] [Google Scholar]
- Tamura Y, Kataoka Y, Cui Y, Takamori Y, Watanabe Y, Yamada H. Multi-directional differentiation of doublecortin- and NG2-immunopositive progenitor cells in the adult rat neocortex in vivo. Eur J Neurosci. 2007;25:3489–3498. doi: 10.1111/j.1460-9568.2007.05617.x. [DOI] [PubMed] [Google Scholar]
- Wang C, Liu F, Liu YY, Zhao CH, You Y, Wang L, Zhang J, Wei B, Ma T, Zhang Q, Zhang Y, Chen R, Song H, Yang Z. Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res. 2011;21:1534–1550. doi: 10.1038/cr.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZV, Mu J, Schraw TD, Gautron L, Elmquist JK, Zhang BB, Brownlee M, Scherer PE. PANIC-ATTAC: a mouse model for inducible and reversible β-cell ablation. Diabetes. 2008;57:2137–2148. doi: 10.2337/db07-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Wu Y, Capecchi MR. Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo. Development. 2006;133:581–590. doi: 10.1242/dev.02236. [DOI] [PubMed] [Google Scholar]
- Yamasaki TR, Blurton-Jones M, Morrissette DA, Kitazawa M, Oddo S, LaFerla FM. Neural stem cells improve memory in an inducible mouse model of neuronal loss. J Neurosci. 2007;27:11925–11933. doi: 10.1523/JNEUROSCI.1627-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Ninomiya M, Hernández Acosta P, García-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the poststroke adult striatum. J Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]