

Abstract
The Rho/Rho-kinase pathway is considered important in the pathogenesis of sustained smooth muscle cell contraction during cerebral vasospasm after aneurysmal subarachnoid hemorrhage (SAH). The aims of this study were to investigate whether combination treatment, with pitavastatin as an inhibitor of RhoA and fasudil as an inhibitor of Rho-kinase, prevents the cerebral vasospasm. SAH was simulated using the double-hemorrhage rabbit model, and pitavastatin, or fasudil, or both (combination treatment) were administrated. The basilar artery (BA) cross-sectional area only in the combination treatment group was statistically larger than in the SAH group (p < 0.05). BA Rho-kinase, as measured by ELISA, was statistically reduced only in the combination treatment group compared with the SAH group (p < 0.05). In the other two treatment groups, pitavastatin or fasudil treatment group showed larger BA cross-sectional areas and lower value for BA Rho-kinase, but there were no statistically significant differences compared with the SAH group. The expression of endothelial nitric oxide synthase (eNOS), evaluated by immunohistochemistry in the pitavastatin group and the combination group, was higher than in the SAH group. Results indicate that combination treatment could extensively prevent cerebral vasospasm due to the synergic effect of combining pitavastatin and fasudil on the Rho/Rho-kinase pathway and on eNOS.
Keywords: Cerebral vasospasm, Pitavastatin, Fasudil, Rho/Rho-kinase pathway, eNOS
Introduction
Cerebral vasospasm after aneurysmal subarachnoid hemorrhage (SAH), which is thought to be caused by sustained contraction of smooth muscle cells of the major cerebral arteries, induces cerebral ischemia and affects subsequent mortality and morbidity[1, 2]. Many intracellular signaling transduction pathways are considered to be implicated in the sustained contraction of smooth muscle cells during cerebral vasospasm [3]. Of these, the Rho/Rho-kinase pathway has been in focus as one of the main pathways [4, 5].
The G protein-coupled receptor is activated by several clot-derived substances that alter the subarachnoid space after SAH and that induce the activation of RhoA followed by activation of Rho-kinase [6]. Rho-kinase inhibits the activity of myosin light chain (MLC) phosphatase, resulting in the activity of MLC kinase predominating over that of MLC phosphatase. This leads to phosphorylation of MLC and promotes the mutual sliding of actin and myosin [7, 8].
This point of view suggests that the Rho-kinase inhibitor, fasudil (hydroxyfasudil), could be efficacious in preventing cerebral vasospasm. However, clinical studies have revealed that this efficacy is not remarkable [9, 10].
On the other hand, statin has recently been shown to inhibit RhoA[11]. Statin inhibits HMG-CoA reductase, an enzyme promoting the conversion of HMG-CoA to mevalonic acid, which results in the inhibition of cholesterol biosynthesis. In addition, mevalonic acid, which is reduced due to statin, leads to a decrease in the production of isopentenyl pyrophosphate and geranylgeranyl pyrophosphate (GGPP). GGPP geranylgeranylates Rho-GDP, which is activated by Rho-GEF. Therefore, decreased GGPP induces the inhibition of RhoA [12, 13].
Statin has already been used in the prevention of vasospasm, with the anticipation that its action would upregulate endothelial nitric oxide synthase (eNOS) [14]. To date, this effort has shown no evident efficacy in clinical studies [15].
Although neither fasudil nor statin show remarkable effects in terms of preventing cerebral vasospasm as single use, a combination of these drugs might show sufficient effect through the strong suppression of the Rho/Rho-kinase pathway, due to inhibition of RhoA by statin and the inhibition of Rho-kinase by fasudil.
In order to elucidate any synergic effect in the prevention of cerebral vasospasm, we performed an experimental study using the rabbit SAH model.
Materials and Methods
Animal Population
All experimental protocols were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, from the National Institutes of Health (NIH) and approved by the Hirosaki University Animal Research Committee (M11-019).
All Japanese white rabbits used in the present study were purchased from the Kitayama Labes Co., Ltd (Nagano, Japan). They were maintained on standard pellets at the Institute for Animal Experiments, Hirosaki University School of Medicine. The temperature in both the feeding and the operating room was maintained at about 25 °C.
Animal Experimental Design
Forty 12-week-old female Japanese white rabbits weighing from 2.50 to 2.99 kg were operated. The animals were assigned randomly to five groups; (1) sham group, physiological saline injected into the cisterna magna twice and animals killed on day 5 (n = 8); (2) SAH group, SAH produced as described later and animals killed on day 5 (n = 8); (3) pitavastatin group, after production of SAH, animals treated with pitavastatin (0.8 mg kg−1 day−1) by oral administration once a day from days 0 to 5 and killed on day 5 (n = 8); (4) fasudil group, after production of SAH, animals treated with fasudil (hydroxyfasudil; 3.0 mg/kg) by intravenous administration twice a day from days 0 to 5 and then killed on day 5 (n = 8); (5) combination group: after the production of SAH, animals received combination treatment by oral administration of pitavastatin (0.8 mg kg−1 day−1) once a day and intravenous administration of fasudil (hydroxyfasudil, 3.0 mg/kg) twice a day from days 0 to 5 and were then killed on day 5 (n = 8).
In total, 59 animals were operated in this study, 8 in sham group, 15 in SAH group, 12 in pitavastatin group, 13 in fasudil group, and 11 in combination group. The mortality rate due to SAH was about 47 % in SAH group, 33 % in pitavastatin group, 38 % in fasudil group, and 27 % in combination group, respectively.
Four rabbits in each group were killed using the perfusion–fixation method for histological and immunohistochemical evaluation of the basilar arteries. The other four rabbits in each group were killed without perfusion–fixation and the basilar arteries were immediately removed and stored at −80 °C for protein analysis by ELISA.
Induction of Experimental SAH
Experimental SAH was produced according to the two-hemorrhage method as previously described [16]. The rabbits were anesthetized with an intravenous injection of pentobarbital (30 mg/kg) and an intramuscular injection of ketamine (20 mg/kg). After anesthesia, under spontaneous breathing a 23-gauge butterfly needle was inserted percutaneously into the cisterna magna. After aspiration of 1.5 ml cerebrospinal fluid, the same amount of nonheparinized arterial blood from the femoral artery was slowly injected into the cisterna magna for 1 min under aseptic technique. Rabbits were then placed in a 30° head-down position for 30 min. After recovery from anesthesia, they were returned to the feeding room. Forty-eight hours afterwards (day 2), the second experimental SAH was performed in the same manner as the first. In the sham group, the same technique was applied, with the injection of sterile saline instead of blood.
Perfusion–Fixation
On day 5, four rabbits in each group were deeply anesthetized with an intravenous injection of high-dose pentobarbital (300 mg/kg) and an intramuscular injection of ketamine (100 mg/kg). Perfusion-fixation was then performed. After the thorax was opened, a cannula was immediately inserted into the ascending aorta via the left ventricle. Subsequently, the descending aorta was clamped, and the right atrium was opened. Perfusion was begun with 500 ml heparinized physiological saline (5000 U/500 ml) at 37 °C, followed by 500 ml of phosphate-buffered 4 % paraformaldehyde (pH 7.4) under a perfusion pressure of 75 mmHg. Finally, the whole brain was carefully removed so as not to stretch or injure the basilar artery.
Measurement of Basilar Artery Cross-section Area
The degree of cerebral vasospasm was evaluated by measuring basilar artery lumen cross-sectional area. The formalin-fixed and paraffin-embedded basilar artery sections (6 μm thick) were deparaffinized, hydrated, washed, and stained with hematoxylin and eosin. Micrographs of the basilar arteries were analyzed by using an Image J® version 1.45 (NIH). Cross-sectional areas of basilar arteries were calculated from the perimeter of the luminal border and the area contained within the boundaries of the internal elastic lamina was neglected. For each vessel, three sequential sections (midpoint of proximal, middle, and distal) were taken, measured, and averaged. The mean ± SEM value obtained for each artery was used as the final value for a particular vessel.
Immunohistochemical Analysis for eNOS
Immunohistochemistry was performed on formalin-fixed paraffin-embedded sections to determine the immunoreactivity of eNOS. Sections were deparaffinized and rehydrated in graded concentrations of ethanol to distilled water. Sections were placed in EDTA buffer (pH 9.0), and heated in a microwave oven for 15 min, then cooled at room temperature for 20 min and rinsed in PBS. Endogenous peroxidase activity was blocked with 3 % H2O2 for 5 min, followed by a brief rinse in distilled water and a 15-min wash in PBS. Nonspecific protein binding was blocked by 5 % horse serum. Sections were incubated with primary anti-eNOS antibody (1:200 dilution, BD Biosciences) for 1 h at room temperature, followed by a 15-min wash in PBS. Sections were incubated with goat anti-rabbit IgG (1:500 dilution) for 60 min at room temperature. Diaminobenzidine was used as the chromogen and counterstaining was performed with hematoxylin.
ELISA Analysis for Rho A and Rho Kinase
The other four rabbits in each group were euthanized using high dose pentobarbital (300 mg/kg) and intramuscular injection of ketamine (100 mg/kg), and the basilar artery was immediately removed, frozen in liquid nitrogen, and stored at −80 °C until analysis.
When proteins were extracted after thawing at room temperature and the vessel surface was cleaned in order to remove the SAH, we disrupted the vessels by sonication, taking care to avoid heat denaturation, with RIPA Lysis Buffer (sc-24948, Santa Cruz). The debris was pelleted with 2,000×g centrifugation for 10 min, and the supernatant was further centrifuged at 15,000×g for 15 min at 4 °C.
Using a protein assay kit (500-0002JA, BIO-RAD), protein concentration was measured by the standard microplate assay method (bovine serum albumin for the calibration curve included with the kit). After we uniformly revised protein density for each group, we confirmed the density of each group by measuring the concentration again. RhoA was measured by the G-LISA™ RhoA Activation Assay Biochem Kit™ (Luminescence Based, cat No. BK121, Cytoskeleton; control RhoA is included with the kit) at 490 nm. Rho-kinase was measured by the CycLex Rho-kinase Assay Kit™ (Absorbance Based, cat No. CY-1160, CycLex; control Rho-kinase is cat No. CY-E1160-1, CycLex, sold separately) at 450 nm. The measurements were carried out three times for both RhoA and Rho-kinase, confirming reproducibility.
Statistical Analysis
Statistical analysis was performed using the JMP® (Version 8; SAS Institute Inc., Cary, NC). Data are expressed as mean ± SEM. In order to compare the two groups expressed as the mean ± SEM, normal distribution is not paired; we used the Kruskal–Wallis test, significance determined at p < 0.05.
Results
Evaluation of Cerebral Vasospasm and Vascular Patency
The cross sections of the basilar artery in each group, determined by staining with hematoxylin and eosin and observed under the light microscope, are shown in Fig. 1.
Fig. 1.
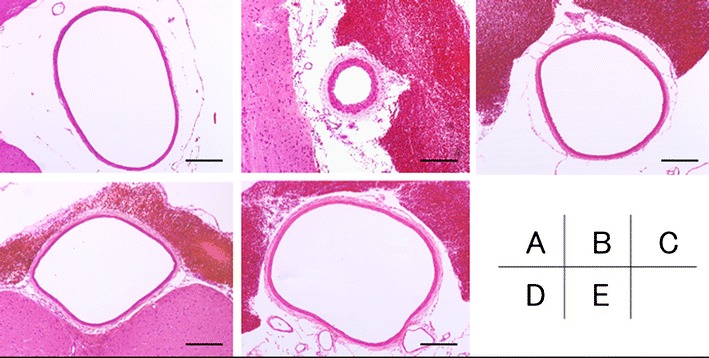
A Sham, B SAH, C pitavastatin, D fasudil, and E combination treatment. Histopathological findings of the basilar artery were evaluated with HE staining. In (B), corrugation of the internal elastic lamina was found. In contrast, in all treatment groups (C–E), there was faint corrugation of the internal elastic lamina around the wall. Scale bar = 200 μm
The cross-sectional areas of the basilar artery in the SAH group were 67 ± 19 μm2, which were statistically significantly less as compared with those in the sham group (Fig. 2). The cross-sectional areas of the basilar artery in the pitavastatin group, fasudil group, and combination group were 230 ± 67, 220 ± 15, and 320 ± 51 μm2, respectively. Although the cross-sectional areas in the combination group showed a maximum value, there was no statistically significant difference among the three treated groups. The cross-sectional areas in the combination treatment group was significantly larger (p < 0.05) compared with those in the SAH group (Fig. 2).
Fig. 2.
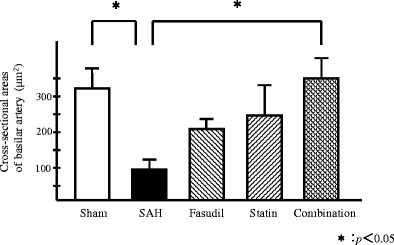
The cross-sectional areas of the basilar artery. In the SAH group, the mean ± SEM cross-sectional areas of the basilar artery were statistically significantly reduced 4 days after SAH versus combination treatment group among all the treatment groups (p < 0.05, n = 4 each). The cross-sectional areas of the combination group were about the same as the sham group
Evaluation of Rho-A/Rho-kinase
RhoA in the SAH group were 1.34 ± 0.1 ng/μl. The SAH group showed a statistically significant increase in RhoA as compared with the sham group (Fig. 3). RhoA in the pitavastatin group, fasudil group, and combination group were 0.82 ± 0.106, 1.16 ± 0.12, and 1.02 ± 0.072 ng/μl, respectively. RhoA was significantly decreased in the pitavastatin group as compared with the SAH group (p < 0.05). The fasudil group and the combination group showed some decreases in RhoA as compared with the SAH group, but statistically significant differences were not seen. And there were no statistically significant differences among all three treatment groups.
Fig. 3.

RhoA expression. Expression of RhoA was significantly inhibited in the vascular smooth muscle cells by ELISA (p < 0.05; versus SAH group, n = 4 each) only in the pitavastatin group among all the treatment groups. RhoA for the fasudil group showed no inhibitory effect and the same degree of absorbance as the SAH group. Although RhoA for the combination group was inhibited as compared with the SAH group, a significant difference did not result
Rho-kinase in the SAH group were 0.354 ± 0.067 units/μl. The SAH group showed a statistically significant increase in Rho-kinase as compared with the sham group (Fig. 4). Rho-kinase in the pitavastatin group, fasudil group, and combination group were 0.257 ± 0.12, 0.227 ± 0.057, and 0.146 ± 0.058 units/μl, respectively. Only in the combination treatment group showed a statistically significant compared with the SAH group (p < 0.05). The pitavastatin group and the fasudil group showed some decreases in Rho-kinase as compared with the SAH group, but statistically significant differences were not seen, and there were no statistically significant differences among all three treatment groups.
Fig. 4.
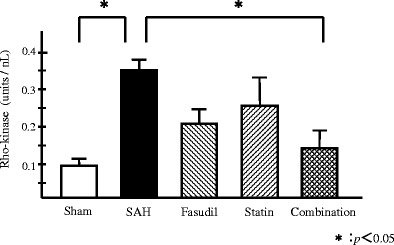
Rho-kinase expression. The expression of Rho-kinase was significantly inhibited in the vascular smooth muscle cells by ELISA (p < 0.05; versus SAH group, n = 4 each) only in the combination group. Although Rho-kinase for the fasudil group was inhibited as compared with the SAH group, a significant difference did not result
Evaluation of eNOS
In comparison to the sham group (Fig. 5A), no or less staining of eNOS in the endothelial cell was seen in the SAH group (Fig. 5B). In the pitavastatin (Fig. 5C) and combination groups (Fig. 5E), eNOS staining increased as compared with the SAH group. However, eNOS staining was not seen in the fasudil group (Fig. 5D).
Fig. 5.
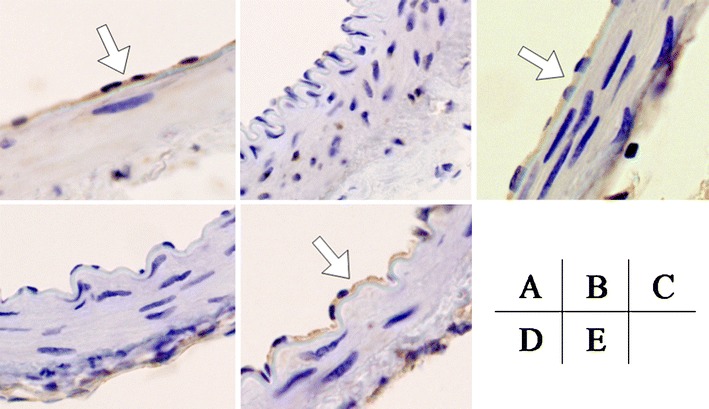
A Sham, B SAH, C pitavastatin, D fasudil, and E combination treatment. Histopathological findings for the basilar artery were evaluated with eNOS staining (the same expansion). In the pitavastatin and combination treatment group, eNOS increased to the same extent as the sham group (see arrow, eNOS expressed in brown by endothelium) in comparison to the SAH group
Discussion
The Rho/Rho-kinase pathway has been considered as playing an important role in sustained contraction during cerebral vasospasm after aneurysmal SAH [5]. After SAH, it is thought that the Rho/Rho-kinase pathway is activated by the following process.
Initially, the G protein-coupled receptor is stimulated by several substances present in the subarachnoid space after SAH [17]. These include thrombin [18, 19], endothelin-1 [20, 21], oxyhemoglobin [17], sphingosine-1 phosphate [22, 23], PDGF [24, 25], TXA2 [26], etc. Stimulated Gα13, among the G protein-coupled receptors, activates Rho-GEF (guanine nucleotide exchange factor), which activates RhoA by converting RhoA-GDP to RhoA-GTP. In turn, phosphorylated RhoA results in the activation of Rho-kinase [26]. Phosphorylated Rho-kinase inactivates MLC phosphatase through phosphorylation of MLC phosphatase target subunit 1 (MYPT1) and MLC phosphatase-specific inhibitor, the 17 kDa PKC-potentiated protein phosphatase 1 inhibitor protein (CPI-17) [27, 28]. Furthermore, it has recently been indicated that phosphorylated Rho-kinase can phosphorylate MLC kinase at the Ser19 residue, which is the site phosphorylated by Ca2+/calmodulin-dependent MLC kinase [29]. Both inactivation of MLC phosphatase and activation of MLC kinase result in sustained contraction of smooth muscle cells.
Inhibition of the Rho/Rho-kinase pathway is therefore considered to represent potential therapy preventing cerebral vasospasm. Fasudil (hydroxyfasudil), a selective inhibitor of Rho-kinase, was introduced in clinical use in 1995 in Japan. In its original clinical randomized double blind trials using placebo as control, it was revealed that fasudil significantly reduced angiographically demonstrable vasospasm by 38 % (from 61 % in the placebo group to 38 % in the fasudil group), and symptomatic vasospasm by 30 % (from 50 to 35 %) after SAH surgery[30]. In addition, fasudil is indicated to have pleomorphic (pleiotropic) effects for cerebral vasospasm, such as protection from endothelial cell damage and anti-inflammatory effects [31]. Several clinical studies have shown that fasudil suppresses cerebral vasospasm and the associated cerebral ischemic symptoms; however, it has also been demonstrated as failing to show sufficient effect in terms of preventing cerebral vasospasm as single use [9, 32].
On the other hand, the hydroxymethylglutaryl-CoA reductase inhibitor, or statin, which is known to have a cholesterol lowering effect, has recently been tried in the prevention of cerebral vasospasm. In experimental studies, statin was shown to ameliorate cerebral vasospasm by upregulating eNOS [33]. Statin activates Akt, which upregulates the activity of eNOS and enhances NO release from endothelial cells and, as a result, statin ameliorates cerebral vasospam [34]. However, in clinical studies, its effects in terms of cerebral vasospasm have been controversial [35–37].
In addition to eNOS upregulation, in the fields of angiology, it has been indicated that statin has several pleiotropic actions that are considered beneficial in preventing cerebral vasospasm, such as inhibiting endothelin-1, inflammation, NADPH oxidase [38], and the caveolin-1 signaling pathway, and, recently, RhoA was also shown to be inhibited by statin [12].
Therefore, we speculated that a combination treatment of statin, for RhoA and eNOS, and fasudil, for Rho-kinase, might suppress the Rho/Rho-kinase pathway more intensely than the single use of statin or fasudil alone.
In this study, the factors examined were phosphorylated RhoA and phosphorylated Rho-kinase, which were essential and important in evaluating the degree of activation of the Rho/Rho-kinase pathway [4, 5], and of eNOS, which increases the production of NO by endothelial cells and is thought to be enhanced by statin or fasudil [10, 34].
As a result, RhoA was suppressed statistically significantly by the administration of statin alone, which corresponds to the results shown by Rashid and Rattan. However, in the combination group, the reduction of RhoA was greater than in the fasudil group but less than in the statin group, and no statistically significant difference was shown as compared with the SAH group. The reason for this is not clarified by this study, but decreased activity of Rho-kinase induced by fasudil may play a role as a feedback to reduce RhoA activity [39]. These interactions between RhoA and Rho-kinase should be clarified in detail in the future.
On the other hand, Rho-kinase activity was inhibited by combination treatment. The combination group showed a statistically significant depression activity for Rho-kinase, suggesting that a combination of statin and fasudil could achieve a synergic effect. Its reduction in activity by statin may be increased by the depression of RhoA activity [12, 13], and its reduction in activity by fasudil was due to the direct effect of fasudil on Rho-kinase [10].
Although eNOS expression after SAH is controversial [40, 41], our study showed a reduction of eNOS expression in the endothelial cells after SAH, and eNOS expression was enhanced in the combination group as well as in the statin group, although its expression was not enhanced in the fasudil group.
Overall, prevention of cerebral vasospasm is superior in the combination therapy group, reflecting its superior effect in inhibiting Rho-kinase activity and maintaining the same enhancement of eNOS as statin. Therefore, despite the incomplete effects of statin or fasudil alone in the prevention of cerebral vasospasm, a combination of these two drugs can achieve adequate effects that should be tested in a clinical study in the near future.
If, as suspected, combination treatment can greatly suppress the Rho/Rho-kinase pathway and induce remarkable amelioration of cerebral vasospasm, the Rho/Rho-kinase pathway will prove to be one of the main pathways inducing sustained contraction of smooth muscle cells during cerebral vasospasm.
Acknowledgments
Disclosure/Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Rabinstein AA, Friedman JA, Weigand SD, McClelland RL, Fulgham JR, Manno EM, et al. Predictors of cerebral infarction in aneurysmal subarachnoid hemorrhage. Stroke J Cereb Circ. 2004;35(8):1862–1866. doi: 10.1161/01.STR.0000133132.76983.8e. [DOI] [PubMed] [Google Scholar]
- 2.Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3(5):256–263. doi: 10.1038/ncpneuro0490. [DOI] [PubMed] [Google Scholar]
- 3.Kai Y, Maeda Y, Sasaki T, Kanaide H, Hirano K. Basic and translational research on proteinase-activated receptors: the role of thrombin receptor in cerebral vasospasm in subarachnoid hemorrhage. J Pharmacol Sci. 2008;108(4):426–432. doi: 10.1254/jphs.08r11fm. [DOI] [PubMed] [Google Scholar]
- 4.Chrissobolis S, Sobey CG. Recent evidence for an involvement of rho-kinase in cerebral vascular disease. Stroke J Cereb Circ. 2006;37(8):2174–2180. doi: 10.1161/01.STR.0000231647.41578.df. [DOI] [PubMed] [Google Scholar]
- 5.Sato M, Tani E, Fujikawa H, Kaibuchi K. Involvement of Rho-kinase-mediated phosphorylation of myosin light chain in enhancement of cerebral vasospasm. Circ Res. 2000;87(3):195–200. doi: 10.1161/01.res.87.3.195. [DOI] [PubMed] [Google Scholar]
- 6.Takai Y, Sasaki T, Tanaka K, Nakanishi H. Rho as a regulator of the cytoskeleton. Trends Biochem Sci. 1995;20(6):227–231. doi: 10.1016/s0968-0004(00)89022-2. [DOI] [PubMed] [Google Scholar]
- 7.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271(34):20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 8.Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273(5272):245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Zhou D, Guo J, Ren Z, Zhou L, Wang S, et al. Effect of fasudil hydrochloride, a protein kinase inhibitor, on cerebral vasospasm and delayed cerebral ischemic symptoms after aneurysmal subarachnoid hemorrhage. Neurol Med Chir. 2006;46(9):421–428. doi: 10.2176/nmc.46.421. [DOI] [PubMed] [Google Scholar]
- 10.Satoh S, Takayasu M, Kawasaki K, Ikegaki I, Hitomi A, Yano K, et al. Antivasospastic effects of hydroxyfasudil, a Rho-kinase inhibitor, after subarachnoid hemorrhage. J Pharmacol Sci. 2012;118(1):92–98. doi: 10.1254/jphs.11075fp. [DOI] [PubMed] [Google Scholar]
- 11.Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H. Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J Off J Japan Circ Soc. 2009;73(2):361–370. doi: 10.1253/circj.cj-08-0817. [DOI] [PubMed] [Google Scholar]
- 12.Rattan S. 3-Hydroxymethyl coenzyme A reductase inhibition attenuates spontaneous smooth muscle tone via RhoA/ROCK pathway regulated by RhoA prenylation. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G962–G969. doi: 10.1152/ajpgi.00034.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Jiang J, Yin H, et al. Atorvastatin inhibits myocardin expression in vascular smooth muscle cells. Hypertens. 2012;60(1):145–153. doi: 10.1161/HYPERTENSIONAHA.112.195644. [DOI] [PubMed] [Google Scholar]
- 14.Sabri M, Ai J, Knight B, Tariq A, Jeon H, Shang X, et al. Uncoupling of endothelial nitric oxide synthase after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2011;31(1):190–199. doi: 10.1038/jcbfm.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugawara T, Ayer R, Jadhav V, Chen W, Tsubokawa T, Zhang JH. Mechanisms of statin treatment in cerebral vasospasm. Acta Neurochir. 2011;110(Pt 2):9–11. doi: 10.1007/978-3-7091-0356-2_2. [DOI] [PubMed] [Google Scholar]
- 16.Foley PL, Caner HH, Kassell NF, Lee KS. Reversal of subarachnoid hemorrhage-induced vasoconstriction with an endothelin receptor antagonist. Neurosurg. 1994;34(1):108–112. [PubMed] [Google Scholar]
- 17.Hansen-Schwartz J, Vajkoczy P, Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm: looking beyond vasoconstriction. Trends Pharmacol Sci. 2007;28(6):252–256. doi: 10.1016/j.tips.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Kai Y, Hirano K, Maeda Y, Nishimura J, Sasaki T, Kanaide H. Prevention of the hypercontractile response to thrombin by proteinase-activated receptor-1 antagonist in subarachnoid hemorrhage. Stroke J Cereb Circ. 2007;38(12):3259–3265. doi: 10.1161/STROKEAHA.107.487769. [DOI] [PubMed] [Google Scholar]
- 19.Maeda Y, Hirano K, Kai Y, Hirano M, Suzuki SO, Sasaki T, et al. Up-regulation of proteinase-activated receptor 1 and increased contractile responses to thrombin after subarachnoid haemorrhage. Br J Pharmacol. 2007;152(7):1131–1139. doi: 10.1038/sj.bjp.0707435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ide K, Yamakawa K, Nakagomi T, Sasaki T, Saito I, Kurihara H, et al. The role of endothelin in the pathogenesis of vasospasm following subarachnoid haemorrhage. Neurol Res. 1989;11(2):101–104. doi: 10.1080/01616412.1989.11739870. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto Y, Ikegaki I, Sasaki Y, Uchida T. The protein kinase inhibitor fasudil protects against ischemic myocardial injury induced by endothelin-1 in the rabbit. J Cardiovasc Pharmacol. 2000;35(2):203–211. doi: 10.1097/00005344-200002000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Takuwa Y, Okamoto Y, Yoshioka K, Takuwa N. Sphingosine-1-phosphate signaling in physiology and diseases. BioFactors (Oxford, England). 2012;doi: 10.1002/biof.1030 [DOI] [PubMed]
- 23.Tosaka M, Okajima F, Hashiba Y, Saito N, Nagano T, Watanabe T, et al. Sphingosine 1-phosphate contracts canine basilar arteries in vitro and in vivo: possible role in pathogenesis of cerebral vasospasm. Stroke J Cereb Circ. 2001;32(12):2913–2919. doi: 10.1161/hs1201.099525. [DOI] [PubMed] [Google Scholar]
- 24.Kishi H, Bao J, Kohama K. Inhibitory effects of ML-9, wortmannin, and Y-27632 on the chemotaxis of vascular smooth muscle cells in response to platelet-derived growth factor-BB. J Biochem. 2000;128(5):719–722. doi: 10.1093/oxfordjournals.jbchem.a022806. [DOI] [PubMed] [Google Scholar]
- 25.Maeda Y, Hirano K, Hirano M, Kikkawa Y, Kameda K, Sasaki T, et al. Enhanced contractile response of the basilar artery to platelet-derived growth factor in subarachnoid hemorrhage. Stroke J Cereb Circ. 2009;40(2):591–596. doi: 10.1161/STROKEAHA.108.530196. [DOI] [PubMed] [Google Scholar]
- 26.Kozasa T, Hajicek N, Chow CR, Suzuki N. Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J Biochem. 2011;150(4):357–369. doi: 10.1093/jb/mvr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Yazbi AF, Johnson RP, Walsh EJ, Takeya K, Walsh MP, Cole WC. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J Physiol. 2010;588(Pt 10):1747–1762. doi: 10.1113/jphysiol.2010.187146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kikkawa Y, Matsuo S, Kameda K, Hirano M, Nakamizo A, Sasaki T, et al. Mechanisms underlying potentiation of endothelin-1-induced myofilament Ca(2+) sensitization after subarachnoid hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2012;32(2):341–352. doi: 10.1038/jcbfm.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Q, Gensch C, Liao JK. Rho-associated coiled-coil-forming kinases (ROCKs): potential targets for the treatment of atherosclerosis and vascular disease. Trends Pharmacol Sci. 2011;32(3):167–173. doi: 10.1016/j.tips.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, et al. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo-controlled double-blind trial. J Neurosurg. 1992;76(4):571–577. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- 31.Ma Z, Zhang J, Du R, Ji E, Chu L. Rho kinase inhibition by fasudil has anti-inflammatory effects in hypercholesterolemic rats. Biol Pharm Bull. 2011;34(11):1684–1689. doi: 10.1248/bpb.34.1684. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki Y, Shibuya M, Satoh S, Sugiyama H, Seto M, Takakura K. Safety and efficacy of fasudil monotherapy and fasudil-ozagrel combination therapy in patients with subarachnoid hemorrhage: sub-analysis of the post-marketing surveillance study. Neurol Med Chir. 2008;48(6):241–247. doi: 10.2176/nmc.48.241. [DOI] [PubMed] [Google Scholar]
- 33.McGirt MJ, Lynch JR, Parra A, Sheng H, Pearlstein RD, Laskowitz DT, et al. Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke J Cereb Circ. 2002;33(12):2950–2956. doi: 10.1161/01.str.0000038986.68044.39. [DOI] [PubMed] [Google Scholar]
- 34.Sugawara T, Ayer R, Jadhav V, Chen W, Tsubokawa T, Zhang JH. Simvastatin attenuation of cerebral vasospasm after subarachnoid hemorrhage in rats via increased phosphorylation of Akt and endothelial nitric oxide synthase. J Neurosci Res. 2008;86(16):3635–3643. doi: 10.1002/jnr.21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tseng MY. Summary of evidence on immediate statins therapy following aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2011;15(2):298–301. doi: 10.1007/s12028-011-9596-6. [DOI] [PubMed] [Google Scholar]
- 36.Vergouwen MDI, Meijers JCM, Geskus RB, Coert BA, Horn J, Stroes ESG, et al. Biologic effects of simvastatin in patients with aneurysmal subarachnoid hemorrhage: a double-blind, placebo-controlled randomized trial. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2009;29(8):1444–1453. doi: 10.1038/jcbfm.2009.59. [DOI] [PubMed] [Google Scholar]
- 37.Sabri M, Macdonald RL. Statins: a potential therapeutic addition to treatment for aneurysmal subarachnoid hemorrhage? World Neurosurg. 2010;73(6):646–653. doi: 10.1016/j.wneu.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 38.Kaneyuki U, Ueda S, Yamagishi S, et al. Pitavastatin inhibits lysophosphatidic acid-induced proliferation and monocyte chemoattractant protein-1 expression in aortic smooth muscle cells by suppressing Rac-1-mediated reactive oxygen species generation. Vasc Pharmacol. 2007;46(4):286–292. doi: 10.1016/j.vph.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Tang AT, Campbell WB, Nithipatikom K. ROCK1 feedback regulation of the upstream small GTPase RhoA. Cell Signal. 2012;24(7):1375–1380. doi: 10.1016/j.cellsig.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vellimana AK, Milner E, Azad TD, Harries MD, Zhou M-L, Gidday JM, et al. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage-induced cerebral vasospasm. Stroke J Cereb Circ. 2011;42(3):776–782. doi: 10.1161/STROKEAHA.110.607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osuka K, Watanabe Y, Usuda N, Atsuzawa K, Yoshida J, Takayasu M. Modification of endothelial nitric oxide synthasea through AMPK after experimental subarachnoid hemorrhage. J Neurotrauma. 2009;26(7):1157–1165. doi: 10.1089/neu.2008.0836. [DOI] [PubMed] [Google Scholar]