

Abstract
Objectives
Radiolabeled amino acids have proven utility for imaging brain tumors in humans, particularly those that target system L amino acid transport. We have prepared the novel phenylalanine analogue, α-[18F]fluoromethyl phenylalanine (FMePhe, 9), as part of an effort to develop new system L tracers that can be prepared in high radiochemical yield through nucleophilic [18F]fluorination. The tumor imaging properties of both enantiomers this new tracer were evaluated through cell uptake, biodistribution and microPET studies in the mouse DBT model of high grade glioma.
Methods
The non-radioactive form of 9 and the cyclic sulfamidate labeling precursor were prepared from commercially available racemic α-benzylserine. Racemic [18F]9 was prepared from the labeling precursor in two steps using standard [18F]fluoride nucleophilic reaction conditions followed by acidic deprotection. The individual enantiomers [18F]9a and [18F]9b were isolated using preparative chiral HPLC. In vitro uptake inhibition assays were performed with each enantiomer using DBT cells. Biodistribution and microPET/CT studies were performed with each enantiomers in male BALB/c mice at approximately 2 weeks after implantation of DBT tumor cells.
Results
Radiolabeling of the cyclic sulfamidate precursor 5 provide racemic [18F]9 in high radiochemical yield (60–70%, n = 4) and high radiochemical purity (>96%, n = 4). In vitro uptake assays demonstrate that both [18F]9a and [18F]9b undergo tumor cell uptake through system L transport. The biodistribution studies using the single enantiomers [18F]9a and [18F]9b demonstrated good tumor uptake with lower uptake in most normal tissues, and [18F]9a had higher tumor uptake than [18F]9b. MicroPET imaging demonstrated good tumor visualization within 10 min of injection, rapid uptake of radioactivity, and tumor to brain ratios of approximately 6:1 at 60 min postinjection.
Conclusions
The novel PET tracer, [18F]FMePhe, is readily synthesized in good yield from a cyclic sulfamidate precursor. Biodistribution and microPET studies in the DBT model demonstrate good tumor to tissue ratios and tumor visualization, with enantiomer [18F]9a having higher tumor uptake. However, the brain availability of both enantiomers was lower than expected for system L substrates, suggesting the [18F]fluorine group in the β-position affects uptake of these compounds by system L transporters.
Keywords: amino acid, glioma, fluorine-18, positron emission tomography, system L
1. Introduction
Amino acid transport is upregulated in many cancers, and a number of amino acid transport systems including system L, system A, system ASC, and cationic amino acid transporters have been targeted with radiolabeled amino acids. System L transport substrates such as O-(2-[18F]fluoroethyl)-L-tyrosine (FET), 6-[18F]fluoro-3,4-dihydroxy-L-phenylalanine (FDOPA) and 3-[123I]iodo-α-methyl-L-tyrosine (IMT) are effective tumor imaging agents in neuro-oncology patients. More recently, other 18F-labeled phenylalanine derivatives including [18F]fluoroalkyl derivatives of phenylalanine and tryptophan have been prepared through nucleophilic substitution reactions. In This class of tracers targeting system L transport provides more accurate estimates of gross tumor volume, better visualization of non-enhancing tumors, and may provide an earlier and more sensitive measure of response to therapy than conventional contrast-enhanced magnetic resonance imaging (MRI) and 2-[18F]fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET). System L transport is sodium-independent, selective for amino acids with large neutral amino acids such as leucine, isoleucine, phenylalanine, tyrosine, valine and tryptophan, and active at the blood brain barrier (BBB). The system L amino acid transporter 1 (LAT1), is upregulated in gliomas and many other cancers, and higher LAT1 protein levels are associated with worse prognosis.
Previous work has demonstrated that the 18F-labeled α,α-dialkyl amino acids 2-amino-3-[18F]fluoro-2-methyl propanoic acid (FAMP) and its N-methyl analogue MeFAMP can be labeled in very high yield (up to ~80% decay corrected yield) and radiochemical purity through cyclic sulfamidate precursors. These tracers are analogues of α-aminoisobutyric acid and are substrates for system A transport. Both [18F]FAMP and [18F]MeFAMP have shown promising tumor imaging properties in rodent models of glioma. Additionally, System L transporters are known to recognize α,α-disubstituted substrates including [123I]IMT. Based on these results, we devised a strategy to radiolabel a novel candidate tracer for system L transport, α-[18F]fluoromethyl phenylalanine (FMePhe, 9) to take advantage the favorable radiochemistry of cyclic sulfamidate precursors.
The development of [18F]9 had two primary motivations. First, the ability to prepare 18F-labeled system L transport substrates in very high radiochemical yield through a simple and reliable radiosynthesis using nucleophilic [18F]fluorination would facilitate batch productions and dissemination of these tracers to other sites. Second, using the α-fluoromethyl group as the site of labeling frees the phenyl ring for structural modifications. Compound 9 could serve as a lead compound to explore the effects of phenyl ring substituents on selectivity for specific transporters within the system L family and on tracer biodistribution and kinetics. Additionally, iodophenyl derivatives could be prepared suitable for both PET imaging with F-18 and for radiotherapy with I-131. In this work, we describe the preparation of a cyclic sulfamidate precursor and radiosynthetic methods for the preparation of [18F]9. We also evaluated the in vitro transport profile, biodistribution, and microPET imaging properties of both enantiomers of [18F]9 in the mouse DBT model of high grade gliomas to assess their ability to image tumors through system L transport.
2. Experimental
All reagents and materials were purchased from commercially available sources. Chemicals were purchased from Aldrich Chemicals Co. (Milwaukee, WI USA) and Sigma Chemical Co. (St. Louis, MO USA) unless otherwise specified, and solvents were purchased from Aldrich Chemicals and Products (Pittsburgh, PA USA). Flash chromatography was carried out using Fisher Scientific Products silica gel 60 (230–400 mesh). Thin-layer chromatography (TLC) analyses were performed with 250 μm UV254 silica gel backing on glass plates (EMD Millipore USA). The TLC plates were developed with ninhydrin and/or Potassium Permanganate stains. Alumina and C-18 SepPak cartridges were purchased from Waters, Inc. (Milford, MA USA).
Melting points were measured with an IA9000 Series digital melting point apparatus (ThermoFisher Scientific, USA) in capillary tubes and are uncorrected. 1H NMR spectra were recorded on a Varian 300 MHz spectrometer, and chemical shifts (δ values) are reported as parts per million (ppm) downfield from tetramethylsilane (TMS), and coupling values in Hz. Elemental analyses were performed by Atlantic Microlabs, Inc. (Norcross, GA USA) and were within ± 0.4 % of the theoretical values. The phrase “usual work up” refers to removal of residual water with anhydrous magnesium sulfate followed by rotary evaporation.
Chemistry
N-(tert-butoxycarbonyl)-α-hydroxymethyl-DL-phenylalanine (2)
Benzylserine 1 (5.13 mmol, 1.0 g) was dissolved in 10:1 methanol:triethylamine (40 mL), and water (6 mL) was added in to this solution to obtain a homogeneous solution followed by di-tert-butyl dicarbonate (7.7 mmol, 1.5 g). The solution was stirred overnight at room temperature. TLC showed no starting material, and the solvent was evaporated under reduced pressure. The residue was extracted with ethyl ether (3 × 20 mL) and followed by usual workup. The crude residue was purified by silica gel flash chromatography (dichloromethane: methanol=1:1) to yield desired product 2 (870 mg, 57%): colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.25–7.29 (m, 3 H), 7.14–7.17 (m, 2 H), 5.67 (br, s, 2H), 5.36 (br, s, 1H), 4.15 (d, 1H, J = 11.4 Hz), 3.97 (d, 1H, J = 12.3 Hz), 3.33 (d, 1H, J = 13.2 Hz), 3.11 (d, 1H, J = 13.8 Hz), 1.46 (s, 9 H). Anal. (C15H21NO5) C,H,N. elemental analysis cal. for: C 61.00, H 7.17, N 4.74, found: C 61.12, H 7.21, N 4.75.
N-(tert-butoxycarbonyl)-α-hydroxymethyl-DL-phenylalanine tert-butyl ester (3)
The N-Boc acid 2 (1 mmol, 296 g) was dissolved in anhydrous toluene (5 mL), and N,N-dimethylformamide di-tert-butyl acetal 3 (5 mmol, 1.02 g) was added to this solution. Under nitrogen, the reaction mixture was stirred for 2 hr at 90°C followed by stirring at room temp overnight. TLC showed no starting material, and the solvent was evaporated. The residue was subjected to silica gel flash chromatography (hexanes: dichloromethane: diethyl ether 2:1:2) to yield the desired product 3 (270 mg, 77%): colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.26–7.32 (m, 5 H), 5.42 (br, s, 1H), 4.22 (dd, 1H, J =5.7, 11.1 Hz), 3.86 (dd, 1H, J = 7.8, 10.8 Hz), 3.38 (d, 1H, J = 14.4 Hz), 3.05 (d, 1H, J = 10.8 Hz), 1.47 (s, 9 H), 1.46 (s, 9 H). Anal. (C19H29NO5) C,H,N.elemental analysis cal. for: C 64.93, H 8.32, N 3.99, found: C 65.01, H 8.35, N 4.05.
Compound 4 (4R,S)-3-(tert-butoxycarbonyl)-4-benzyl-1,2,3-oxathiazolidine-4-carboxylic acid tert-butyl ester 2-oxide. (4)
A solution of compound 3 (0.71 mmol, 250 mg) in anhydrous acetonitrile (4 mL) was cooled to −40°C, and thionyl chloride (1.78 mmol, 129 uL) in 1mL of acetonitrile was added to this solution followed by pyridine (5.0 eq, 286 μL). Under nitrogen, the solution was stirred for 15 min, and then the cold bath was removed. After 15 more min, the TLC showed no starting material, and the solvent was evaporated. A 20 mL portion of anhydrous dichloromethane was added to the residue, and the resulting solution was passed through a short pack of celite and silica gel (5 gram). The solvent was removed under reduced pressure to yield the desired product 4 (200 mg, 71%) which was used in the next reaction without further purification: colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.25–7.39 (m, 5 H), 4.86–5.06 (m, 1H), 4.61–4.74 (m, 1H), 3.49–3.77 (m, 1H), 3.13–3.32 (m, 1H), 1.59 (s, 9 H), 1.50 (s, 9 H). HRMS: calculated for C19H27NO6S: 397.1559, found: [M+H+] 398.1639.
(4R,S)-3-(tert-butoxycarbonyl)-4-benzyl-1,2,3-oxathiazolidine-4-carboxylic acid tert-butyl ester 2,2-dioxide. (5)
A solution of compound 4 (0.5 mmol, 200 mg) in acetonitrile (3 mL) was cooled to 0°C, and RuO2 (5 mol%, 3.3 mg) and NaIO4 (1.1 eq., 118 mg) were added to this solution followed by 1 mL of water. The solution was stirred for 10 min, and then the ice bath was removed. After 30 min of stirring at room temperature, the TLC showed no starting material. Saturated NaHCO3 solution (10 mL.) was added to the reaction mixture followed by extraction with diethyl ether (3 × 10 mL) and usual workup. Purification by silica gel flash chromatography (hexanes: dichloromethane: diethyl ether 2:1:2) provided the desired cyclic sulfamidate 5 (170 mg, 82%): colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.16–7.34 (m, 5 H), 4.51 (d, 2H, J = 9.3 Hz), 4.45 (d, 2H, J = 9.6 Hz), 4.42 (s, 2H), 4.67 (d, 1H, J = 14.7 Hz), 3.23 (d, 1H, J = 15 Hz), 3.13–3.32 (m, 1H), 1.60 (s, 9 H), 1.52 (s, 9 H). HRMS: calculated for C19H27NO7S: 413.1508, found: [M+Na+] 436.1408.
(4R,S)-3-(tert-butoxycarbonyl)-4-benzyl-1,2,3-oxathiazolidine-4-carboxylic acid methyl ester 2,2-dioxide
The same procedure for the synthesis of compound 5 was applied by using the known compound N-(tert-butoxycarbonyl)-α-hydroxymethyl-DL-phenylalanine tert-butyl ester to yield the product 7: colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.19–7.38 (m, 5 H), 4.86–5.06 (m, 1H), 4.61–4.74 (m, 1H), 3.88 (s, 3H), 3.70 (d, 1H, J = 14.7 Hz), 3.30 (d, 1 H, J = 14.4 Hz), 1.57 (s, 9 H). Anal. (C16H21NO7S) C,H,N.elemental analysis cal. for: C 51.74, H 5.70, N 3.77, found: C 51.78, H 5.75, N 3.73.
α-fluoromethyl-DL-phenylalanine methyl ester (8)
To a solution of compound 7 (0.061 mmol, 23 mg) in DMF (1 mL) was added tetrabutylammonium fluoride (1M, 1.5 eq, 90 uL). The reaction mixture was stirred for 2 hr at room temperature followed by the addition of 6M HCl (1.3 mL). After stirring at room temperature overnight, the reaction mixture was brought to pH = 7 using a solution of saturated sodium bicarbonate. The mixture was extracted with diethyl ether (3 × 20 mL) followed by usual work up to yield the desired product 8 (5 mg, 38%): colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.12–7.33 (m, 5 H), 4.72 (dd, 1H, J =11.7, 46.5 Hz), 4.40 (dd, 1H, J =11.4, 47.4 Hz), 3.74 (s, 3H), 3.05 (d, 1H, J =13.2 Hz), 2.77 (d, 1H, J =13.2 Hz). Anal. (C11H14FNO2) C,H,N. elemental analysis cal. for: C 62.55, H 6.68, N 6.63, found: C 62.70, H 6.78, N 6.69.
α-fluoromethyl-DL-phenylalanine hydrochloride salt
Compound 8 (0.023 mmol, 5 mg) was dissolved in 6M HCl (1.0 mL) and stirred overnight at 100°C. The solvent was removed under reduced pressure to provide the desired product 9 (5 mg, 90%): white solid, mp 205.0°C–207.0°C:; 1H NMR (300 MHz, CD3OD) δ 7.24–7.40 (m, 5 H), 4.81–5.03 (m, 1H), 4.61–4.78 (m, 1H), 3.32 (d, 1H, J =14.4 Hz), 3.06 (d, 1H, J =15.4 Hz). Anal. (C10H13ClFNO2) C,H,N.; elemental analysis cal. for: C 51.40, H 5.61, N 5.99, found: C 51.12, H 5.52, N 6.01.
Radiosynthesis of both enantiomers of [18F]9
[18F]fluoride was produced using an RDS-11 cyclotron in the Washington University Cyclotron Facility using [18O]water and the 18O(p,n)18F reaction. Typical radiosyntheses used approximately 50 mCi (1.8 GBq) of [18F]fluoride. The [18F]fluoride was dried by azeotropic distillation using CH3CN (3 × 1 mL) in the presence of K2CO3 (0.75 mg) and K222 (5 mg) at 105 °C under a flow of N2, and then a solution of 5 (1 mg) in CH3CN (500 μL) was added. Incorporation of 18F was achieved by heating the reaction mixture in an oil bath (105 °C) for 15 min. Incorporation of [18F]fluoride was estimated using radiometric thin layer chromatography (radio-TLC) analysis with silica gel TLC plates developed using, 1:1 methanol:water. Then 4 N HCl (250 μL) was added to the reaction mixture followed by microwave irradiation (60 W) for 60 sec. The solution was then dried with a flow of N2 at 105 °C, and then 1 mL of water was added to the vessel prior to HPLC injection. The racemic final product [18F]9 was purified by chiral HPLC using an Chirobiotic Tag column (5μm × 25cm × 10.0mm) eluted with 92% CH3CN, 8% water for 10 min, then changed to 85% CH3CN, 15% water at a flow rate of 3 mL/min. The radioactive peaks corresponding to the single enantiomers of [18F]9b were collected at retention times of approximately 23 min as [18F]9a (1st eluting) and 28 min [18F]9b (2nd eluting). Both enantiopure isomers (>99%) were concentrated under a flow of N2 with heating at 105°C and then formulated in sterile water for cell uptake studies or 0.9% saline for biodistribution studies. As for the imaging studies, single enantiopure isomer [18F]9a (>99%) and isomer [18F]9b (80%) were used for microPET/CT studies. The relatively lower radio-purity of isomer [18F]9b for microPET imaging studies was due to non-optimized preparative HPLC separation.
The radiochemical and enantiomeric purity and specific activity of the final product were assessed using chiral analytical HPLC (Chirobiotic Tag column 150 mm × 4.6 mm, 20 μ, 80:20 CH3CN: water, 1 mL/min, 220 nm). The mass associated with the 18F-labeled product determined by comparison of the integrated UV absorbance with a calibrated mass/UV absorbance standard curve of non-radioactive (R,S)-9. The identities of the enantiomers of [18F]9a and [18F]9b were confirmed by coinjection of the non-radioactive standard (R,S)-9 on the analytical HPLC system.
Cell Uptake Assays
Cell uptake assays were performed using the cluster tray method as reported in the literature\. Mouse DBT gliomas cells were cultured at 1×105 per 24 well (Corning, NY, USA) for 48 hours in a 5% CO2 atmosphere in 10% FBS DMEM culture medium. Two buffer conditions with and without sodium were used for the assays. The phosphate-buffered saline solution contained 105 mM sodium chloride, 3.8 mM potassium chloride, 1.2 mM potassium bicarbonate, 25 mM sodium phosphate dibasic, 0.5 mM calcium chloride dihydrate, 1.2 mM magnesium sulfate, and 5.6 mM D-glucose. The sodium-free phosphate-buffered choline solution was identical to the phosphate-buffered saline solution except choline chloride and choline phosphate dibasic were substituted for sodium chloride and sodium phosphate dibasic, respectively.
The following inhibitors were used for the cell uptake assays: N-methyl-α-aminoisobutyric acid (MeAIB, 10 mM), a mixture of L-alanine/L-serine/L-cysteine (ASC, 3.3 mM of each amino acid) and (R,S)-(endo,exo)-2-aminobicyclo(2,2,1)-heptane-2-carboxylic acid (BCH, 10 mM). The control conditions contained 10 mM of sucrose to maintain consistent osmolality. The assays were performed as described previously at pH 7.40 with each condition performed in quadruplicate. Briefly, cells were washed two times with 37°C assay buffer (2 mL) prior to initiating the assay. For each tracer ([18F]9a or [18F]9b), solutions containing approximately 2.0 mCi/mL (64 MBq/ml) were prepared in the appropriate assay buffer, and then 20 μl of the radioactive tracer was added to appropriate buffer with or without inhibitors. Cells were incubated with radiotracer in assay buffer (0.4 mL total volume) under control or inhibitor conditions for 30 seconds at 37°C. The assay buffer was then discarded from each well followed by washes (3 x mL) ice-cold buffer to remove extracellular radiotracer. The cells were lysed using cells with a 0.2%SDS/0.2 M sodium hydroxide (0.2 mL) at room temperature for 30 min. A 100 μL portion of the lysate from each well was counted to determine the amount of radioactivity taken up by the cells, and 3 × 20 uL potions were used for determination of protein content using the bicinchoninic acid method (Pierce, BCA Protein Assay Kit). Standard dilutions of each assay condition were counted to determine the amount of activity added to each well.
The amount of radioactivity per well was normalized based on the amount of radioactivity added and the protein content of each cell. The uptake data were expressed as percent of uptake relative to control, and each plate was analyzed with a one-way analysis of variance (ANOVA) with Tukey posttests using GraphPad Prism software (GraphPad Software, La Jolla, CA) with p values ≤ .05 considered statistically significant.
DBT Tumor Model
All animal experiments were conducted following the Institutional Animal Care and Use Committee-approved protocols in compliance with the Guidelines for the Care and Use of Research Animals established by the Washington University Medical School Animal Studies Committee. For intracranial tumors, the DBT tumor cells (1×104 cells suspended in a volume of 8 μL) were implanted in the right midcerebrum of male BALB/C mice (20–25 g) as described previously. For subcutaneous tumors, the DBT cells (5 × 105 cells suspended in a volume of 50 μL) were injected subcutaneously into the flanks of male BALB/C mice (20–25 g). Tumor-bearing animals were used for imaging and biodistribution studies approximately 14 days after implantation.
Biodistribution studies with [18F]9a and [18F]9b in mice with subcutaneous DBT tumors
Biodistribution studies were performed using male BALB/C mice with subcutaneous DBT tumors at 14 days after implantation. Mice were anesthetized with 1% isoflurane/oxygen and 20–30 μCi (0.74 – 1.1 MBq) of [18F]9a or [18F]9b was administered via tail vein injection. The animals were euthanized in groups of 4 animals at 5, 30 and 60 minutes post injection. The tumors, organs, and tissues of interest were dissected, weighed and the amounts of activity were using an automated Beckman Gamma 8000 well counter. Additionally, radioactive standards from the dosages were counted. The percentage of dose per gram (%ID/g) of radioactivity in the tumors and normal organs/tissues as well as the tumor to normal organ/tissue ratios were calculated. Uptake in tumor, skeletal muscle, and brain tissue were compared through Tukey posttests using GraphPad Prism software with p values ≤ .05 considered statistically significant.
MicroPET/CT imaging studies
Male BALB/c mice (n=3) with intracranial DBT tumors underwent dynamic microPET imaging from 0 to 60 min after intravenous tail injection of 200–300 μCi (7.4 – 11 MBq) of [18F]9a or [18F]9b at day 14 after tumor implantation using INVEON and MicroPET Focus 220 systems. Dynamic scanning time is 0–60 min. Computed tomography (CT) images were also acquired with the INVEON system. At the conclusion of the imaging studies, the animals were euthanized and their brains were fixed in 4% paraformaldehyde systems for histological analysis with hematoxylin and eosin staining. The microPET data were analyzed by manually drawing regions of interest (ROIs) over the areas of tumor identified on the PET and over normal brain tissues in the contralateral hemisphere to generate time activity curves (TACs). The standardized uptake values and the average tumor to brain ratios at 50–60 min post-injection for both enantiomer of [18F]9a and [18F]9b were compared using 1-way ANOVAs with Tukey posttests (p ≤ 0.05 considered significant).
Results and Discussion
Chemistry
Commercially available DL-benzylserine 1 was used as the starting material. The amino group of 1 was first protected by a Boc group to yield product 2 which was then esterified using the commercially available reagent N,N-dimethylformamide di-tert-butyl acetal in toluene to obtain compound 3. This reagent selectively protected the carboxylic acid functional group by a tert-butyl group in the presence of the unprotected hydroxyl group. Reaction of compound 4 with thionyl chloride in pyridine provided the five-membered ring cyclic sulfamidite 4, which was then oxidized to the cyclic sulfamidate radiolabeleing precursor 5 (Figure 1).
Figure 1.

Preparation of the non-radioactive and 18F-labeled forms of 9.
The non-radioactive racemic 9 standard was synthesized from compound 7, a methyl ester protected cyclic sulfamidate, which was reacted with tetra-n-butylammonium fluoride (TBAF) and followed by hydrolysis to form FMePhe standard compound 9. The methyl ester was used for this reaction series because of the relative stability of the methyl ester compared to the N-Boc group to acid hydrolysis which facilitated isolation and purification of the intermediate 8. However, the stability of the methyl ester is undesirable during radiosynthesis, and thus the t-butyl ester was utilized for the radiolabeling precursor.
Radiosynthesis
The total synthesis and purification time was 90 min and provided the racemic mixture of products in a decay-corrected yield of 65 ± 11%. This high yield is typical of cyclic sulfamidate precursors used for 18F-labeling. Radiochemical purity was >99%, and specific activity > 1Ci/μmol at the end of synthesis. After optimized chiral HPLC purification, the first eluting enantiomer [18F]9a was isolated in more than 99% ee, and the second eluting enantiomer [18F]9b in 98% ee (Figure 2 and Figure 3). Our initial purification method used the addition of potassium carbonate followed by intermittent gentle shaking using tongs to neutralize the aqueous hydrochloric acid prior to injection of the crude racemic product on the chiral HPLC system, but this method led to intermittent contamination of [18F]9b by small but significant amounts of the first eluting enantiomer [18F]9a. This problem was solved by evaporating the crude product under nitrogen gas flow and heating followed by dissolving the radiolabeled product in water prior to HPLC purification as described in the Experimental section.
Figure 2.
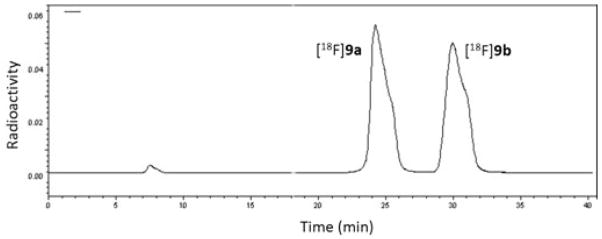
Preparative chiral HPLC separation of the single enantiomers [18F]9a and [18F]9b. Refer to the methods section for HPLC conditions.
Figure 3.

Analytical HPLC co-injections of a mixture of non-radioactive 9a and 9b with [18F]9a and [18F]9b. The retention time of [18F]9b differs slightly that of the non-radioactive form in this figures as these chromatograms were obtained from separate HPLC coinjections with only one non-radioactive chromatogram shown for simplicity. For both [18F]9a and [18F]9b, only one enantiomer was detectable.
Of note, minor decomposition of the 18F-labeled intermediate occurred during acid deprotection which was attributed to defluorination. Based on radiometric TLC monitoring, the fluorine-18 incorporation step showed >90% yield of the presumed 18F-labeled N-sulfamate intermediate. However, when microwave-assisted deprotection was performed under acidic conditions, a minor impurity at the baseline with the same Rf as [18F]fluoride grew in size as final product was formed. In contrast the isolated final product [18F]9a was stable under the same conditions, indicating that decomposition of the intermediate rather than the final product occurred.
Cell uptake assays
The results of the in vitro cell uptake assays are displayed in Figures 4a and 4b. The uptake assays were performed with a short 30 sec uptake time to evaluate the initial influx of [18F]FMePhe and minimize the potential for efflux which occur with many AA transporters including system L and system ASC.
Figure 4.
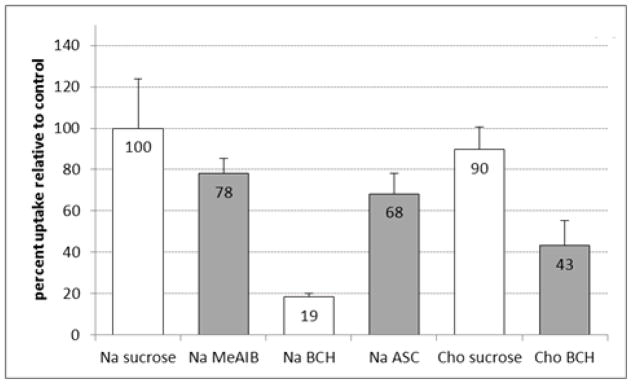
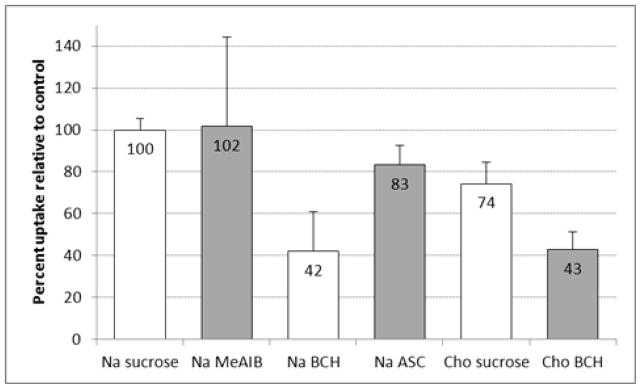
Figure 4a. In vitro uptake of [18F]9a by DBT glioma cells in the presence and absence of amino-acid transport inhibitors. The uptake data are normalized based on the amount of activity added to each well and the total amount of protein in each well. The data are expressed as percentage uptake relative to the sodium control condition, and the values for each condition are noted in the appropriate bars. To provide a consistent osmolarity compared to the inhibitory conditions, Na control and Cho controls contain 10 mM of sucrose. Na = assay buffer containing sodium ions; Cho = assay buffer containing choline ions; MeAIB = 10 mM N-methyl α-aminoisobutryic acid (system A inhibitor); BCH = 10 mM 2-aminobicyclo(2,2,1)-heptane-2-carboxylic acid (system L inhibitor).
Figure 4b. In vitro uptake of [18F]9b by DBT glioma cells in the presence and absence of amino-acid transport inhibitors. The uptake data are normalized based on the amount of activity added to each well and the total amount of protein in each well. The data are expressed as percentage uptake relative to the sodium control condition, and the values for each condition are noted in the appropriate bars. To provide a consistent osmolarity compared to the inhibitory conditions, Na control and Cho controls contain 10 mM of sucrose. Na = assay buffer containing sodium ions; Cho = assay buffer containing choline ions; MeAIB = 10 mM N-methyl α-aminoisobutryic acid (system A inhibitor); BCH = 10 mM 2-aminobicyclo(2,2,1)-heptane-2-carboxylic acid (system L inhibitor).
The majority of uptake of [18F]9a was significantly inhibited by 10 mM of the system L inhibitor BCH in both the presence and absence of sodium (81 ± 2 % and 57 ± 12 % inhibition, respective; p < 0.001). Replacing the sodium ions in the medium with sodium-free choline media did not significantly inhibit uptake of [18F]9a relative to control. The system A transport inhibitor MeAIB did not cause significant inhibition of uptake of [18F]9a, and a minor component of inhibition was observed by the mixture of the small, neutral amino acids Ala, Ser, and Cys (ASC) (32 ± 10%; p < 0.05). The other enantiomer, [18F]9b, was also inhibited by BCH in the presence and absence of sodium (58 ± 18 % and 57 ± 9 %, p < 0.05). The MeAIB and ASC inhibitor conditions as well as the use of sodium-free choline media did not lead to statistically significant inhibition of uptake of [18F]9b. These data are consistent with both [18F]9a and [18F]9b undergoing cellular uptake mediated primarily by system L transport in DBT cells in vitro, although minor contributions from other transport systems may be present as well.
Biodistribution in mice with intracranial DBT tumors
The results of biodistribution studies with each enantiomer of [18F]9 in mice with subcutaneous DBT tumors are shown in Table 1 and Table 2. In general, [18F]9a demonstrated superior tumor imaging properties compared to [18F]9b in terms of tumor uptake and retention. Additionally, the uptake of [18F]9a was higher and more persistent in the pancreas, a prototypic target organ for radiolabeled amino acids, compared to [18F]9b. Together, these data suggest that [18F]9a is a better substrate in vivo for amino acid transport than [18F]9b, although both compounds showed relatively rapid washout of tumor and normal tissues and lower brain availability than expected with system L amino acid transport substrates.
Table 1.
Biodistribution of [18F]9a in BALB/c mice with subcutaneous DBT tumors. Mice were anesthetized with 1% isoflurane/oxygen and 20–30 μCi (0.74 – 1.1 MBq) of [18F]9a was administered via tail vein injection. The animals were euthanized in groups of 4 animals at 5, 30 and 60 minutes post injection.
| 5 min | 30 min | 60 min | |
|---|---|---|---|
|
| |||
| blood | 4.53 ± 0.34 | 1.21 ± 0.17 | 0.28 ± 0.09 |
| bone | 1.54 ± 0.26 | 1.49 ± 1.68 | 0.44 ± 0.23 |
| brain | 0.33 ± 0.07 | 0.25 ± 0.04 | 0.09 ± 0.03 |
| fat | 0.89 ± 0.19 | 0.24 ± 0.09 | 0.17 ± 0.16 |
| heart | 3.09 ± 0.46 | 1.20 ± 0.15 | 0.30 ± 0.11 |
| kidney | 15.93 ± 1.57 | 4.39 ± 0.48 | 1.11 ± 0.34 |
| liver | 4.11 ± 0.19 | 1.12 ± 0.09 | 0.29 ± 0.11 |
| lung | 3.81 ± 0.19 | 1.06 ± 0.16 | 0.26 ± 0.10 |
| muscle | 2.26 ± 0.36 | 1.35 ± 0.27 | 0.52 ± 0.27 |
| pancreas | 21.03 ± 2.23 | 5.99 ± 1.60 | 1.64 ± 0.72 |
| salivary gland | 3.44 ± 0.31 | 0.92 ± 0.14 | 0.25 ± 0.10 |
| spleen | 3.79 ± 0.32 | 1.07 ± 0.15 | 0.28 ± 0.12 |
| thyroid | 2.93 ± 0.50 | 0.97 ± 0.13 | 0.35 ± 0.12 |
| tumor | 3.28 ± 0.82 | 2.17 ± 0.36 | 0.82 ± 0.37 |
The data are expressed as %ID/g with the exception of the thyroid which is expressed as %ID/organ. Error is expressed as standard deviation. n = 3 or 4 for each value.
Table 2.
Biodistribution of [18F]9b in BALB/c mice with subcutaneous DBT tumors. Mice were anesthetized with 1% isoflurane/oxygen and 20–30 μCi (0.74 – 1.1 MBq) of [18F]9b was administered via tail vein injection. The animals were euthanized in groups of 4 animals at 5, 30 and 60 minutes post injection.
| 5 min | 30 min | 60 min | |
|---|---|---|---|
|
| |||
| blood | 4.06 ± 0.35 | 0.44 ± 0.07 | 0.15 ± 0.04 |
| bone | 1.08 ± 0.11 | 0.42 ± 0.05 | 0.30 ± 0.06 |
| brain | 0.24 ± 0.08 | 0.08 ± 0.01 | 0.04 ± 0.01 |
| fat | 1.12 ± 0.32 | 0.17 ± 0.11 | 0.09 ± 0.04 |
| heart | 2.29 ± 0.16 | 0.67 ± 0.05 | 0.27 ± 0.15 |
| kidney | 18.09 ± 1.46 | 2.07 ± 0.37 | 0.55 ± 0.07 |
| liver | 4.12 ± 0.33 | 0.49 ± 0.04 | 0.17 ± 0.02 |
| lung | 3.66 ± 0.23 | 0.44 ± 0.07 | 0.13 ± 0.01 |
| muscle | 1.52 ± 0.17 | 0.66 ± 0.04 | 0.34 ± 0.09 |
| pancreas | 8.90 ± 0.96 | 1.08 ± 0.18 | 0.23 ± 0.07 |
| salivary gland | 2.29 ± 0.17 | 0.30 ± 0.03 | 0.10 ± 0.02 |
| spleen | 2.77 ± 0.14 | 0.93 ± 0.73 | 0.17 ± 0.03 |
| thyroid | 2.23 ± 0.29 | 0.51 ± 0.05 | 0.20 ± 0.03 |
| tumor | 2.05 ± 0.48 | 0.90 ± 0.16 | 0.42 ± 0.16 |
The data are expressed as %ID/g with the exception of the thyroid which is expressed as %ID/organ. Error is expressed as standard deviation. n = 3 or 4 for each value.
The tumor uptake of activity of [18F]9a after injection was rapid with 3.3 ± 0.8 %ID/g at 5 min. There was a relatively fast washout of the activity from the tumor with uptake values of 2.2 ± 0.4 %ID/g at the 30 min time point and 0.8 ± 0.4 %ID/g at 60 min. The tumor uptake of [18F]9b was lower at 5 min (2.1 ± 0.4 %ID/g, p = 0.04) and at 30 min (0.9 ± 0.2 %ID/g, p = 0.003) after injection with a trend towards lower uptake at 60 min (0.4 ± 0.2 %ID/g, p = 0.09) as well. Similarly, the normal brain uptake was higher with [18F]9a than with [18F]9b at 30 min postinjection (0.25 ± 0.04 versus 0.08 ± 0.01 %ID/g, p < 0.001) with a trend towards higher uptake with [18F]9a at 60 min as well (0.09 ± 0.03 versus 0.04 ± 0.01 %ID/g, p = 0.06). Tumor to brain ratios were high at 30 and 60 min postinjection for both compounds (9:1 to 10:1). However, the uptake of activity in the normal brain with both of these tracers was substantially lower than observed in this model with the well-characterized system L substrate [18F]FET (2.2 ± 0.4 %ID/g at 30 min and 1.8 ± 0.2%ID/g; unpublished data).
Outside the brain, [18F]9a demonstrated average tumor to tissue ratios of at least 1.6:1 at 30 and 60 min postinjection in all normal tissues studied except the kidneys and pancreas. The [18F]9b enantiomer showed similar results but lower %ID/g values and a greater degree of washout at 60 min after injection. High pancreatic and renal uptake is characteristic of many radiolabeled amino acids with varying mechanisms of transport. The relatively low levels of activity in the bone indicate that no substantial in vivo defluorination occurred during the 60 min time course of the study.
The low brain uptake and high tumor to brain ratios observed in vivo with both enantiomers of [18F]9 are not consistent with results observed with other system L substrates such as [18F]FET. This discrepancy between in vitro and in vivo results may be due to low affinity transport of [18F]9a and [18F]9b by system L which allows transport in the amino acid free in vitro setting but reduces uptake in vivo due to competition with endogenous amino acids. These results are surprising given the system L transport and good tumor imaging properties of a-methyl substituted amino acids like [123I]IMT, but the presence of the fluoro group in the β-position of [18F]9 may change the basicity of the amino group and transport properties of these compounds. Loss of recognition by system A amino acid transporters has been reported for di- and trifluoromethyl analogues of AIB. The reason for this difference between [18F]9a and [18F]9b is likely due in part to differential recognition of L- and D-isomers of amino acid by transport systems. Many transport systems including system L have higher rates of transport of substrates with the L-configuration at the α-carbon, although system L transporters also transport D-isomers of aromatic amino acids which have also been explored as tumor imaging agents. Our data suggest that the [18F]9a enantiomer may correspond to the L-configuration of standard aromatic amino acids based on its higher uptake by both tumors and normal brain tissue, but the absolute configurations of [18F]9a and [18F]9b were not determined. The relatively fast washout of both [18F]9a and [18F]9b may be due to rapid efflux from cells after initial uptake, although this hypothesis has not been tested.
MicroPET studies in mice with intracranial DBT tumors
The results of microPET corroborate the distinct uptake and kinetic patterns observed in the biodistribution studies with [18F]9a and [18F]9b in intracranial DBT tumors and in normal brain. Time activity curves showing average SUVs and tumor to brain ratios obtained with [18F]9a and [18F]9b are presented in Figures 5 and 6, and representative microPET/CT images are shown in Figure 7. Overall, [18F]9a possessed better tumor imaging properties, although the amount of uptake in normal brain was lower than expected for system L substrates.
Figure 5.
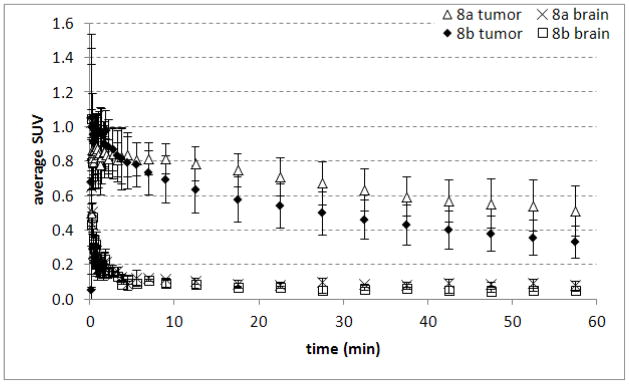
Time-activity curves from intracranial DBT tumors and contralateral normal brain obtained with microPET after the injection of [18F]9a and [18F]9b. Mice were anesthetized with 1% isoflurane/oxygen and 200–300 μCi (7.4 – 11 MBq) of [18F]9a or [18F]9b was administered via tail vein injection at day 14 after tumor implantation. The data are displayed as average SUVs, and each time point represents the mean of 3 animals with standard deviation. Dynamic scanning time is 0–60 min.
Figure 6.
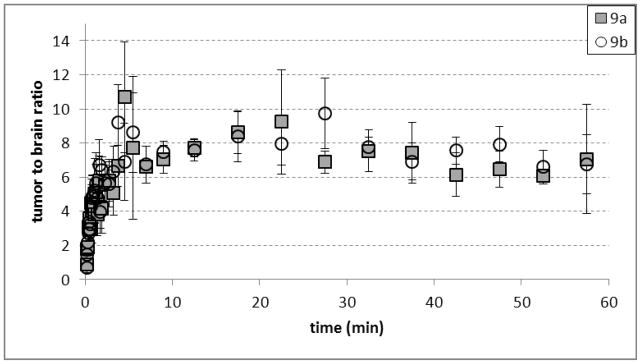
Average tumor-to-brain ratios observed in microPET studies with [18F]9a and [18F]9b. The data were obtained in 3 mice with intracranial DBT tumors. Mice were anesthetized with 1% isoflurane/oxygen and 200–300 μCi (7.4 – 11 MBq) of [18F]9a or [18F]9b was administered via tail vein injection at day 14 after tumor implantation using INVEON and MicroPET Focus 220 systems. Dynamic scanning time is 0–60 min.
Figure 7.
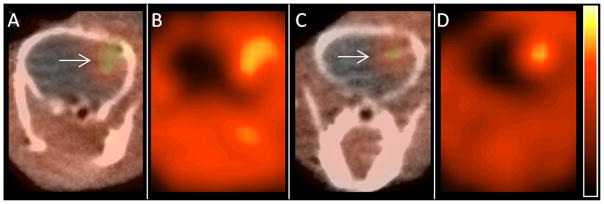
Representative microPET images obtained with [18F]9a and [18F]9b. Images A (PET/CT) and B (PET only) were acquired using [18F]9a, and images C (PET/CT) and D (PET only) were acquired with [18F]9b. The location of the tumor is designated on panels A and C with white arrows. Mice were anesthetized with 1% isoflurane/oxygen, and 200–300 μCi (7.4 – 11 MBq) of [18F]9a or [18F]9b was administered via tail vein injection approximately 2 weeks after tumor implantation. Images were acquired using INVEON and MicroPET Focus 220 systems. Dynamic scanning was performed for 0–60 min, and the displayed images are from summed data at 45–60 min after injection.
The TACs for both [18F]9a and [18F]9b demonstrated that the peak tumor uptake of radioactivity occurred within 10 min after injection followed by washout from tumor and normal brain over the remainder of the 60 min study. The brain uptake of both [18F]9a and [18F]9b also peaked within 1 min after injection and decreased rapidly over time. Tumor visualization was possible with both two enantiomers, although the absolute amount of uptake in tumor tissue was approximately 40–50% higher with [18F]9a at 30–60 min after injection. The tumor to brain ratios were similar for both compounds and on the order of 7:1 in the later portions of the microPET studies. These tumor to brain ratios were somewhat lower than those observed in the biodistribution studies which may be due to partial volume averaging in the brain tumors. As in the biodistribution studies, the very low uptake in normal brain is not consistent with system L transport and may limit the use of these tracers for visualizing the non-enhancing portions of gliomas. These findings are surprising given the brain availability and good tumor imaging properties of α-methyl substituted aromatic amino acids such as [123I]IMT and 3-[18F]fluoro-α-methyl-L-phenylalanine (FMT). These results also suggest α-fluoromethyl substituted aromatic amino acids may be not be suitable lead compounds for developing radiolabeled amino acids targeting system L amino acid transport which may be related to the electronegativity of the fluoro substituent.
Conclusions
The new 18F-labeled phenylalanine structural analogue, [18F]FMePhe 9, has been prepared in good radiochemical yield using cyclic sulfamidate precursors, and the individual enantiomers have been separated through chiral HPLC for biological evaluation. In vitro cell uptake studies demonstrate that both of these isomers are substrates for system L type amino acid transport as expected from their structures. Biodistribution studies demonstrated better tumor imaging properties with the first eluting enantiomer [18F]9a, with tumor to brain ratios of approximately 10:1 at 30 and 60 min after injection and good tumor to normal tissue ratios outside the brain. Both enantiomers of FMePhe had relatively low brain uptake and relatively rapid washout from both tumor and normal brain tissues. The low brain uptake of these tracers suggests that they are relatively poor system L substrates in vivo which may be due to the β-fluoro substituent may limit their utility for brain tumor imaging.
Acknowledgments
The authors thank the personnel of the Washington University Cyclotron Facility for [18F]fluoride production. We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, Mo., for the use of the Small Animal Cancer Imaging Core, which provided biodistribution and microPET services. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA91842. This research was funded by the National Cancer Institute (K08CA154790) and the Mallinckrodt Institute of Radiology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langen KJ, Pauleit D, Coenen HH. 3-[123I]Iodo-alpha-methyl-L-tyrosine: uptake mechanisms and clinical applications. Nucl Med Biol. 2002;29:625–31. doi: 10.1016/s0969-8051(02)00328-1. [DOI] [PubMed] [Google Scholar]
- 2.Langen KJ, Hamacher K, Weckesser M, Floeth F, Stoffels G, Bauer D, et al. O-(2-[18F]fluoroethyl)-L-tyrosine: uptake mechanisms and clinical applications. Nucl Med Biol. 2006;33:287–94. doi: 10.1016/j.nucmedbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Sutinen E, Jyrkkio S, Alanen K, Nagren K, Minn H. Uptake of [N-methyl-11C]alpha-methylaminoisobutyric acid in untreated head and neck cancer studied by PET. Eur J Nucl Med Mol Imaging. 2003;30:72–7. doi: 10.1007/s00259-002-1010-3. [DOI] [PubMed] [Google Scholar]
- 4.McConathy J, Yu W, Jarkas N, Seo W, Schuster DM, Goodman MM. Radiohalogenated nonnatural amino acids as PET and SPECT tumor imaging agents. Med Res Rev. 2011;26:20250. doi: 10.1002/med.20250. [DOI] [PubMed] [Google Scholar]
- 5.Qu W, Zha Z, Ploessl K, Lieberman BP, Zhu L, Wise DR, et al. Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J Am Chem Soc. 2011;133:1122–33. doi: 10.1021/ja109203d. [DOI] [PubMed] [Google Scholar]
- 6.McConathy J, Zhou D, Shockley SE, Jones LA, Griffin EA, Lee H, et al. Click Synthesis and Biologic Evaluation of (R)- and (S)-2-Amino-3-[1-(2-[18F]fluoroethyl)-1H-[1,2,3]Triazol-4-yl]Propanoic Acid for Brain Tumor Imaging with Positron Emission Tomography. Mol Imaging. 2010;9:329–42. [PubMed] [Google Scholar]
- 7.Weber WA, Wester HJ, Grosu AL, Herz M, Dzewas B, Feldmann HJ, et al. O-(2-[18F]fluoroethyl)-L-tyrosine and L-[methyl-11C]methionine uptake in brain tumours: initial results of a comparative study. Eur J Nucl Med. 2000;27:542–9. doi: 10.1007/s002590050541. [DOI] [PubMed] [Google Scholar]
- 8.Lau EWF, Drummond KJ, Ware RE, Drummond E, Hogg A, Ryan G, et al. Comparative PET study using F-18 FET and F-18 FDG for the evaluation of patients with suspected brain tumour. Journal of Clinical Neuroscience. 2010;17:43–9. doi: 10.1016/j.jocn.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Pauleit D, Floeth F, Tellmann L, Hamacher K, Hautzel H, Muller HW, et al. Comparison of O-(2-18F-fluoroethyl)-L-tyrosine PET and 3-123I-iodo-alpha-methyl-L-tyrosine SPECT in brain tumors. J Nucl Med. 2004;45:374–81. [PubMed] [Google Scholar]
- 10.Hellwig D, Romeike B, Ketter R, Moringlane J, Kirsch C-M, Samnick S. Intra-individual comparison of p-[123I]-iodo-L-phenylalanine and L-3-[123I]-iodo-α-methyl-tyrosine for SPECT imaging of gliomas. European Journal of Nuclear Medicine and Molecular Imaging. 2008;35:24–31. doi: 10.1007/s00259-007-0514-2. [DOI] [PubMed] [Google Scholar]
- 11.Pauleit D, Floeth F, Herzog H, Hamacher K, Tellmann L, Muller HW, et al. Whole-body distribution and dosimetry of O-(2-[18F]fluoroethyl)-L-tyrosine. Eur J Nucl Med Mol Imaging. 2003;30:519–24. doi: 10.1007/s00259-003-1118-0. [DOI] [PubMed] [Google Scholar]
- 12.Jeong SY, Lim SM. Comparison of 3′-deoxy-3′-[(18)F]fluorothymidine PET and O-(2-[(18)F]fluoroethyl)-L-tyrosine PET in patients with newly diagnosed glioma. Nucl Med Biol. 2012;39:977–81. doi: 10.1016/j.nucmedbio.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Kersemans K, Mertens J, Caveliers V. Radiosynthesis of 4-[F-18]fluoromethyl-L-phenylalanine and [F-18]FET via a same strategy and automated synthesis module. Journal of Labelled Compounds & Radiopharmaceuticals. 2010;53:58–62. [Google Scholar]
- 14.Wang L, Lieberman BP, Plossl K, Qu W, Kung HF. Synthesis and comparative biological evaluation of L- and D-isomers of 18F-labeled fluoroalkyl phenylalanine derivatives as tumor imaging agents. Nucl Med Biol. 2011;38:301–12. doi: 10.1016/j.nucmedbio.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 2005;15:254–66. doi: 10.1016/j.semcancer.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Li R, Younes M, Frolov A, Wheeler TM, Scardino P, Ohori M, et al. Expression of neutral amino acid transporter ASCT2 in human prostate. Anticancer Res. 2003;23:3413–8. [PubMed] [Google Scholar]
- 17.Shennan DB, Thomson J. Inhibition of system L (LAT1/CD98hc) reduces the growth of cultured human breast cancer cells. Oncol Rep. 2008;20:885–9. [PubMed] [Google Scholar]
- 18.Nawashiro H, Otani N, Shinomiya N, Fukui S, Ooigawa H, Shima K, et al. L-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors. Int J Cancer. 2006;119:484–92. doi: 10.1002/ijc.21866. [DOI] [PubMed] [Google Scholar]
- 19.McConathy J, Martarello L, Malveaux EJ, Camp VM, Simpson NE, Simpson CP, et al. Radiolabeled amino acids for tumor imaging with PET: radiosynthesis and biological evaluation of 2-amino-3-[18F]fluoro-2-methylpropanoic acid and 3-[18F]fluoro-2-methyl-2-(methylamino)propanoic acid. J Med Chem. 2002;45:2240–9. doi: 10.1021/jm010241x. [DOI] [PubMed] [Google Scholar]
- 20.Yu W, McConathy J, Williams L, Camp VM, Malveaux EJ, Zhang Z, et al. Synthesis, radiolabeling, and biological evaluation of (R)- and (S)-2-amino-3-[(18)F]fluoro-2-methylpropanoic acid (FAMP) and (R)- and (S)-3-[(18)F]fluoro-2-methyl-2-N-(methylamino)propanoic acid (NMeFAMP) as potential PET radioligands for imaging brain tumors. J Med Chem. 2010;53:876–86. doi: 10.1021/jm900556s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gazzola GC, Dall’Asta V, Franchi-Gazzola R, White MF. The cluster-tray method for rapid measurement of solute fluxes in adherent cultured cells. Anal Biochem. 1981;115:368–74. doi: 10.1016/0003-2697(81)90019-1. [DOI] [PubMed] [Google Scholar]
- 22.Jost SC, Collins L, Travers S, Piwnica-Worms D, Garbow JR. Measuring brain tumor growth: combined bioluminescence imaging-magnetic resonance imaging strategy. Mol Imaging. 2009;8:245–53. [PMC free article] [PubMed] [Google Scholar]
- 23.Jost SC, Wanebo JE, Song SK, Chicoine MR, Rich KM, Woolsey TA, et al. In vivo imaging in a murine model of glioblastoma. Neurosurgery. 2007;60:360–70. doi: 10.1227/01.NEU.0000249264.80579.37. discussion 70-1. [DOI] [PubMed] [Google Scholar]
- 24.Benrabh H, Lefauconnier JM. Blood-endothelial cell and blood-brain transport of L-proline, alpha-aminoisobutyric acid, and L-alanine. Neurochem Res. 1996;21:1227–35. doi: 10.1007/BF02532400. [DOI] [PubMed] [Google Scholar]
- 25.Stout DB, Huang SC, Melega WP, Raleigh MJ, Phelps ME, Barrio JR. Effects of large neutral amino acid concentrations on 6-[F-18]Fluoro-L-DOPA kinetics. J Cereb Blood Flow Metab. 1998;18:43–51. doi: 10.1097/00004647-199801000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Christensen HN, Oxender DL. The Acid Strength of the Amino Group as a Factor in the Transport of Amino Acids. Biochim Biophys Acta. 1963;74:386–91. doi: 10.1016/0006-3002(63)91382-9. [DOI] [PubMed] [Google Scholar]
- 27.Tsukada H, Sato K, Fukumoto D, Nishiyama S, Harada N, Kakiuchi T. Evaluation of D-isomers of O-11C-methyl tyrosine and O-18F-fluoromethyl tyrosine as tumor-imaging agents in tumor-bearing mice: comparison with L- and D-11C-methionine. J Nucl Med. 2006;47:679–88. [PubMed] [Google Scholar]
- 28.Tsukada H, Sato K, Fukumoto D, Kakiuchi T. Evaluation of D-isomers of O-18F-fluoromethyl, O-18F-fluoroethyl and O-18F-fluoropropyl tyrosine as tumour imaging agents in mice. Eur J Nucl Med Mol Imaging. 2006;33:1017–24. doi: 10.1007/s00259-006-0076-8. [DOI] [PubMed] [Google Scholar]
- 29.Kersemans V, Cornelissen B, Bacher K, Kersemans K, Thierens H, Dierckx RA, et al. In vivo evaluation and dosimetry of 123I-2-iodo-D-phenylalanine, a new potential tumor-specific tracer for SPECT, in an R1M rhabdomyosarcoma athymic mouse model. J Nucl Med. 2005;46:2104–11. [PubMed] [Google Scholar]
- 30.Langen KJ, Broer S. Molecular transport mechanisms of radiolabeled amino acids for PET and SPECT. J Nucl Med. 2004;45:1435–6. [PubMed] [Google Scholar]
- 31.Inoue T, Shibasaki T, Oriuchi N, Aoyagi K, Tomiyoshi K, Amano S, et al. 18F alpha-methyl tyrosine PET studies in patients with brain tumors. J Nucl Med. 1999;40:399–405. [PubMed] [Google Scholar]